Excited-state double-proton transfer dynamics of deuterated 7-azaindole dimers in a free jet studied by hole-burning spectroscopy
Kenji Sakota, Akihiko Hara and Hiroshi Sekiya*
Department of Chemistry, Faculty of Sciences, Graduate School of Molecular Chemistry, Faculty of Science, Kyushu University, 6-10-1 Hakozaki, Higashi-ku, Fukuoka, 812-8581, Japan. E-mail: hsekiscc@mbox.nc.kyushu-u.ac.jp
First published on 27th November 2003
Abstract
Fluorescence excitation and hole-burning spectra have been measured for deuterated 7–azaindole dimers [(7AI)2] in a free jet in order to investigate their excited-state double-proton transfer (ESDPT) dynamics. Only one transition system is observed in the S1–S0 region of the excitation spectrum of (7AI)2-dd, where two hydrogen atoms of the NH groups are deuterated. Two transition systems are observed in the spectrum of (7AI)2-hd in which one of the hydrogen atom of the NH groups is deuterated. The two systems have been ascribed to the S1–S0 transitions of (7AI)2-h*d and (7AI)2-hd*. In these molecules one monomer moiety, 7AI-h or 7AI-d, is excited in the S1 state. The separation of the two electronic origins has been determined to be 21 cm−1. In contrast to (7AI)2-hd, two monomer moieties must be simultaneously excited in the S1(1Bu) states of (7AI)2-dd and (7AI)2-hh. These findings can be consistently explained by considering that (7AI)2-dd and (7AI)2-hh in the S1 state have C2h symmetry, whereas (7AI)2-h*d and (7AI)2-hd* have Cs symmetry. The bandwidth for one quantum of the intermolecular stretching vibration of (7AI)2–h*d (4.1 cm−1) in the excitation spectrum is greater than 2.4 cm−1 for the stretching vibration of (7AI)2-hd*, indicating that the rate of the ESDPT reaction depends significantly on the excited site. These results support a concerted mechanism for proton transfer in (7AI)2-dd and (7AI)2-hh. We will discuss the reason for the observation of bi-exponential decays detected by photo-excitation of vibronic bands of (7AI)2 in a molecular beam with a femtoseond laser (A. Douhal, S. K. Kim and A. H. Zewail, Nature, 1995, 378, 260) on the basis of the symmetry of the 7AI dimers and vibrational mode-specific proton transfer.
1. Introduction
The excited-state double-proton transfer (ESDPT) reaction has been the subject of numerous spectroscopic and theoretical studies. The ESDPT reaction has been considered to play an important role in photochemical processes in DNA bases. The doubly hydrogen-bonded dimer of 7-azaindole (7AI)2 is a prototypical system that undergoes photo-induced double-proton transfer. The 7AI dimer [(7AI)2] has been recognized as a good model molecule of the hydrogen-bonded DNA base pairs. The investigation of the dynamics of (7AI)2 may provide information about the mutation of DNA induced by UV radiation. The structure and the mechanism of ESDPT in (7AI)2 have been extensively studied by various spectroscopic techniques.1–17 Fuke and Kaya measured the fluorescence excitation spectrum of (7AI)2 by monitoring visible emission from the tautomer.5 The bandwidth for the intermolecular bending fundamental is narrower than that for the 0–0 transition, but those of the intermolecular stretching and the combination vibrations with the intermolecular bending vibration are broader than that for the 0–0 transition. These results clearly indicate that the ESDPT reaction is accelerated by the excitation of the intermolecular stretching mode, but the reaction is suppressed by the excitation of the intermolecular bending mode. Thus, the ESDPT reaction of (7AI)2 occurs in a highly vibrational-mode specific fashion as in the case of proton tunneling in tropolone and 9-hydroxyphenalenone.18–20Recent studies of (7AI)2 have concentrated on the ESDPT mechanism, i.e., whether the two protons move simultaneously (concerted mechanism) or stepwise (sequential mechanism). In Scheme 1 a concerted double proton transfer (DPT) and a sequential single-proton transfer (SPT) is shown. Douhal et al. observed femtosecond transients of the (7AI)2 ions and also the ions of deuterated species in the gas phase.8 Bi-exponential decays with two time constants, 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∼650 fs and 1.6–3.3 ps, were observed as a function of the vibrational excess energy in the region ΔE=0–1.5 kcal mol−1.8 The fast decay component was attributed to proton transfer from the initial state to an intermediate state, and the slow component to proton transfer from the intermediate state to the tautomer. Similar bi-exponential decays were observed in deuterated (7AI)2-hd and (7AI)2-dd, where hd indicates that one 7AI moiety is N-deuterated, while dd indicates that both moieties are N-deuterated. On the basis of these results, a sequential mechanism was proposed for the ESDPT reaction. Essentially the same conclusion was obtained from a femtosecond time-resolved Coulomb explosion study.13
∼650 fs and 1.6–3.3 ps, were observed as a function of the vibrational excess energy in the region ΔE=0–1.5 kcal mol−1.8 The fast decay component was attributed to proton transfer from the initial state to an intermediate state, and the slow component to proton transfer from the intermediate state to the tautomer. Similar bi-exponential decays were observed in deuterated (7AI)2-hd and (7AI)2-dd, where hd indicates that one 7AI moiety is N-deuterated, while dd indicates that both moieties are N-deuterated. On the basis of these results, a sequential mechanism was proposed for the ESDPT reaction. Essentially the same conclusion was obtained from a femtosecond time-resolved Coulomb explosion study.13
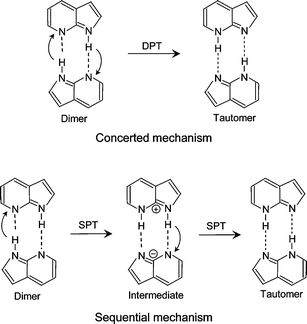 | ||
| Scheme 1 | ||
The mechanism of ESDPT proposed from time-resolved study of (7AI)2 in solution is inconsistent with that in the gas phase. Two groups observed bi-exponential decay profiles with time constants of ∼0.2 ps and ∼1.1 ps by photo-excitation of (7AI)2 in solution.12,14,17 Fiebig et al.17 ascribed the two decay constants to the sequential mechanism. However, Takeuchi and Tahara12c measured decay profiles from higher energy region to the red-edge of the dimer absorption (218–313 nm). They showed that the fast component is due to internal conversion from the 1Lb-type state to the 1La-type state, while the slow component is due to the simultaneous double-proton transfer reaction. This assignment was confirmed by observing a single exponential decay profile on exciting the red-edge of the absorption. The results of Takeuchi and Tahara supported the concerted mechanism.12
The geometry of undeuterated (7AI)2-hh in the electronic excited state and the potential energy surfaces have been calculated to investigate the mechanism of ESDPT.9,16,21–23 Douhal et al. carried out CIS geometry optimization and showed that the most stable structure in the S1 state has Cs symmetry, and that one monomer moiety is excited in the S1 state.21 The locally excited nature of (7AI)2-hh is suggested to be important for the sequential proton transfer. On the other hand, Catalan et al. proposed that (7AI)2-hh has C2h symmetry, therefore two 7AI moieties must be simultaneously excited.22 In contrast to the Cs-symmetry potential energy surface obtained by Douhal et al.21 no local minimum exists on the potential energy surface obtained from CIS calculations by Catalan et al.22 Thus, theoretical results suggest that the investigation of the symmetry of the excited state is crucial to understand the ESDPT dynamics of (7AI)2.
In the present work, we have measured the electronic spectra of deuterated (7AI)2. The vibronic transitions of deuterated species are superimposed with those of (7AI)2-hh. The application of UV–UV hole-burning spectroscopy enabled us to separate the transitions of different species. We have determined the symmetry of the S1 states of the three isotopomers (7AI)2-dd, (7AI)2-hh, and (7AI)2-hd. It has been found that the ESDPT rate depends significantly on the excited site of (7AI)2-hd, in which one of the two monomer moieties, 7AI-h or 7AI-d, could be selectively excited. We will provide a new interpretation for the bi-exponential decay behavior detected by Douhal et al.8 on the basis of the vibrationally resolved hole-burning spectra. The results in the present work are consistent with the concerted mechanism for (7AI)2–dd and (7AI)2–hh.
2. Experimental
The experimental apparatus used in this study has been described elsewhere.24 Briefly, the vacuum chamber was evacuated with a 10 in diffusion pump backed by an oil rotary pump. The fluorescence excitation spectrum was measured for jet-cooled samples using helium as a carrier gas. The sample mounted in the nozzle housing was heated to 80°C. The backing pressure was 2 atm. The pulsed valve (General Valve Series 9, 0.5 mm diam.) was operated at 5 Hz. The fluorescence excitation spectra of (7AI)2 and deuterated isotopomers were measured by using a frequency-doubled dye-laser (Lumonics HT-1000 and Lumonics HD-300) pumped by a third harmonic of the Nd3+:YAG laser (Spectra Physics GCR 170), and only visible emission was detected with a Toshiba Y45 glass filter. The UV–UV hole-burning spectra were recorded by monitoring depletions of fluorescence from particular transitions. The frequency-doubled dye-laser (Inrad Autotracker III and Spectra Physics PDL-3) pumped by a second harmonic of the Nd3+:YAG laser (Spectra Physics GCR 18) was used as a pump laser. The laser system used for the probe laser was essentially the same as that used for the the fluorescence excitation spectrum. The outputs of the pump and probe lasers were introduced to the chamber from opposite directions. A typical delay time between the pump and probe lasers was about 800 ns. The signal from the photomultiplier (Hamamatsu 1P28A) was fed into a digital oscilloscope (LeCroy 9310A), and the averaged signal was stored on a PC (NEC PC9801) for further analyses. 7AI was purchased from TCI and was used without purification. Deuterated 7AI dimers were produced by introducing a few drops of D2O into the nozzle housing.3. Results
Fig. 1a shows the fluorescence excitation spectrum of a mixture of undeuterated and dueterated 7AI dimers. The introduction of D2O generates hydrogen-bonded complexes of 7AI with D2O. Fluorescence from the hydrogen-bonded 7AI–D2O complexes was eliminated by using a cut-off filter to detect only the tautomer fluorescence from 7AI dimers. The vibronic bands of the deuterated dimers are superimposed on the vibronic bands of undeuterated (7AI)2-hh in the excitation spectrum (Fig. 1a). In order to separate the vibronic bands of isotopomers, the hole-burning spectra are measured by probing the bands at 32293 and 32472 cm−1. These two bands are marked with arrows in Fig. 1a. It is clear that the hole-burning spectra in Figs. 1b and 1c must be due to deuterated (7AI)2. The intensities of vibronic bands marked with the open circles in the fluorescence excitation spectrum increased when the nozzle housing involving D2O was heated for a long time (ca. a day). However, the intensities of these bands are much weaker than those of the deuterated species assigned to (7AI)2-dd (Fig. 2). Therefore, we infer that the vibronic bands marked with the open circles are due to deuterated 7AI dimers that have C–D bond(s). Such species have been observed in the time-of-flight spectrum measured by Douhal et al.8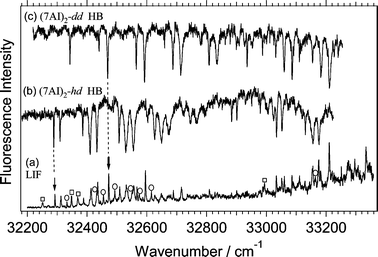 | ||
| Fig. 1 Fluorescence excitation spectrum of a mixture of (7AI)2-hh marked with the open squares and deuterated dimers in a free jet obtained by monitoring visible emission (a). The vibronic bands marked with the open-circles are due to the bands of the 7AI dimers which have C–D bond(s). UV–UV hole-burning spectra measured by probing the vibronic band at 32293 cm−1
(b) and the band at 32472 cm−1
(c). The positions of these bands are indicated with the arrows in Fig. 1a. | ||
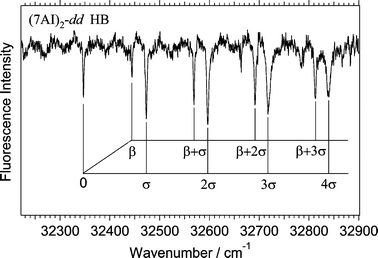 | ||
| Fig. 2 Hole-burning spectrum of deuterated (7AI)2 obtained by probing the vibronic band at 32472 cm−1. The spectrum has been assigned to (7AI)2-dd
(see text). | ||
An enlarged feature of the hole-burning spectrum in Fig. 1c is shown in Fig. 2. The vibronic pattern in the hole-burning spectrum in Fig. 2 is very similar to that in the excitation spectrum of (7AI)2-hh,5 although the bandwidths for vibronic bands in Fig. 2 are much narrower than those for (7AI)2–hh. By analogy with the excitation spectrum of (7AI)2-hh, the 98 cm−1 and 128 cm−1 vibrations are assigned to the intermolecular bending (β) and stretching (σ) fundamentals, respectively. The spectrum consists of a progression of the intermolecular stretching vibration and a progression of the combination bands due to the intermolecular stretching and bending vibrations.
Fig. 3 shows the hole-burning spectrum measured by probing the band at 32293 cm−1 that is marked with an arrow in Fig. 1a. Fig. 3 is an enlarged feature of the hole-burning spectrum in Fig. 1b. The vibronic pattern in the hole-burning spectrum in Fig. 3 is apparently very different from that in Fig. 2. The lowest frequency band (A) at 32293 cm−1 is assigned to the electronic origin of the S1–S0 transition. A prominent feature in the spectrum is the observation of a transition at 32314 cm−1
(B), which is located only 21 cm−1 above band A. It is clear that the two transition systems are simultaneously observed in the hole-burning spectrum. The intermolecular bending (β) fundamentals are the same (96 cm−1) for the two systems within the resolution of the spectrum, while those of the intermolecular stretching (σ)
(121 and 123 cm−1) are slightly different between the two systems. The vibrational assignment for the two systems is summarized in Table 1 together with the bandwidths measured from the fluorescence excitation spectrum. The excitation spectrum of Fig. 1a is shown in Fig. 4 on an enlarged scale to distinguish the band positions and the widths of vibronic bands of undeuterated and deuterated species. Previously, the bands at 32293 cm−1 and 32314 cm−1 labeled A and B were assigned to the S1–S0 origins of (7AI)2-hd and (7AI)2-dd, respectively.5b However, the hole-burning spectrum in Fig. 3 clearly shows that these two bands originate from the same electronic ground state.
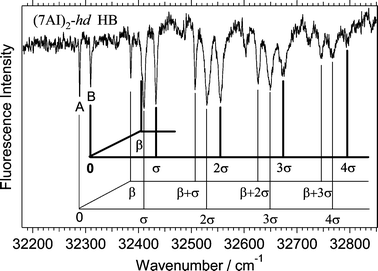 | ||
| Fig. 3 Hole-burning spectrum of a deuterated (7AI)2 obtained by probing the vibronic band at 32293 cm−1. The spectrum has been ascribed to (7AI)2-hd
(see the text). | ||
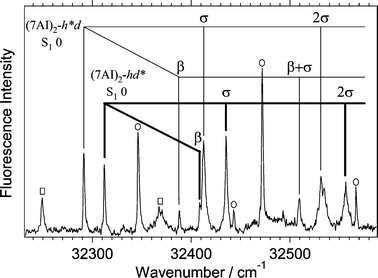 | ||
| Fig. 4 Fluorescence excitation spectrum of a mixture of (7AI)2-hd, (7AI)2–dd, and (7AI)2-hh near the electronic origins. The assignment of the intermolecular vibrations of (7AI)2-hd is given in the figure. The vibronic bands of (7AI)2-hh and (7AI)2-dd are indicated with open squares and solid circles, respectively. The electronic origin of (7AI)2-dd at 32348 cm−1 overlaps with the bending fundamental of (7AI)2-hh at 32350 cm−1. | ||
| Species | Δν/cm−1a | Bandwidth/cm−1 | Assignmentd |
|---|---|---|---|
| a Relative wavenumber from the electronic origin. The number in the parentheses indicates the wavenumber of the electronic origin.b Bandwidth was not determined due to overlapping of bands.c Ref. 5(b).d β and σ denote the intermolecular bending and stretching vibrations, respectively. | |||
| (7AI)2-dd | 0 (32348) | b | Origin |
| 98 | b | β | |
| 124 | 2.6 | σ | |
| 222 | 1.7 | β+σ | |
| 247 | 2.6 | 2σ | |
| 344 | 1.7 | β+2σ | |
| 368 | 5.1 | 3σ | |
| 465 | 2.6 | β+3σ | |
| 487 | 5.1 | 4σ | |
| (7AI)2-h*d | 0 (32293) | 1.6 | Origin |
| 96 | 1.7 | β | |
| 121 | 4.1 | σ | |
| 216 | 2.8 | β+σ | |
| 239 | b | 2σ | |
| (7AI)2-hd* | 0 (32314) | 1.6 | Origin |
| 96 | b | β | |
| 123 | 2.4 | σ | |
| 243 | 5.2 | 2σ | |
| (7AI)2-hhc | 0 (32252) | 5 | Origin |
| 98 | 3 | β | |
| 120 | 10 | σ | |
| 215 | 7 | β+σ | |
| 240 | 3 | 2σ | |
It is reasonable to assign the species observed in Figs. 2 and 3 to (7AI)2-dd and (7AI)2-hd, respectively, on the basis of the following reasons; (i) The relative blue shift of the origin band of (7AI)2-hd may be smaller than that of (7AI)2–dd, (ii) the bandwidth for the same vibrational mode may be broader for (7AI)2–hd than for (7AI)2–dd due to a smaller tunneling reduced mass for (7AI)2–hd, and (iii) two transition systems are observed in Fig. 3, whereas only one system is observed in Fig. 2 as well as in the excitation spectrum of (7AI)2–hh. The reason for the observation of the two systems in the spectrum of (7AI)2–hd will be discussed in the following section.
4. Discussion
4.1 Symmetry of the excited state
We have observed the two transition systems in the electronic spectrum of (7AI)2-hd, whereas only one system is observed in the spectra of (7AI)2-hh and (7AI)2-dd. The observation of the two systems in the spectrum of (7AI)2-hd could be explained by a lowering of the symmetry due to the deuteration. We assume that the ground state and the excited state of (7AI)2-hh and (7AI)2-dd belong to the C2h symmetry group. The ground-state and excited-state wave functions, Ψ0 and Ψ±, for (7AI)2-hh and (7AI)2-dd are described by| Ψ0=|g1〉|g2〉 | (1) |
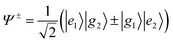 | (2) |
←g transition is electric-dipole allowed for one-photon absorption, while the g←g and u←u transitions are forbidden. Therefore, only the 1Bu←1Ag transition should appear in the one-photon excitation spectra of (7AI)2-hh and (7AI)2-dd.(7AI)2-hd in the S1 state may have Cs symmetry, and two A′ states of (7AI)2-hd correlate to the Ag and Bu states of (7AI)2–hh and (7AI)2-dd. The electronic transitions into the two A′ states become allowed for one-photon absorption due to a lowering of the symmetry. This argument is consistent with the observation of the two transition systems in Fig. 3. Each monomer unit could be distinguished in (7AI)2–hd, and bands A and B should correspond to the transitions into the zero-point levels of the locally excited S1 states of (7AI)2-h*d and (7AI)2-hd*. This assignment is based on the consideration that the blue-shift of the electronic origin band of (7AI)2-hd* relative to that of (7AI)2-hh may be larger than that of (7AI)2-h*d. Thus, nature of the lowest excited state of (7AI)2-hd is quite different from those of (7AI)2-hh and (7AI)2-dd. A similar result has been obtained from the study of the 2-pyridone dimer.31,32,34
Two groups have calculated the excited-state geometry of (7AI)2-hh and the potential energy surface along the proton transfer coordinate.21,22 Catalan et al. treated the symmetry of (7AI)2 as C2h, and showed that no local minimum exists along the reaction coordinate.22 In contrast, Moreno et al. concluded that the excited-state geometry of (7AI)2 is Cs from the optimization of the stable structure.21 They obtained a local minimum suggesting the existence of an intermediate state, which is responsible for the sequential mechanism. The barrier height along the proton transfer coordinate remarkably depends on the symmetry of the excited state. The calculated barrier heights are ca. 8 and 17 kcal mol−1 for the C2h and Cs symmetry groups, respectively.21,22 Thus, controversial results have been reported about the symmetry of (7AI)2. The experimental results in this work support the C2h symmetry proposed by Catalan et al.22
4.2 Effect of localization of excitation on ESDPT in (7AI)2–hd
The most prominent finding from the electronic spectrum of jet-cooled (7AI)2–hh is vibrational-mode specific proton transfer.5 Fuke and Kaya showed that the excitation of the intermolecular stretching mode (σ) enhances the ESDPT reaction, but the excitation of the intermolecular bending mode (β) suppresses the ESDPT reaction.5 This argument is based on the changes in the widths of the vibronic bands in the excitation spectrum. The comparison of the widths of the vibronic bands of the three species (7AI)2-hh, (7AI)2-dd, and (7AI)2-hd shown in Table 1 reveals that vibrational-mode-specific ESDPT occurs in (7AI)2-dd and (7AI)2-hd as well as in (7AI)2–hh. Deuteration of the transferred proton decreases the proton tunneling probability remarkably because of the substantial increase in the reduced mass. The bandwidths of the intermolecular stretching and bending vibrations of (7AI)2-dd are much narrower than those of the corresponding vibrations of (7AI)2-hh, in agreement with the proposed tunneling mechanism of ESDPT.5In the spectrum of (7AI)2-hd, two systems have been observed. These systems have been ascribed to transitions into the S1 state of (7AI)2-h*d and (7AI)2-hd*. The widths of the electronic origin bands for (7AI)2-h*d and (7AI)2-hd* are the same (1.6 cm−1) within experimental error. The excitation of the stretching vibration (σ) increases the bandwidth for both (7AI)2-h*d and (7AI)2-hd*. It should be noted that the bandwidth for (7AI)2-h*d (4.1 cm−1) is significantly larger than that (2.4 cm−1) for (7AI)2-hd*. The difference in the bandwidth between (7AI)2-h*d and (7AI)2-hd* may originate from the localization of excitation on the monomer site, therefore the mechanism of ESDPT in (7AI)2-h*d should be different from that in (7AI)2-hh and (7AI)2-dd.
4.3 Vibrational mode-specific ESDPT and excited-state decay profiles
The mechanism of ESDPT reaction of (7AI)2 is of considerable interests. Here, we focus on the mechanism in the gas phase. The data in the frequency domain obtained in this work may correspond to the time-domain data. Femtosecond pump–probe spectroscopy has been applied to investigate the mechanism of ESDPT in (7AI)2-hh, (7AI)2-dd, and (7AI)2-hd in a molecular beam by Douhal et al.8 They measured the femtosecond transients as a function of the excess vibrational energy of about ΔE=0, 1.0, 1.5 kcal mol−1. Bi-exponential transients of (7AI)2 were observed on electronic excitation of each species. The fast component was ascribed to single proton transfer to an intermediate state, and the slow component to the single proton transfer from the intermediate state to the tautomer state as shown in Scheme 1.On the other hand, the widths of vibronic bands in the excitation spectrum of this work provide information on the ESDPT dynamics. For a vibrational-mode-specific ESDPT reaction occuring in (7AI)2, the decay time should depend on the excited vibrational state.5 The excitation spectra measured in this work indicate that closely spaced vibronic states have comparable Franck–Condon factors for one-photon absorption. For example, in the spectrum of (7AI)2-dd two closely spaced vibronic bands assigned to β+2σ and 3σ are detected at Δν(ν−ν00)=344 and 368 cm−1, for which the bandwidths are 1.7 and 5.1 cm−1, respectively. When these two bands are simultaneously excited with a femtosecond pump laser the observed decay profile may be fitted by a bi-exponential function. In the femtosecond transients of (7AI)2-dd excited at ΔE=1.0 kcal mol−1
(about ΔE=350 cm−1), bi-exponential decays with time constants of 3 ps and 25 ps were detected. In our experiment the bandwidth of the pump laser should be narrow enough to resolve the β+2σ and 3σ vibrations, which are separated by 24 cm−1 in the spectrum of (7AI)2-dd. However, the bandwidth of the pump laser used for the measurement of the femtosecond transients of (7AI)2 might be much larger than that needed for the excitation of a single vibronic band, since the cross correlation of the pump and probe pulses was reported to be ∼150 fs.8 Therefore, the pump laser might excite more than two vibronic bands in the experiment at ΔE=0, 1.0, and 1.5 kcal mol−1. When three or four bands are simultaneously excited the resulting decay profile is a multi-exponential function. The fast and slow components in the decay profile originate from the broad stretching and the narrow bending modes. Consequently, even for a multi-exponential decay an approximately bi-exponential decay profile may be obtained. Thus, we suggest that the bi-exponential decays detected by the femtosecond transients of (7AI)2 ions are caused by simultaneous excitation of vibronic states.
On the basis of the above discussion of the decay profiles and vibrational mode-specific proton transfer together with the symmetry of (7AI)2, this work supports the concerted mechanism for (7AI)2-hh and (7AI)2-dd. However, the mechanism for (7AI)2-hd should be different; A sequential ESDPT reaction may occur in (7AI)2-hd due to photo-excitation of the locally excited state. An experiment to measure picosecond transients in the gas phase is in progress in order to obtain the decay profiles when a single vibronic state is excited.
4. Conclusion
We have measured the UV–UV hole-burning spectra of (7AI)2-dd and (7AI)2-hd. The vibronic transitions of these species are well separated from those of (7AI)2-hh. On the basis of the data we have revised the previous assignment of the electronic origins of (7AI)2-dd and (7AI)2-hd. The vibronic pattern in the spectrum of (7AI)2-dd is very similar to that of (7AI)2-hh, while the widths of vibronic bands for (7AI)2-dd are much narrower than those for (7AI)2-hh due to the increase in the tunneling reduced mass. Two transition systems have been observed in the spectrum of (7AI)2-hd. We ascribe the two systems to the S1←S0 transitions of (7AI)2-h*d and (7AI)2–hd* with an observed splitting of the two electronic origins of 21 cm−1. The observed spectra are consistently explained when we take into account that the excited electronic states of (7AI)2-hh and (7AI)2-dd have C2h symmetry, but (7AI)2-hd has Cs symmetry. It has been found that the bandwidths for the intermolecular stretching vibrations differ significantly between (7AI)2-h*d and (7AI)2-hd*. We suggest that the bi-exponential decays in the femtosecond time-resolved study of Douhal et al.8 is resulting from an excitation of closely spaced vibronic bands of different lifetime. Our results support the concerted mechanism for (7AI)2–hh and (7AI)2–dd.Acknowledgements
The authors thank Dr Tahei Tahara (Institute of Physical and Chemical Research) and Prof. Takakazu Nakabayashi (Hokkaido University) for valuable discussions. This work was supported in part by a Grant-in-Aid Scientific Research No.15250015 from the Japanese Ministry of Education, Science, Sports, and Culture.References
- (a) C. A. Taylor, M. A. El-Bayoumi and M. Kasha, Proc. Natl. Acad. Sci. USA, 1969, 63, 253 CAS; (b) K. C. Ingham, M. Abu-Elgheit and M. A. El-Bayoumi, J. Am. Chem. Soc., 1971, 93, 5023 CrossRef CAS; (c) K. C. Ingham and M. A. El-Bayoumi, J. Am. Chem. Soc., 1974, 96, 1674 CrossRef CAS.
- W. M. Hetherrington III, R. H. Micheels and K. B. Eisenthal, Chem. Phys. Lett., 1979, 66, 230 CrossRef.
- H. Bulska, A. Grabowski, B. Pakula, J. Sepiol, J. Waluk and U. P. Wild, J. Lumin., 1984, 29, 65 CrossRef CAS.
- (a) K. Tokumura, Y. Watanabe and M. Itoh, Chem. Phys. Lett., 1984, 111, 379 CrossRef CAS; (b) K. Tokumura, Y. Watanabe, M. Udagawa and M. Itoh, J. Am. Chem. Soc., 1987, 109, 1346 CrossRef CAS.
- (a) K. Fuke, H. Yoshiuchi and K. Kaya, J. Phys. Chem., 1984, 88, 5840 CrossRef CAS; (b) K. Fuke and K. Kaya, J. Phys. Chem., 1989, 93, 614 CrossRef CAS.
- P. Share, M. Pereira, M. Sarisky, S. Repinec and R. M. Hochstrasser, J. Lumin., 1991, 48/49, 204 CrossRef.
- Y. Chen, R. L. Rich, F. Gai and J. W. Petrich, J. Phys. Chem., 1993, 97, 1770 CrossRef CAS.
- A. Douhal, S. K. Kim and A. H. Zewail, Nature, 1995, 378, 260 CrossRef CAS.
- A. Douhal, V. Guallar, M. Moreno and J. M. Lluch, Chem. Phys. Lett., 1996, 256, 370 CrossRef CAS.
- A. Nakajima, M. Hirano, R. Hasumi, K. Kaya. H. Watanabe, C. C. Carter, J. M. Williamson and T. A. Miller, J. Phys. Chem., 1997, 101, 392 Search PubMed.
- R. Lopez-Martens, P. Long, D. Sogaldi, B. Soep, J. Syage and P. Millie, Chem. Phys. Lett., 1997, 273, 219 CrossRef CAS.
- (a) S. Takeuchi and T. Tahara, Chem. Phys. Lett., 1997, 277, 340 CrossRef CAS; (b) S. Takeuchi and T. Tahara, J. Phys. Chem. A, 1998, 102, 7740 CrossRef CAS; (c) S. Takeuchi and T. Tahara, Chem. Phys. Lett., 2001, 347, 108 CrossRef CAS.
- (a) D. E. Folmer, L. Poth, E. S. Wisniewski and A. W. Castleman Jr., Chem. Phys. Lett., 1998, 287, 1 CrossRef CAS; (b) D. E. Folmer, E. S. Wisniewski and A. W. Castleman Jr., Chem. Phys. Lett., 2000, 318, 637 CrossRef CAS.
- M. Chachisvilis, T. Fiebig, A. Douhal and A. H. Zewail, J. Phys. Chem. A, 1998, 102, 669 CrossRef CAS.
- S. Mente and M. Maroncelli, J. Phys. Chem. A, 1998, 102, 3860 CrossRef CAS.
- V. Guallar, V. Batista and W. H. Miller, J. Chem. Phys., 1999, 110, 9922 CrossRef CAS.
- T. Fiebig, M. Chachisvilis, M. Manger, A. H. Zewail, A. Douhal, I. Garcia-Ochoa and A. De la Hoz Ayuso, J. Phys. Chem. A, 1999, 103, 7419 CrossRef CAS.
-
(a) H. Sekiya, Y. Nagashima and Y. Nishimura, J. Chem. Phys., 1990, 92, 5761 CrossRef CAS;
(b) H. Sekiya, Y. Nagashima, T. Tsuji, Y. Nishimura, A. Mori and H. Takeshita, J. Phys. Chem., 1991, 95, 10311 CAS.
- (a) K. Nishi, H. Sekiya, H. Kawakami, A. Mori and Y. Nishimura, J. Chem. Phys., 1998, 109, 1589 CrossRef CAS; (b) K. Nishi, H. Sekiya, H. Kawakami, A. Mori and Y. Nishimura, J. Chem. Phys., 1999, 111, 3961 CrossRef CAS; (c) K. Nishi, H. Sekiya, T. Mochida, T. Sugawara and Y. Nishimura, J. Chem. Phys., 2000, 112, 5002 CrossRef CAS.
- H. Mori, H. Sekiya, E. Miyoshi, K. Mogi and Y. Sakai, J. Chem. Phys., 2003, 119, 4159 CrossRef CAS.
- (a) A. Douhal, M. Moleno and J. M. Lluch, Chem. Phys. Lett., 2000, 324, 75 CrossRef CAS; (b) A. Douhal, M. Moleno and J. M. Lluch, Chem. Phys. Lett., 2000, 324, 81 CrossRef CAS; (c) M. Moreno, A. Douhal and J. M. Luch, J. Phys. Chem. A, 2001, 105, 3887 CrossRef CAS.
- (a) J. Catalan, J. C. Del. Valle and M. Kasha, Proc. Natl. Acad. Sci. USA, 1999, 96, 8338 CrossRef CAS; (b) J. Catalan, J. C. Del. Valle and M. Kasha, Chem. Phys. Lett., 2000, 318, 629 CrossRef CAS; (c) J. C. Del. Valle, M. Kasha and J. Catalan, Int. J. Quantum Chem., 2000, 77, 118 CrossRef.
- L. Serrano-Andres, M. Merchan, A. Carlos Borin and J. Stalring, Int. J. Quantum Chem., 2001, 84, 181 CrossRef CAS.
- K. Sakota, K. Nishi, K. Ohashi and H. Sekiya, Chem. Phys. Lett., 2000, 322, 407 CrossRef CAS.
- D. P. Craig and P. C. Hobbins, J. Chem. Soc., 1955, 539 RSC.
- D. S. McClure, Can. J. Chem., 1958, 36, 59 CAS.
- R. L. Fulton and M. Gouterman, J. Chem. Phys., 1961, 35, 1059 CrossRef CAS.
- R. L. Fulton and M. Gouterman, J. Chem. Phys., 1964, 41, 2280 CAS.
- A. S. Davydov, Theory of Molecular Excitons, McGraw-Hill, 1962 Search PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1999, 103, 8516.
- K. O. Bornsen, H. L. Selzle and E. W. Schlag, J. Chem. Phys., 1986, 85, 1726 CrossRef.
- A. Held and D. W. Pratt, J. Am. Chem. Soc., 1990, 112, 8629 CrossRef CAS.
- A. Held and D. W. Pratt, J. Am. Chem. Soc., 1992, 96, 4869 CrossRef CAS.
- A. Muller, F. Talbot and S. Leutwyler, J. Chem. Phys., 2002, 116, 2836 CrossRef CAS.
- N. Kanamaru, submitted for publication.
| This journal is © the Owner Societies 2004 |