Potential energy surface of the CO2− anion
Thomas
Sommerfeld
*,
Hans-Dieter
Meyer
and
Lorenz S.
Cederbaum
Theoretische Chemie, Universität Heidelberg, Im Neuenheimer Feld 229, 69120Heidelberg, Germany. E-mail: Thomas.Sommerfeld@urz.uni-heidelberg.de
First published on 25th November 2003
Abstract
Electron attachment to CO2 is studied by exploring the parts of the potential energy surface where an electron can be bound in the fixed-nuclei picture. High level ab initio methods are used, and in particular the question of an adequate description of the threshold region, where the attachment energy goes to zero and the electronically bound anionic states become unstable, is discussed. Since the relevant coordinates are the symmetric stretching and the bending mode, we consider a two dimensional cut (C2v symmetry constraint) through the three dimensional nuclear coordinate space, and specifically C–O bond lengths between 1.1 and 1.6 Å and bending angles between 180 and 125° are examined. Three bound CO2− states are identified in this region. The minimum associated with the long-lived CO2− state is localised on the lowest surface of the anion, and only a small barrier separates it from the region where the anion becomes unstable. Using earlier results from R-matrix calculations and our findings we can then answer the question which of the short-lived scattering states connects to the long-lived CO2− species. All three states of the CO2− anion are vibronically coupled, and the implications for the nuclear dynamics of these states are discussed, in particular in view of the recently observed vibrational excitation spectra.
I. Introduction
Despite the long standing interest in electron scattering from and electron attachment to carbon dioxide, there is still a number of open questions, in particular, pertaining to the ‘connection’ of the short-lived scattering states with the long-lived anions observed in mass spectrometry.Long-lived CO2− anions (lifetimes in the μs and even ms range) can be produced in various different processes including electron collisions with cyclic anhydrides1 or CO2 clusters,2 double electron transfer to CO2+ ions,3,4 and sputter techniques.5 While high quartet and sextet states have been invoked to explain the ms lifetimes,6,7 the CO2− species showing μs lifetimes have been rationalised in terms of a bent minimal energy structure (C2v symmetry) on the lowest potential energy surface (PES) of CO2− at which the anion is stable with respect to vertical electron loss.8–14 In contrast, direct attachment leads only to short-lived species, notably a 2Σ+g virtual state and a 2Πu resonance, which are the key ingredients for understanding electron scattering from neutral CO2 (see, e.g.,15–17 and references therein). The virtual state is attributed18–20 to cause the prominent zero-energy peak, which appears at electron scattering energies below 1 eV, and the 2Πu shape resonance produces a characteristic peak in the cross section at about 3.8 eV.17,21–24
Until recently, the short-lived and the long-lived regimes have largely remained separate issues. Connections of the vertically stable C2v anion with the 2Πu resonance and the 2Σ+g virtual state have been discussed for some time,16,25–29 but only in Allan’s current vibrational excitation measurements,28 has a larger region of the CO2− PES been probed in a short-lifetime experiment. In these experiments excitation of odd bending mode quanta are clearly seen, whereas these excitations are excluded by symmetry in resonance scattering models, e.g. the boomerang model used in the most elaborate resonance scattering calculations to date.24 This clearly demonstrates that something basic is missing in our current understanding of the CO2− anion.
The present article concentrates on an ab initio investigation of the lowest electronic states of CO2− as a function of the symmetric stretch distance R and the binding angle θ. Ab initio scattering calculations on CO2− have recently been performed by Morgan,16 focusing on the angular dependence of the virtual state pole, and by Rescigno et al.,17,23 focusing on the 2Πu shape resonance pole. Here we attack the problem from a different direction and study the CO2− surface in the region where it represents a stable electronic state. In this way we can treat electron correlation at a very high level, which is expensive in scattering computations. Using a similar approach and decreasing the bond angle from the bent minimum, the surface of CO2− had been predicted to increase smoothly until it crosses the neutral PES and is turned into a resonance.11,14 Yet, the methods employed in these two papers have been demonstrated to be inappropriate in the threshold region where the attachment energy becomes small.29 Moreover, as we will show, the situation is more complicated as there are three close lying electronic states, which behave differently at threshold.
II. Electronic structure considerations
In this work a two-dimensional section of the CO2− PES is studied where the molecule is constrained to C2v symmetry and the C–O bond length R and the O–C–O bond angle θ are the variables. Neutral CO2 is a linear closed-shell molecule showing a bond length of R0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =1.161 Å and its electronic configuration is
=1.161 Å and its electronic configuration is | (core)6(σg)2(σu)2(σg)2(σu)2(πu)4(πg)4. | (2.1) |
Note that upon bending the molecule (and placing it in the y–z plane) the orbital symmetries change in the following way: σg→a1, σu→b2, πg→a2+b2, and πu→a1+b1. Since CO2 possesses two low lying empty valence orbitals of σg and πu symmetry, one expects two low lying anionic states. At the linear equilibrium geometry of neutral CO2 one finds accordingly a virtual 2Σ+g state and a 2Πu resonance, which in C2v symmetry correspond to two 2A1 and one 2B1 states. Thus, studying the CO2− PES does effectively involve examining three vibronically coupled electronic states.
Here we investigate only those parts of the three anionic PES that lie below the PES of the neutral, i.e., those parts where the additional electron is bound in the fixed-nuclei picture. At these geometries the anionic states can in principle be treated using standard quantum chemistry techniques, yet so-called single-reference methods that are based on a one-determinant self-consistent field (SCF) wavefunction will break down in two cases. First, if the bond length R is increased, both neutral CO2 and the three CO2− states will show increasing multi-configuration character, and second, in the regions where the attachment energy becomes very small single-reference based methods do fail for the lowest anionic state29 and may be equally inappropriate for the higher lying ones. In the next paragraphs we will consider these two issues and describe then the methods that have been used in our calculations.
To study the multi-configuration character of the wavefunctions for increased bond lengths we have performed a series of full-valence complete-active-space SCF (CASSCF) calculations for linear CO2 (16 electrons in 12 orbitals). Up to a bond length of 1.3 Å the wavefunction has clearly one-determinant character with the closed-shell configuration (see eqn. (2.1)) showing a weight of over 90%. Increasing the bond length to 1.6 Å the weight of the closed-shell configuration drops to 80% indicating that a single-reference description is getting increasingly inadequate, and for even longer C–O distances multi-configuration wavefunctions are needed.
The multi-configuration character of the anionic wavefunction in the region close to the intersection seam between the PES of the neutral and the anion has been discussed in ref. 29. In this region the electron binding energy is entirely due to electron correlation effects (dispersion), and any SCF procedure fails to describe the anionic wavefunction. Starting from the orbitals of neutral CO2, the lowest 2A1 state of the anion is, contrary to the usual situation, not described by the configuration where the additional electron occupies the lowest unoccupied orbital (LUMO), but by several configurations corresponding to occupation of higher lying virtual orbitals. Thus, single-reference configuration interaction (CI) or coupled cluster (CC) methods based on the SCF orbitals of the neutral are bound to fail. The same is true for single-reference methods based on unrestricted of open-shell restricted Hartree–Fock (HF) calculations of the anion, since the open-shell HF wavefunctions are qualitatively wrong in this region.29 For example, approaching the intersection seam from a region with a large binding energy, unrestricted HF based CC calculations with single double and non-iterative triple excitations (CCSD(T)) will initially repair the wrong HF behaviour and show a qualitatively correct PES. Yet, from a certain point well before the attachment energy goes to zero, the CCSD iterations will converge on a CO2-and-free-electron state rendering this approach useless for localising the intersection with the PES of the neutral.
Here we employ the equation-of-motion coupled-cluster with single and double excitations method (EOM-CCSD) (for an overview of this and related methods see30). This method yields directly the electron attachment energies to the neutral closed-shell CO2 molecule, and thus the seam of intersection is also obtained directly. In contrast, the PES of the anionic states are only indirectly accessible by adding the computed attachment energies to the PES of the neutral. Since the calculations are based on a single-reference description of neutral CO2 we can only consider bond lengths of say up to 1.6 Å, but the region of intersection is correctly described, and the method possesses a build-in balance between the neutral and the anionic states.30 Note that in contrast to approaches where the PES of the neutral and the anion are computed in two separate calculations, the issue of a balanced treatment does not imply balance between two calculations, but a balanced description within a single calculation.
As mentioned above, to compute the PES of the anionic states the PES of neutral CO2 is needed, and the best choice is the most accurate surface available (see also ref. 29). We use the CCSD(T) method and Dunning’s augmented correlation-consistent valence triple-ζ basis set (aug-cc-pVTZ)31 to compute the PES of neutral CO2 on a grid between R=1.1 and 1.6 Å and between θ=180 and 125° in 0.1 Å and 2.5° steps, respectively (1173 grid points). At the same grid points the lowest electron attachment energies have been computed using the EOM-CCSD method and the Aug-cc-pVTZ basis set further augmented with a [4s4p2d] set of diffuse functions on all atoms (even scaled exponents starting from the lowest respective exponents in the aug-cc-pVTZ basis, scaling factor 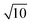 ). As shown in ref. 29 these extra diffuse functions are crucial to obtain the correct behaviour at threshold. For all computations the computer code ACES II was used.32
). As shown in ref. 29 these extra diffuse functions are crucial to obtain the correct behaviour at threshold. For all computations the computer code ACES II was used.32
III. Ab initio results
In this section we first discuss the computed electron attachment energies and consider then the associated PES of the CO2− anion. In Fig. 1 the EOM-CCSD results for the lowest two 2A1 and the lowest 2B1 states are shown. In the investigated nuclear coordinate range only these three states show negative attachment energies, i.e., the electron is bound and its binding energy is positive; all other states are electronically unstable in the entire examined range. All three states are unstable at the equilibrium structure of neutral CO2, but either on stretching the C–O bond or on bending the molecule the lowest 2A1 state is quickly turned into a bound state (Fig. 1). In contrast, the second 2A1 state becomes only bound on stretching, whereas bending has very little influence on its attachment energy, and the 2B1 state becomes only stable for extended bond lengths and small bending angles (Fig. 1).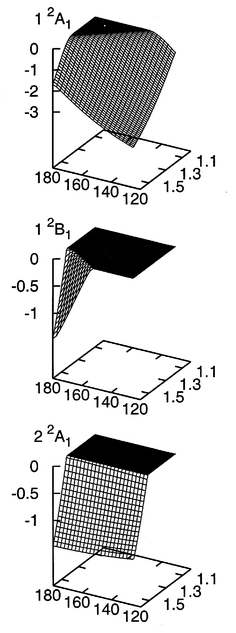 | ||
| Fig. 1 Electron attachment energy (in eV) of the lowest two 2A1 and the lowest 2B1 states of CO2− as a function of the bond angle θ (in deg.) and the C–O bond length R (in Å). The data have been obtained at the EOM-CCSD level of theory, and in the regions where the electron cannot be bound the attachment energy has been set to zero. | ||
Viewed as a function of R and θ the electron binding energies have a remarkably simple structure: In the region where the electron is bound the binding energies are essentially linear functions, and simple expansions such as Eb(R,θ)=E0b+aR+bθ will yield very reasonable approximations. Only close to threshold deviates the binding energy of the lowest 2A1 state markedly from linearity and goes very smoothly to zero, i.e., the lowest PES of the anion almost merges with the PES of the neutral before it is dissolved in the continuum. In contrast, the binding energy of the 2B1 state is, at least at the scale investigated here, linear at threshold, and the PES of the 2B1 state crosses the PES of the neutral.
This behaviour can be seen more clearly in the cuts shown in Fig. 2, where the electron binding energies have been converted into PES using the CCSD(T) PES of neutral CO2. In linear geometry the 2Πu state is bound for R>1.44 and crosses into the continuum, while the 2Σ+g state is bound down to R≈1.36 and merges with the PES of the neutral before the anion becomes unstable. This difference is even more apparent in the second cut at a fixed bond angle of 170°. The curve corresponding to the 2B1 component of the 2Πu state crosses, whereas the two curves of the 2A1 state merge with the PES of neutral, though the higher 2A1 state to a much lesser extent. For completeness we show in Fig. 3 a cut along the bending angle; the two 2A1 states strongly repel each other when the molecule is bend.
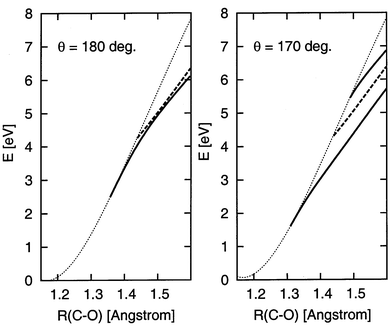 | ||
| Fig. 2 Cuts through the computed CO2 and CO2− PES at fixed bond angles of 180° and 170°. The CCSD(T) PES of CO2 is shown as a dotted line in both panels, and its minimum is taken as the energy origin. Left panel, θ=180°, full curve: 2Σ+g state, dashed curve: 2Πu state. Right panel, θ=170°, full curves: 2A1 states, dashed curve: 2B1 state. | ||
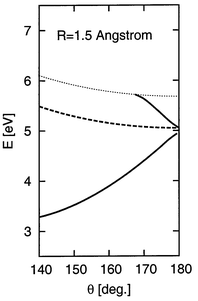 | ||
| Fig. 3 Cut through the computed CO2 and CO2− PES at a fixed bond length of R=1.5 Å. The CCSD(T) PES of CO2 is shown as a dotted line, and its minimum is taken as the energy origin. Full curves: 2A1 states, dashed curve: 2B1 state. | ||
A more complete picture of the lowest adiabatic CO2− surface including the seam of intersection with the PES of the neutral is shown in Fig. 4. The displayed surface has been obtained by adding the computed attachment energy or, in the region where the anion is unstable, zero to the PES of the neutral. Thus, in the region where the anion is stable (large bond lengths and/or angles) the surface of the anion is shown, and on the other side of the indicated intersection seam the plotted surface is that of the neutral that should however be understood as the surface of CO2− with one electron at a far distance. Owing to the merging behaviour of the anionic state, the surface obtained in this way is smooth and can, e.g., be used to study long-lived states of CO2−. Note that the simple functional form of the attachment energy is reflected in an essentially linear intersection seam.
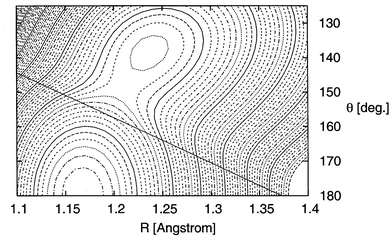 | ||
| Fig. 4 Contour plot of the CO2−/CO2 PES as a function of the C–O bond length R
(in Å) and the bond angle θ
(in degrees). The displayed surface has been obtained by adding the computed attachment energy or, in the region where the anion is unstable, zero to the PES of the neutral, and the intersection between the two regions is indicated by a straight full line. The energy origin is the minimum of neutral CO2
(R=1.17; θ=180°), and the energy difference between two contour lines is 0.05 eV. The dash-dotted contour around the CO2 minimum corresponds to 0.05 eV, and the dotted contour around the CO2− minimum (R=1.24; θ=138°) corresponds to 0.6 eV. The “neutral” part of the surface should be interpreted as a CO2− surface with one atom at a large distance. | ||
The lowest adiabatic CO2− surface shows a shallow minimum at R=1.24 Å and θ=138° in satisfying agreement with direct calculations of the CO2− equilibrium geometry.14 Moreover the adiabatic EA of 0.58 eV obtained from our surface (uncorrected for zero-point effects) is close to the value obtained at the CCSD(T) level using the Aug-cc-pVTZ basis set (0.63 eV). Yet, the CO2− minimum is separated only by a small barrier of about 45 meV (350 cm−1) from the region of instability suggesting that at best one vibrational state, if any, can be trapped in the minimum. In view of the tiny barrier let us reemphasise that we use a CO2 ground state surface computed with a single-reference method as well as the EOM-CCSD method for the attachment energy that is also based on a single-reference method for neutral CO2. Therefore, both methods are most accurate close to the equilibrium position of the neutral, and more elaborate (and far more costly) calculations will, on the one hand, yield a more open CO2 PES, and may, on the other hand, yield a steeper increase of the attachment energy function. While we can only speculate on the effects of a more accurate attachment energy, a more accurate CO2 PES will lead to a somewhat deeper minimum for CO2−. We do not expect any large effects, but in view of the small barrier even small changes may be crucial. Be this as it may, only nuclear dynamics calculations on the CO2− surface will show whether the lifetimes of the vibrational states living in the CO2− minimum can explain the experimental observations. In any case our findings suggest that the lifetime of any long-lived CO2− states is not governed by the Franck–Condon overlaps with the vibrational states of the neutral, but is determined by crossing of or tunnelling through the barrier and a subsequent fast electron loss in the metastable region.
IV. Discussion and conclusions
Our results for the three CO2− PES in the region where these states are stable with respect to electron loss together with the work of Morgan et al.16,26 allow us to answer the question about the connection of the long-lived CO2− anion with the short-lived scattering states. Moreover, we can explain the occurrence of vibrational excitation peaks with odd quanta in the bending mode which were observed recently in electron scattering experiments.28So far it had been known from R-matrix calculations that upon bending the virtual 2Σ+g state becomes stable and connects to the long-lived CO2− minimum.16,26 On the other hand, scattering calculations show that upon stretching in linear geometry the 2Πu resonance becomes sharper and finally bound.17 Together with the results from bound state calculations11,14 the latter results suggest that along the symmetric stretching coordinate the 2Πu resonance connects to the long-lived CO2− minimum (cf. introduction of ref. 27), yet, in view of our findings this conclusion is wrong. From the cuts in Fig. 2 it is immediately clear that both short-lived states become stable when the CO2 molecule is stretched, and that the 2Σ+g becomes bound first, that is, at smaller C–O distances and at considerably lower energies. Thus, the lowest adiabatic 2A1 PES is connected, independently of the path taken, with the virtual 2Σ+g state. Yet, departing from linearity, the 2A1 component of the 2Πu state will mix with the 2A1 state connected to the 2Σ+g curve (cf.Fig. 3) and the two 2Πu components are Renner–Teller coupled, and one can therefore argue that the lowest 2A1 state will acquire some π character. For a correct treatment, the CO2− anion must be viewed as a three state vibrionic coupling problem.
As just mentioned, two 2Πu components of the CO2− PES are Renner–Teller coupled. The Renner–Teller vibronic coupling problem is rather well understood and its coupling elements – originating from the Coriolis coupling within the kinetic energy operator – are known. McCurdy et.al. have very recently performed an extensive study24 on the vibrational excitation cross sections of CO2 by electron impact in the vicinity of the 2Πu resonance. They included the two surfaces which correlate to 2Πu, accounted for the Renner–Teller effect when performing the nuclear dynamics, and evaluated the cross sections within the local complex potential (or boomerang) model.33 As the authors emphasise, their treatment cannot describe excitations of non-totally symmetric levels from the ground state, i.e. transition to odd quanta bending modes (with an odd quanta vibrational angular momentum around the symmetry axis) are forbidden. On the other hand, such transitions are clearly observed in Allan’s measurements.28 We expect that the inclusion of the missing 2A1 surface, which is the ground state of CO2−, will resolve this problem. A vibrionic coupling between Σ and Π states is known to lift the selection rule.34 Such an approach, however, requires the knowledge of the non-adiabatic coupling elements. Moreover, it is likely that a local theory for the dynamics is not sufficient and a non-local treatment will become necessary, because the lower 2A1 state turns into a virtual state rather than into a resonance close to linearity. This renders the e−–CO2 scattering problem a hard one.
Acknowledgements
We gratefully acknowledge stimulating discussions with Michael Allan, Hartmut Hotop, and Sven Feuerbacher. This work was supported by the Deutsche Forschungsgemeinschaft (Forschergruppe Niederenergetische Elektronenstreuprozesse).References
- C. D. Cooper and R. N. Compton, Chem. Phys. Lett., 1972, 14, 29 CrossRef CAS.
- A. Knapp, O. Echt, D. Kreisle, T. D. Mark and E. Recknagel, Chem. Phys. Lett., 1986, 126, 225 CrossRef CAS.
- D. Schröder, C. A. Schalley, J. N. Harvey and H. Schwartz, Int. J. Mass Spectrom., 1999, 185–187, 25 CrossRef.
- M. K. Raarup, H. H. Andersen and T. Andersen, J. Phys. B, 1999, 32, L659 CrossRef CAS.
- R. Middleton and J. Klein, Phys. Rev. A, 1999, 60, 3786 CrossRef CAS.
- A. Dreuw, T. Sommerfeld and L. S. Cederbaum, Theor. Chem. Acc., 1998, 100, 60 CrossRef CAS.
- A. Dreuw and L. S. Cederbaum, J. Phys. B, 1999, 32, L665 CrossRef CAS.
- M. Krauss and D. Neumann, Chem. Phys. Lett., 1972, 14, 26 CrossRef CAS.
- C. D. Cooper and R. N. Compton, J. Chem. Phys., 1973, 59, 3550 CrossRef CAS.
- R. N. Compton, P. W. Reinhardt and C. D. Cooper, J. Chem. Phys., 1975, 63, 3821 CrossRef CAS.
- D. G. Hopper, Chem. Phys., 1980, 53, 85 CrossRef CAS.
- W. B. England, Chem. Phys. Lett., 1981, 78, 607 CrossRef CAS.
- Y. Yoshioka, H. F. Schaefer III and K. D. Jordan, J. Chem. Phys., 1981, 75, 1040 CrossRef CAS.
- G. L. Gutsev, R. J. Bartlett and R. N. Compton, J. Chem. Phys., 1998, 108, 6756 CrossRef CAS.
- F. A. Gianturco and F. Schneider, J. Phys. B, 1996, 29, 1175 CrossRef CAS.
- L. A. Morgan, Phys. Rev. Lett., 1998, 80, 1873 CrossRef CAS.
- T. N. Rescigno, D. A. Byrum, W. A. Isaacs and C. W. McCurdy, Phys. Rev. A, 1999, 60, 2186 CrossRef CAS.
- M. A. Morrison, Phys. Rev. A, 1982, 25, 1445 CrossRef CAS.
- H. Estrada and W. Domcke, J. Phys. B, 1985, 18, 4469 CrossRef CAS.
- S. Mazevet, M. A. Morrison, L. A. Morgan and R. K. Nesbet, Phys. Rev. A, 2001, 64, 40701 CrossRef CAS.
- M. A. Morrison, N. F. Lane and L. Collins, Phys. Rev. A, 1977, 15, 2186 CrossRef CAS.
- C.-H. Lee, C. Winstead and V. McKoy, Chem. Phys., 1999, 111, 5056 CrossRef CAS.
- T. N. Rescigno, W. A. Isaacs, A. E. Orel, H. -D. Meyer and C. W. McCurdy, Phys. Rev. A, 2002, 65, 32716 CrossRef.
- C. McCurdy, W. A. Isaacs, H. -D. Meyer and T. Rescigno, Phys. Rev. A, 2003, 67, 42708 CrossRef .
- W. Domcke and L. S. Cederbaum, in Electron-Atom,Electron-Molecule Collisions, ed. J. Hinze, Plenum PressNew York, 1973, p. 255 Search PubMed.
- J. Tennyson and L. A. Morgan, Philos. Trans. R. Soc. London, Ser. A, 1999, 357, 1161 Search PubMed.
- W. Vanroose, C. W. McCurdy and T. N. Rescigno, Phys. Rev. A, 2002, 66, 32720 CrossRef.
- M. Allan, J. Phys. B, 2002, 35, L387 CrossRef CAS.
- T. Sommerfeld, J. Phys. B, 2003, 36, L127 CrossRef CAS.
- F. Mertins and J. Schirmer, Phys. Rev. A, 1996, 53, 2140 CrossRef CAS.
- R. A. Kendall, T. H. Dunning, Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6769 CrossRef.
- Computer code ACES II, Quantum Theory Project, University of Florida, 1998.
- D. T. Birtwistle and A. Herzenberg, J. Phys. B, 1971, 4, 53 CrossRef CAS.
- H. Köppel, W. Domcke and L. S. Cederbaum, Adv. Chem. Phys., 1984, 57, 59.
| This journal is © the Owner Societies 2004 |