Reactions of hydroxyl radicals with trichloroethene and tetrachloroethene in argon matrices at 12 K
Karen S. Wiltshire, Matthew J. Almond and Philip C. H. Mitchell
School of Chemistry, University of Reading, Whiteknights, Reading, UK RG6 6AD
First published on 2nd December 2003
Abstract
Irradiation of argon matrices at 12 K containing hydrogen peroxide and tetrachloroethene using the output from a medium-pressure mercury lamp gives rise to the carbonyl compound trichloroacetyl chloride (CCl3CClO). Similarly trichloroethene gives dichloroacetyl chloride (CCl2HCClO) – predominantly in the gauche form – under the same conditions. It appears that the reaction is initiated by homolysis of the O–O bond of H2O2 to give OH radicals, one of which adds to the double bond of an alkene molecule. The reaction then proceeds by abstraction of the H atom of the hydroxyl group and Cl-atom migration. This mechanism has been explored by the use of DFT calculations to back up the experimental findings. The mechanism is analogous to that shown by the simple hydrocarbon alkenes.
Introduction
The chemistry of the troposphere is dominated by the oxidation of hydrocarbons and during daytime the hydroxyl radical is the most important oxidant. It is formed by photolysis of ozone to yield O(1D) atoms which then react with water to give hydroxyl radicals:-1–4O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +hν→O(1D)+O2 +hν→O(1D)+O2O(1D) |
Compared to the information available for simple alkenes much less is known about the chloroalkenes. However, these man-made pollutants have been identified in urban areas and have several industrial and non-industrial sources. They have been found in samples taken from points adjacent to a municipal incinerator, a waste collection centre and a sewage treatment plant.9 Gas-phase measurements show that the initial step of the reaction is analogous to that of the hydrocarbon alkenes i.e. addition of the OH radical to give an energy-rich OH-haloalkene complex,3 which may either decompose back to reactants, or be collisionally stabilised to give the hydroxyalkyl radical:-
OH+CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCl=HOC2H3Cl* CHCl=HOC2H3Cl*HOC2H3Cl* |
| HOC2H3Cl*→H2CCH(OH)+Cl. |
Decomposition of the hydroxychloroalkyl radicals certainly occurs in the troposphere but the mechanisms are uncertain. The reactions probably proceed by further oxidation.3 Product analysis has shown that HC(O)Cl is formed in the reaction of vinyl chloride or trichloroethene and OH radicals, and that phosgene (COCl2) is formed in the reaction of trichloroethene or tetrachloroethene and OH radicals.10 These C1 products are unlikely to be primary products, however. Accordingly, we have carried out a study of the reactions of trichloroethene and tetrachloroethene with hydroxyl radicals in argon matrices at 12 K. In this way we hoped to identify products of these reactions and hence to learn something of the reaction mechanisms. In particular we wished to search for and identify chlorine-containing carbonyl products and to obtain evidence that, like the hydrocarbon alkenes, the reaction within the matrix is initiated by addition of the OH radical to the double bond of the alkene.
Experimental section
The matrix-isolation apparatus used at Reading has been described in detail elsewhere.11,12 The experiments were carried out in a very similar manner to previous experiments on hydrocarbon alkenes.6 Samples of trichloroethene (Aldrich, 99.5% pure), tetrachloroethene (Aldrich, >99% pure), dichloroacetyl chloride (Aldrich, 99% pure) and trichloroacetyl chloride (Aldrich, 99% pure) were used as supplied after freeze–pump–thaw purification. Gas phase mixtures of the alkene under investigation with argon were made up on a vacuum line using standard manometric techniques. Hydrogen peroxide vapour was obtained from the H2O2–urea adduct13 and was co-condensed with the alkene/argon mixture. Matrices were photolysed using the broad-band ultraviolet output from a medium-pressure mercury lamp. It has been shown elsewhere that this method is an efficient route to generate OH radicals.5–8,13,14 Spectra were recorded on a Perkin-Elmer 983 dispersive spectrophotometer with a typical resolution of 2 cm−1 or better. Data were saved onto a Perkin-Elmer data station, transferred to a computer and manipulated using GRAMS software.Calculations
DFT calculations were carried out using the Gaussian 98 computational package15 with the 6-31G(d) basis sets and the B3-LYP functional. There were no imaginary frequencies generated by any of the calculations.Results and discussion
When a hydrogen peroxide-doped argon matrix (at 12 K) initially containing ca. 1% of tetrachloroethene was subjected to broad-band UV-visible photolysis for a period of 2 h the infrared bands of tetrachloroethene16,17 (916 (vs) 781 (vs) cm−1 with weaker features at 898 (m), 811 (m) and 760 (m) cm−1) and of hydrogen peroxide13 (3582 (s) and 1291 (s) cm−1) were seen to decay. At the same time new features appeared which may be assigned to H2O18–21 (3590 and 1624 cm−1), CO222–24 (2342 and 665 cm−1) and CO25,26 (2143 cm−1). Bands were also observed at 1809, 870 and 852 cm−1. These are assigned to the product trichloroacetyl chloride CCl3CClO.8,15,16,27In order to confirm that the product of this reaction is indeed tricholoroacetyl chloride an authentic sample of this compound was isolated at 1% concentration in an argon matrix at 12 K and the infrared spectrum was recorded. In Table 1 are compared the infrared spectra of the matrix-isolated and liquid27 compound. Unfortunately only three absorptions were observed for the product of the matrix reaction of C2Cl4 with OH radicals. However, these correspond well to the two strongest bands of matrix-isolated CCl3ClO (see Table 1). The band at 1809 is assigned to the ν(CO) vibration and those at 870 and 852 to a matrix-split band (see Fig. 1) arising from the antisymmetric stretch of the CCl3 moiety.27 The match between these spectra, although not entirely unambiguous, does suggest that CCl3ClO is produced by the reaction of C2Cl4 with OH radicals in an argon matrix. When photolysis was continued for further periods of 2 h and 17 h, the spectral changes reported above continue in that bands of the reactants decayed while those of the product trichloroacetyl chloride and of H2O, CO2 and CO increased. Following photolysis the matrix was annealed to 40 K and re-cooled to 12 K for 5 cycles. This had the effect of sharpening the bands but did not have any noticeable effect on their intensities. The behaviour of the spectral features upon photolysis and annealing is detailed in Table 2 and the spectra are illustrated in Fig. 1.
| CCl3ClO (liquid) (ref. 27)a | CCl3ClO (Ar matrix) (this work)b | Assignment |
|---|---|---|
| a No band intensities are given in ref. 27.b s=strong, m=medium, w=weak, v=very. | ||
| 1800 | 1800 (vs) | ν(CO) |
| 1020 | 1023 (ms) | ν(C–C) |
| 850 | 857 (vs) | νas (CCl3) |
| 800 | 792 (s) | νas(CCl3) |
| 735 | 745 (ms) | ν(C–Cl) |
| 620 | 623 (ms) | ρ(CO) |
| 512 | 513 (ms) | δ(OC–Cl) |
| 428 | 427 (m) | νsym(CCl3) |
| 362 | 360 (m) | δ(C–CO) |
| 284 | 278 (w) | δ(CCl3) |
| ν/cm−1 | hν 2 h | hν 4 h | hν 21 h | Anneal | Origin | Assignment |
|---|---|---|---|---|---|---|
| a Definition of symbols: *=appears, ↑=increases in intensity, ↓=decreases in intensity, –=unchanged intensity. | ||||||
| 3590 | * | ↑ | ↑ | – | H2O | ν(OH) |
| 3582 | ↓ | ↓ | ↓ | – | H2O2 | ν(OH) |
| 2342 | * | ↑ | ↑ | – | CO2 | νas(OCO) |
| 2143 | * | ↑ | ↑ | – | CO | ν(CO) |
| 1809 | * | ↑ | ↑ | – | CCl3ClO | ν(CO) |
| 1624 | * | ↑ | ↑ | – | H2O | δ(OH) |
| 1291 | ↓ | ↓ | ↓ | – | H2O2 | ν(O–O) |
| 916 | ↓ | ↓ | ↓ | – | C2Cl4 | ν(C–Cl) |
| 898 | ↑ | – | ↑ | – | CCl3ClO+C2Cl4 | ν(C–Cl) |
| 870 | * | ↑ | ↑ | – | CCl3ClO | ν(C–Cl) |
| 852 | * | ↑ | ↑ | – | CCl3ClO | ν(C–Cl) |
| 811 | ↓ | ↓ | ↓ | – | C2Cl4 | ν(C–Cl) |
| 781 | ↓ | ↓ | ↓ | – | C2Cl4 | ν(C–Cl) |
| 760 | ↓ | ↓ | ↓ | – | C2Cl4 | ν(C–Cl) |
| 665 | * | ↑ | ↑ | – | CO2 | δ(OCO) |
 | ||
| Fig. 1 The region 2000–500 cm−1 of the absorbance infrared spectra of an argon matrix (12 K) initially containing tetrachloroethene and hydrogen peroxide (C2Cl4/H2O2/Ar=1∶1∶100), after (a) deposition, (b) 2 h, (c) 4 h, (d) 21 h broad-band UV-visible photolysis using a medium-pressure mercury lamp, and (e) 5×annealing to 40 K, then re-cooling to 12 K. Symbols: t=tetrachloroethene, h=H2O2, and *=new band. | ||
We have previously carried out similar experiments where hydrogen peroxide is photolysed in matrices containing ethene or other simple alkenes.6–8 It was shown that under the conditions of these experiments photolysis of H2O2 proceeds via O–O bond rupture to yield OH radicals.5–8 Reaction of the OH radical with the alkene substrate yields a hydroxyalkyl radical which may suffer one of two fates. Upon annealing, reaction with a second OH radical in the matrix cage gives the diol, whereas prolonged photolysis causes decomposition of the radical to form a carbonyl product. It seems reasonable to suppose that a similar mechanism (Scheme 1) operates in the case of tetrachloroethene. In this case, however, there is no sign of any bands belonging to a radical intermediate, nor were any major changes detected upon annealing the matrix. It is pertinent, therefore, to discuss briefly why the two systems may behave rather differently.
 | ||
| Scheme 1 Proposed pathway for the reaction of tetrachloroethene with hydrogen peroxide in an argon matrix at 12 K upon irradiation using a medium-pressure mercury lamp. | ||
First is the possibility that it is the absorption properties of the radicals that determines their lifetime within the matrix. Addition of chlorine atoms to a saturated organic compound typically red-shifts the absorbance maximum by some 5–10 nm,16 making photolysis, under the conditions of our experiments, more efficient. We found it impossible to verify this point by specifically excluding certain irradiation wavelengths by using filters. This was because of the long photolysis times required, which become impossibly long when filters are employed and the light intensity is substantially reduced. A second important point concerns the stability of the radical intermediates. Chloroalkenes with one or more vinylic chlorine atoms are stabilised by a mesomeric interaction between the chlorine atom and the CC double bond. When hydroxyl radicals react with the chloroalkene this mesomeric interaction is lost. This destabilisation is reflected in the lower rate constants for the reaction of OH radicals with steadily more chlorinated alkenes as shown in Table 3.3,28,29 Once a carbonyl product is formed, provided an acyl chlorine atom is present, a mesomeric effect with the carbonyl double bond is possible. Thus the radical intermediate is destabilised with respect to both reactant alkene and product carbonyl. Finally it is known that chlorine atom migration in a system like this is more efficient than hydrogen atom migration because a chlorine atom readily forms a cyclical transition state between the two carbon atoms.10 These factors probably all contribute to the destabilisation of the hydroxychloroalkyl radical with respect to the carbonyl product and suggest why the radical was not seen in the matrix in this case.
| Alkene | k/cm3 molecule−1 s−1 |
|---|---|
| Ethene | 8.52×10−12 |
| Chloroethene | 6.60×10−12 |
| 1,1-Dichloroethene | 1.12×10−12 |
| cis-1,2-Dichloroethene | 2.71×10−12 |
| trans-1,2-Dichloroethene | 2.50×10−12 |
| Trichloroethene | 2.35×10−12 |
| Tetrachloroethene | 1.55×10−12 |
One important feature to emerge from our experiments with tetrachloroethene is that no product containing a C–H bond was formed. This counts against an alternative mechanism in which the carbonyl product is formed by Cl-atom elimination or abstraction from the hydroxychloroalkyl radical intermediate. This would yield an enol which could rearrange, upon photolysis, to give a keto product, but in this case the keto product would contain a C–H group:-
| HOCCl2CCl2→HOClCCCl2+Cl HOClC |
It is of interest to note that CCl3CClO has previously been detected on oxidation of C2Cl4 in gaseous mixtures containing O2 and NO.30 Under these conditions it was suggested that this reaction proceeds by addition of a Cl atom to C2Cl4 to yield the C2Cl5 radical which adds to O2. Subsequent loss of an O (by oxidation of NO to NO2) and a Cl atom yields CCl3CClO:-
| Cl+CCl2CC2→CCl3CCl2 CCl3CCl2 CCl3CCl2O2 |
We next turned our attention to the reaction of tricholorethene with hydroxyl radicals. Here the reaction is potentially more complicated because there are two possible radical intermediates and two possible carbonyl products. The possible reaction pathways are shown in Scheme 2.
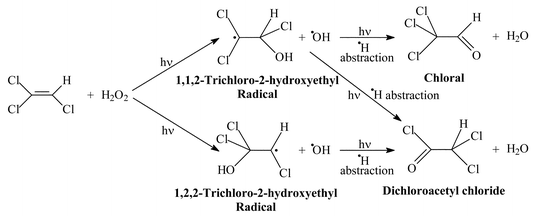 | ||
| Scheme 2 Proposed pathways for the reaction of trichloroethene with hydrogen peroxide in an argon matrix at 12 K upon irradiation using a medium-pressure mercury lamp. | ||
Two hours' irradiation of a hydrogen peroxide-doped argon matrix at 12 K containing ca. 1% of trichloroethene led to a reduction in the intensity of the bands of hydrogen peroxide13 (3583 and 1291 cm−1) and of tricholoroethene16,17 (3094, 1593, 936, 846, 784 and 632 cm−1). At the same time new bands appeared (alongside those of H2O, CO2 and CO) at 2792, 1804, 993, 766 and 587 cm−1. Prolonged photolysis for a total period of 4 or 21 h caused product bands to increase further in intensity. Annealing to 40 K and recooling to 12 K had little effect other than to sharpen the bands slightly. The spectral changes are illustrated in Figs. 2 and 3 and the behaviour of the bands upon photolysis is listed in Table 4.
 | ||
| Fig. 2 Difference of the infrared spectrum of an argon matrix (12 K) initially containing trichloroethene and hydrogen peroxide (C2Cl4/H2O2/Ar=1∶1∶100) after 21 h of photolysis minus the spectrum of the same matrix after deposition but before photolysis. | ||
 | ||
| Fig. 3 The region 2000–500 cm−1 of the absorbance infrared spectra of an argon matrix (12 K) initially containing trichloroethene and hydrogen peroxide (C2Cl3H/H2O2/Ar=1∶1∶100), after (a) deposition, (b) 2 h, (c) 4 h, (d) 21 h broad-band UV-visible photolysis using a medium-pressure mercury lamp, and (e) annealing to 40 K, then re-cooling to 12 K. Symbols: t=trichloroethene, h=H2O2, and *=new band. | ||
| ν/cm−1 | hν 2 h | hν 4 h | hν 21 h | Anneal | Origin | Assignment |
|---|---|---|---|---|---|---|
| a Definition of symbols: *=appears, ↑=increases in intensity, ↓=decreases in intensity, –=unchanged intensity. | ||||||
| 3583 | ↓ | ↓ | ↓ | – | H2O2 | ν(OH) |
| 3398 | * | ↑ | ↑ | – | H2O | ν(OH) |
| 3094 | ↓ | ↓ | ↓ | – | C2Cl3H | ν(CH) |
| 2792 | * | ↑ | ↑ | – | C2Cl3HO | ν(CH) |
| 2342 | * | ↑ | ↑ | – | CO2 | νas(OCO) |
| 2142 | * | ↑ | ↑ | – | CO | ν(CO) |
| 1804 | * | ↑ | ↑ | – | C2Cl3HO | ν(CO) |
| 1602 | * | ↑ | ↑ | – | H2O | δ(OH) |
| 1593 | ↓ | ↓ | ↓ | – | C2Cl3H | δ(CH) |
| 1291 | ↓ | ↓ | ↓ | – | H2O2 | ν(OO) |
| 993 | * | ↑ | ↑ | – | C2Cl3HO | ν(CCl) |
| 936 | ↓ | ↓ | ↓ | – | C2Cl3H | ν(CCl) |
| 846 | ↓ | ↓ | ↓ | – | C2Cl3H | ν(CCl) |
| 784 | ↓ | ↓ | ↓ | – | C2Cl3H | ν(CCl) |
| 766 | * | ↑ | ↑ | – | C2Cl3HO | ν(CCl) |
| 661 | * | ↑ | ↑ | – | CO2 | δ(OCO) |
| 632 | ↓ | ↓ | ↓ | – | C2Cl3H | δ(CCl) |
| 587 | * | ↑ | ↑ | – | C2Cl3HO | δ(CCl) |
The new bands seen upon photolysis clearly belong to a carbonyl product. Assuming that the reaction mechanism is analogous to that of the hydrocarbon alkenes or of tetrachloroethene, the question is, is this product chloral or dicholoroacetyl chloride? In order to attempt to answer this question an authentic sample of dichloroacetyl chloride was isolated at 1% concentration in an argon matrix. Bands were observed for this matrix-isolated compound at 2801 (s), 1815 (vs), 1784 (vs), 1083 (s), 993 (s), 800 (s), 786 (s), 756 (vs) and 583 (s) cm−1. Comparison with literature data for the gaseous and liquid compound show that both syn and gauche forms of this molecule have been isolated. The bands at 2801, 1815, 993, 800, 756 and 583 cm−1 may be assigned to the gauche form, while those at 2801, 1784, 1083 and 786 cm−1 arise from the syn conformer.31 When these spectra are compared with the spectrum obtained by photolysing C2Cl3H and H2O2 in an argon matrix it is suggested that the gauche form of dichloroacetyl chloride is formed predominantly under these conditions. All of the product bands match well with observed bands of gauche-CCl2HCClO (see Table 5).
| CHCl2CClO (ν/cm−1) gauchea | OH+C2HCl3 product (ν/cm−1) | Δb |
|---|---|---|
| a s=strong, m=medium, w=weak, v=veryb Δ is the percentage difference in wavenumber between the position of a band of the OH+C2HCl3 photoproduct and of the corresponding band of the authentic sample of CHCl2CClO.c Band not observed: probably obscured by absorption of C2Cl3H. | ||
| 2801 (s) | 2792 (s) | 0.321 |
| 1815 (vs) | 1804 (s) | 0.606 |
| 993 (s) | 993 (ms) | 0.000 |
| 800 (s) | –c | – |
| 756 (vs) | 766 (ms) | 1.32 |
| 583 (s) | 587 (wm) | 0.686 |
There is no obvious match between our product spectrum and the reported spectrum of chloral (CCl3CHO).32,33 In particular the reported position of ν(CO) for chloral vapour (1778 cm−1) or CCl4 solution (1768 cm−1) does not match well with our observed product band at 1804 cm−1. Other strong bands of chloral vapour are seen at 1030, 987, 857 and 739 cm−1. The band at 987 cm−1 matches one of our product bands quite well but the others do not have any obvious counterparts. It therefore appears that the product of reaction of trichloroethene with OH radicals in an argon matrix is dichloroacetyl chloride, predominantly in the gauche form. We have no detailed mechanistic explanation as to why the gauche form should be favoured but we note that this is the lower-energy form by some 3.5 kJ mol−1.31
DFT calculations were carried out in order to identify the most energetically favourable species with the lowest value of ΔE. Table 6 gives the energy (enthalpy) values obtained for reactants and possible products. Zero point energies were added to all values. The energy values for the various possible reaction steps are illustrated by the diagram in Scheme 3.
| Species | E/Eh | E/kJ mol−1 | ΔE/kJ mol−1a |
|---|---|---|---|
| a Energy changes for reaction steps – see Scheme 3.b Energy of C2Cl3H+H2O2 set at zero.c Includes energy of OH.d Includes energy of H2O. | |||
| C2Cl3H | −1457.3380 | −3826240 | 0b |
| H2O2 | −151.5034 | −397772 | 0b |
| CCl2CClHOH | −1533.0693 | −4025073 | −150.7c |
| CClHCCl2OH | −1533.0794 | −4025100 | −124.2c |
| OH | −75.7147 | −198789 | – |
| CCl3CHO | −1532.5480 | −4023704 | −245.5d |
| CCl2HCClO | −1532.5702 | −4023763 | −303.8d |
| H2O | −76.3869 | −200554 | – |
| CO2 | −188.5429 | −495019 | – |
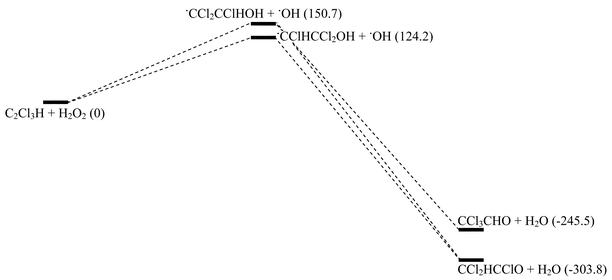 | ||
| Scheme 3 Energy (kJ mol−1) diagram for the reaction of trichloroethene with hydrogen peroxide calculated using the B3-LYP functional with the 6-31G(d) basis set. Numbers in parentheses are the energy changes for the reactions indicated. | ||
These calculations serve to back up the experimental findings in that dichloroacetyl chloride and water are calculated to be the most stable reaction products. The overall energy value of this pathway is −303.8 kJ mol−1. The first step of the reaction, the formation of a hydroxyalkyl radical, is strongly exothermic. The energy to overcome this step is provided by photolysis of the reaction mixture. It is calculated that the 1,2,2-trichloro-2-hydroxyethyl radical is favoured over the isomeric 1,1,2-trichloro-2-hydroxyethyl radical. This is perhaps somewhat surprising in that it might be expected that the radical with the unpaired electron on the CCl2 moiety would be the more stable. The 1,2,2-trichloro-2-hydroxyethyl radical proceeds to the product dichloroacetyl chloride by H-atom abstraction and migration of a Cl atom. The alternative 1,1,2-trichloro-2-hydroxyethyl radical would need to undergo H-atom migration to form dichloroacetyl chloride. As mentioned previously Cl-atom migration is expected to be favoured over H-atom migration. The conversion of radical intermediate to carbonyl product is calculated to be strongly exothermic (−428.0 kJ mol−1) in line with the failure to observe the radical intermediates in our experiments.
Conclusions
Our experiments have identified the products of reaction of tetrachloroethene and of trichloroethene with hydroxyl radicals as being the carbonyl compounds trichloroacetyl chloride and dicloroacetyl chloride respectively. It is likely that the reactions proceed via addition of an OH radical to the double bond of the alkene in a reaction entirely analogous to that of the simple hydrocarbon alkenes. This addition reaction forms a hydroxychloroalkyl radical which reacts further by abstraction of the hydroxyl hydrogen atom. In keeping with the findings from gas phase studies10 chlorine-atom loss from this radical is, at best, a very minor pathway. For trichloroethene, where two carbonyl products are possible, the observed product – dichloroacetyl chloride – has an acyl chlorine atom which gives a mesomeric stabilisation. This product is probably formed by Cl-atom rather than H-atom migration. It is found that this product is formed predominantly in the low-energy gauche form. Our experiments suggest that carbonyl products of the type observed may be important in the atmospheric oxidation of chloroalkenes, just as acetaldehyde is important in ethene oxidation.1,2,34 The use of DFT calculations in support of the experimental results from matrix isolation is shown to be important in exploring these reaction mechanisms.Acknowledgements
We thank EPSRC for a studentship for KSW and the Leverhulme Trust for the award of an Emeritus Fellowship to PCHM.References
- F. J. Comes, Angew. Chem. Int. Ed. Engl., 1994, 33, 1816 CrossRef.
- P. Crutzen, Faraday Discuss., 1995, 100, 1 RSC.
- R. Atkinson, Chem. Rev., 1986, 86, 69 CrossRef CAS.
- A. M. Thompson, Science, 1992, 256, 1157 CAS.
- Y. Kuo, G. Wann and Y. Lee, J. Chem. Phys., 1993, 99, 3272 CrossRef CAS.
- E. J. Feltham, M. J. Almond, G. Marston, K. S. Wiltshire and N. Goldberg, Spectrochim Acta, Part A, 2000, 56, 2589 CrossRef CAS.
- E. J. Feltham, PhD thesis, University of Reading, 2000.
- K. S. Wiltshire, PhD thesis, University of Reading, 2003.
- J. Leach, A. Blanch and A. C. Bianchi, Atmos. Environ., 1999, 33, 4309 CrossRef CAS.
- C. J. Howard, J. Chem. Phys., 1976, 65, 4771 CrossRef CAS.
- M. J. Almond and M. Hahne, J. Chem. Soc., Dalton Trans., 1988, 809 RSC.
- M. J. Almond and R. H. Orrin, J. Chem. Soc., Dalton Trans., 1992, 1229 RSC.
- M. Petterson, S. Tuominen and M. Räsänen, J. Phys. Chem. A, 1997, 101, 1166 CrossRef CAS.
- L. Khriachtchev, M. Pettersson, S. Tuominen and M. Räsänen, J. Chem. Phys., 1997, 107, 7252 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Ciolowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98, Revision A9, Gaussian Inc., Pittsburgh, 1998.
- D. H. Williams and I. Fleming, Spectroscopic Methods in Organic Chemistry, McGraw Hill, Maidenhead, 5th edn., 1995 Search PubMed.
- B. Schrader, Raman/Infrared Atlas of Organic Compounds., VCH, Weinheim, 2nd edn, 1989 Search PubMed.
- M. van Thiel, E. D. Becker and G. C. Pimentel, J. Chem. Phys., 1957, 27, 486 CrossRef CAS.
- E. Catalano and D. E. Milligan, J. Chem. Phys., 1959, 30, 45 CrossRef CAS.
- G. P. Ayers and A. D. E. Pullin, Spectrochim Acta, Part A, 1976, 32, 1629 CrossRef.
- X.-K. Zhang, E. G. Lewars, R. E. March and J. M. Parnis, J. Phys. Chem., 1993, 97, 4320 CrossRef CAS.
- L. Fredin, B. Nelander and G. Ribbergård, J. Mol. Spectrosc., 1974, 53, 410 CAS.
- M. J. Irvine and A. D. E. Pullin, Aust. J. Chem., 1982, 35, 1961 CAS.
- M. J. Irvine and A. D. E. Pullin, Aust. J. Chem., 1982, 35, 1971 CAS.
- H. Kühne, S. Vaccani, T.-K. Ha, A. Bauder and H.-H. Günthard, Chem. Phys. Lett., 1976, 38, 449 CrossRef.
- H. Kühne, M. Forster, J. Hulliger, H. Ruprecht, A. Bauder and H.-H. Günthard, Helv. Chim. Acta, 1980, 63, 1971 CrossRef CAS.
- R. Fausto and J. J. C. Teixera-Dias, J. Mol. Struct., 1986, 144, 241 CrossRef CAS.
- M. D. King, C. E. Canosa-Mas and R. P. Wayne, Phys. Chem. Chem. Phys., 1991, 1, 2231 Search PubMed.
- M. D. King, C. E. Canosa-Mas and R. P. Wayne, Phys. Chem. Chem. Phys., 1991, 1, 2239 Search PubMed.
- B. W. Gay, P. L. Hanst, J. J. Bulfani and R. C. Noonan, Environ. Sci. Technol., 1976, 10, 58.
- L. Breitman and E. W. R. Steacie, Can. J. Chem., 1953, 31, 328 CAS.
- R. J. Bellamy and R. L. Williams, J. Chem. Soc., 1958, 3465 RSC.
- M. C. L. Gerry, W. Lewis-Bevan and N. P. C. Westwood, Can. J. Chem., 1985, 63, 676 CAS.
- R. Atkinson, S. M. Aschmann, J. Arey and B. Shorees, J. Geophys. Res., 1992, 97, 6065 CAS.
| This journal is © the Owner Societies 2004 |