Molecular dynamics simulation of dilute aqueous DMSO solutions. A temperature-dependence study of the hydrophobic and hydrophilic behaviour around DMSO
Ricardo L.
Mancera
*a,
Michalis
Chalaris†
b,
Keith
Refson
c and
Jannis
Samios
*b
aDe Novo Pharmaceuticals Ltd., Compass House, Vision Park, Chivers Way, Histon, Cambridge, UK CB4 9ZR. E-mail: Ricardo.Mancera@denovopharma.com; Fax: +44 (0)1223 238088; Tel: +44 (0)1223 238011
bLaboratory of Physical Chemistry, Department of Chemistry, University of Athens, Panepistimiopolis, 157-71, Athens, Greece. E-mail: isamios@cc.uoa.gr; Fax: +30 17233219; Tel: +30 18065030
cCLRC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire, UK OX11 0QX
First published on 25th November 2003
Abstract
We have studied the temperature dependence of the hydrophobic and hydrophilic hydration properties of dimethyl sulfoxide (DMSO) in water by performing a series of molecular dynamics simulations of such aqueous solutions at a concentration of 0.055 mole fraction at 298, 318 and 338 K. Our results corroborate the existence of an enhancement in the structure of water. There is a well-defined hydration structure around the oxygen atom of DMSO, which establishes strong linear hydrogen-bonds with water molecules. Such interactions increase the lifetime of water–water hydrogen bonds in the vicinity of the OS group. Hydrophobic hydration around the Me groups of DMSO is observed, shown by the formation of an ordered hydration shell around these groups, with strong water–water hydrogen bonds that have longer lifetimes than in the bulk of the solution. We could find no evidence for a temperature-dependent hydrophobic interaction between these groups. All these combined effects reveal that the hydrophilic hydration of DMSO dominates over any hydrophobic effects as the temperature is increased.
1. Introduction
Aqueous solutions of dimethyl sulfoxide (DMSO, or (CH3)2S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) are of special interest because of their unique physico-chemical and biological properties.1,2 These solutions are of particular importance due to their extensive use as a mixed solvent in various chemical technology applications. DMSO is miscible with water in all proportions, and the excess thermodynamic properties of aqueous solutions of DMSO exhibit strong deviations from ideality.3 Extreme deviations from additivity are observed in the density,4,5 viscosity,5,6 adiabatic and isothermal compressibility,7 relative dielectric permittivity,8,9 surface tension,9,10 heats of mixing5,11–13 and others. As an example, a solution of one mole of DMSO in three moles of water has a very low freezing point (−70
O) are of special interest because of their unique physico-chemical and biological properties.1,2 These solutions are of particular importance due to their extensive use as a mixed solvent in various chemical technology applications. DMSO is miscible with water in all proportions, and the excess thermodynamic properties of aqueous solutions of DMSO exhibit strong deviations from ideality.3 Extreme deviations from additivity are observed in the density,4,5 viscosity,5,6 adiabatic and isothermal compressibility,7 relative dielectric permittivity,8,9 surface tension,9,10 heats of mixing5,11–13 and others. As an example, a solution of one mole of DMSO in three moles of water has a very low freezing point (−70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C), compared to 18.6°C for DMSO and 0°C for water in their neat liquids.14 It has been observed that the maximum deviations occur at concentrations of 0.3 to 0.4 mole fraction of DMSO.5,11,12,15 Although these observations are well known from various experimental studies, an unquestioned picture concerning the underlying molecular mechanisms remains still quite challenging.
°C), compared to 18.6°C for DMSO and 0°C for water in their neat liquids.14 It has been observed that the maximum deviations occur at concentrations of 0.3 to 0.4 mole fraction of DMSO.5,11,12,15 Although these observations are well known from various experimental studies, an unquestioned picture concerning the underlying molecular mechanisms remains still quite challenging.
Aqueous DMSO has several interesting biological properties. It can induce cell fusion16 and increase cell permeability,17 and it can act as cryoprotectant for membranes and proteins.18,19 The pharmacological profile of DMSO includes anti-inflammatory, analgesic, anti-viral, anti-bacterial and radioprotectant properties.1,2,20,21 Although these properties have been extensively documented, the molecular mechanism by which they arise is again still poorly understood.
DMSO and its aqueous solutions have received considerable attention from a wide range of experimental techniques, such as X-ray and neutron diffraction studies,22,23 as well as optical,12,24–29 acoustic,7,30–32 NMR33–39 and dielectric40–43 spectroscopies. These studies have suggested that DMSO behaves as a strong structure maker by rigidifying the water structure, possibly through the hydrophobic hydration of the methyl groups of DMSO. However, these observations have required a considerable degree of interpretation of the data to provide structural information.
Computer simulation techniques are well suited for studying the detailed thermodynamics, structure and dynamics of liquids and solutions. Rao and Singh44 calculated the relative differences in the free energy of hydration between methanol and DMSO in water. Vaisman and Berkowitz45 performed MD simulations of dilute solutions of DMSO in water at concentrations of 0.005, 0.04 and 0.2 mole fraction. Their simulations revealed a sharpening in the water–water pair correlation functions at higher DMSO concentrations without loss of hydrogen-bonding energies, as well as the existence of 1(DMSO):2(H2O) hydrogen-bonded aggregates at all three concentrations. Their analysis of the DMSO structure suggested the existence of a hydrophobic association of DMSO molecules.
Luzar and Chandler46 performed MD simulations at concentrations of 0.21 and 0.35 mole fraction. Their simulations revealed that the local tetrahedral structure of water was preserved at all concentrations. The first molecular coordination shells become more structured with increasing DMSO concentration, with a simultaneous decrease in the average number of water–water hydrogen bonds, in agreement with their own neutron diffraction data.47,48 They also found the existence of 1(DMSO):2(H2O) hydrogen-bonded aggregates of a nearly tetrahedral geometry. Their analysis of hydrogen-bond lifetimes showed that DMSO–water are longer lived than water–water hydrogen bonds, which in turn are longer lived than those in pure water. These authors have also reported46,49 that they found no evidence of hydrophobic interactions between the methyl groups of DMSO, although water possessed the typical orientational correlations of the cage-like structure found in the hydrophobic hydration around non-polar groups. They also concluded that the strong DMSO–water correlations observed in these mixtures are due to the stronger hydrogen bonds between DMSO and water.
Borin and Skaf50 have recently performed a series of MD simulations across the entire DMSO composition range in SPC/E water, revealing the existence of two well-defined kinds of hydrogen-bonded aggregates. One of them, the 1(DMSO):2(H2O) previously identified, has a nearly tetrahedral arrangement between the two hydrogen bonds, and predominates in water-rich mixtures (DMSO mole fraction<50%). The other one, a 2(DMSO):1(H2O) aggregate, features a central water molecule making hydrogen bonds to two DMSO molecules and predominates in DMSO-rich mixtures. They also observed an association of a pair of DMSO molecules through their oxygen atoms by the formation of the 2(DMSO):1(H2O) aggregates.
An NPT simulation of a 1(DMSO):3(H2O) mixture at ambient conditions51 confirmed the existence of 1(DMSO):2(H2O) aggregates but not of 1(DMSO):3(H2O) aggregates. Another typical configuration was found, consisting of two DMSO and three water molecules, in which the water molecule was bridged by two DMSO molecules.
Chalaris and Samios52 have more recently carried out MD simulations of the liquid mixture DMSO–water using different effective potential models for pure liquid water and DMSO. Their treatment has been devoted to the estimation of the ability of these models in predicting certain properties of the mixture at various DMSO concentrations and at ambient conditions. They found that the SPC/P2, TIPS2/P2 and TIP4P/P2 combined models yield the most reasonable descriptions of this mixture.
Recent Car–Parrinelo simulations of a DMSO–water solution at a concentration of 0.25 mole fraction53 revealed the existence of transient 1(DMSO):3(H2O) aggregates within a complex hydrogen-bonding network. These simulations also showed that hydrogen atoms of water molecules very close to the methyl groups of DMSO tend to orient hydrophilically (away from the hydrogen atoms of the methyl groups), although the general orientation of water molecules in the vicinity of the methyl groups was hydrophobic. It remains unclear whether all the above observations could be the consequence of the small size of the system simulated (of particular importance for an aqueous system) and/or of the limited hydrogen-bonding analysis performed.
In this work we have carried out for the first time a temperature-dependence study of dilute DMSO aqueous solutions. We report here our analysis of the thermodynamic, structural and hydrogen-bonding properties of aqueous solutions of DMSO at a concentration of 0.055 mole fraction for three temperatures: 298, 318 and 338 K. Our purpose has been to characterise the balance between hydrophilic and hydrophobic effects, namely the balance between the strong hydrogen-bonding interactions between DMSO and water on the one hand, and the hydrophobic structure around the methyl groups of DMSO on the other hand. Since the former would be expected to weaken as temperature increases while the latter (if at all present to a significant degree) could possibly drive hydrophobic aggregation, an analysis of the overall hydration structure is presented and discussed.
2. Simulation potentials and method
A series of molecular dynamics (MD) simulations were performed in the canonical ensemble (NVT) using the program Moldy.54 We used a new version, which implements symplectic integration algorithms for the rigid-body molecule equations of motion55,56 and the Nosé–Poincaré thermostat.57 This gives much greater stability than the previous version, which used the textbook prescription of applying an integrator separately to each quaternion component and performing normalization, every step.54 Consequently, in aqueous simulations, a time step as large as 2.5 fs still gives excellent Hamiltonian conservation and superior long-term stability compared to the largest stable time step of 0.5 fs for the previous integrator.54Cubic periodic boundary conditions were applied throughout, while a cut-off radius of 10.0 Å was applied to short-range interactions, with standard long-range corrections for particles at larger separations. The Ewald sum method was applied to compute long-range electrostatic interactions.58
The intermolecular potentials used in all simulations were the four-site TIP4P model of water59 and the rigid, united-atom P2 model of DMSO.46,48,52 The parameters used for these potentials can be found in Table 1. Cross-interaction parameters were calculated from the Lorentz–Berthelot mixing rules. This approach for treating water–DMSO interactions has been widely used on a number of water–DMSO simulations, yielding good results.45,46,49,50 Although no optimisation of the parameters has been carried out for the mixture, this is a standard first approximation that has been observed to be reasonably accurate for simulating aqueous systems,60 including DMSO.52 Recently a flexible, all-atom potential for DMSO was reported, with similar quality for reproducing the structure of DMSO–water solutions.61
| ε/kJ mol−1 | σ/Å | q | |
|---|---|---|---|
|
a Cross-interaction parameters can be obtained using the Lorentz–Berthelot combination rules: σAB=½
(σA+σB) and εAB=(εA
*
εB)½
b The geometry of DMSO was defined with an S–O bond length of 1.53 Å, an S–C bond length of 1.8 Å, an O–S–C bond angle of 106.75° and a C–S–C bond angle 97.4°.46
c The geometry of water was defined with an OW–H bond length of 0.9572 Å and an H–OW–H bond angle of 104.52°.59 The fictitious site (M) is located at a distance of 0.15 Å from the OW atom down the bisector of the molecule.59
|
|||
| O | 0.29922 | 2.8 | −0.459 |
| S | 0.99741 | 3.4 | 0.139 |
| Me | 1.230 | 3.8 | 0.160 |
| OW | 0.6487 | 3.154 | 0.0 |
| H | 0.0 | 0.0 | 0.52 |
| M | 0.0 | 0.0 | −1.04 |
The simulations were performed at constant experimental densities. The simulation boxes contained 28 DMSO molecules and 472 water molecules, giving rise to a concentration of 0.055 mole fraction. The initial configurations for each of the simulations were obtained from NVT Monte Carlo simulations using the program POLYMC,62 after 3 million trial moves at the required temperature. The required temperatures in the subsequent MD simulations were obtained by using the Nosé–Poincaré thermostat,57 with a temperature mass parameter of 100 kJ mol−1ps−2. A time step of 2.5 fs was used in all the simulations. A period of 10000 steps (25 ps) was allowed for equilibration. After this equilibration period, an additional period of 400000 steps (1.0 ns) in all simulations was allowed for the collection of data. The trajectories generated were stored every 25 fs.
3. Results and discussion
In this paper we analyse the hydration structure of a DMSO aqueous solution at a concentration of 0.055 mole fraction at 298, 318 and 338 K, in an attempt to explore systematically the effect of temperature on the structure of such dilute solutions of DMSO in water. We first describe the average structure of the solutions as revealed by their pair correlation functions. We then analyse the structure of water in different regions of the solution, followed by a description of the hydrogen-bonded network of water and its dynamics.3.1. Bulk thermodynamic properties
A summary of the bulk thermodynamic properties of the simulations can be seen in Table 2. As described earlier, experimental densities were used throughout. The simulated solution at 298 K has a very small pressure, indicating that the simulated and experimental densities match closely. The potential energy of the system is in good agreement with available experimental data at 298 K.52 As the temperature is increased, the simulated densities are likely to be slightly lower than the experimental ones, as revealed by the increased pressure of the system. This is probably a reflection of the fact that both the water and DMSO potentials used have been parametrized at 298 K, and so small deviations from the experimental pressures can be expected at higher temperatures. The potential energy of the system increases as the temperature decreases, as expected.| T/K | ρ/g cm−3 | U/kJ mol−1 | P/MPa |
|---|---|---|---|
|
a
T=temperature, ρ=density, U=average potential energy, P=average pressure. All values in brackets are standard deviations.
|
|||
| 298.1 (5.9) | 1.0238 | −40.86 (0.25) | −2.02 (45.3) |
| 318.1 (6.3) | 1.0146 | −39.62 (0.26) | 11.5 (45.7) |
| 338.0 (9.9) | 1.0032 | −38.41 (0.26) | 21.4 (46.0) |
3.2. Solution structure
Radial distribution functions (RDF) or pair correlation functions g(r) were calculated from the data collected over 1.0 ns after full equilibration. Fig. 1a shows the pair correlations for all intermolecular water sites in the solution: OW–OW, OW–HW and HW–HW. For simplicity we have not labelled individually each of the three temperatures simulated. However, it can be seen that the plot shows that as temperature is increased and thermal energy increases, water in the solutions becomes less structured, as revealed by the broadening of the first peaks. Furthermore, the heights of the peaks confirm the enhancement of water structure with respect to bulk water,63 as reported earlier.45,46,48–50Fig. 1b shows the pair correlations between the oxygen in the sulfonyl group of DMSO and the water sites: OS–OW and OS–HW. As before, the increase in temperature produces a gradual broadening of the peaks of the radial distribution functions. Importantly, all pair correlations show that water molecules establish a linear hydrogen bond to the OS atom in DMSO, as the first peak in the OS–HW g(r) is observed at a distance of 1.55 Å, while the first peak in the OS–OW g(r) is observed at a distance of 2.55 Å. At this concentration of DMSO, it is to be expected that the structure of the solutions corresponds to the existence of predominant 1(DMSO):2(H2O) aggregates. | ||
| Fig. 1 Atomic pair correlations for the various molecular sites in the solutions: (a) water–water pair correlations, (b) OS–water pair correlations, (c) Me–water pair correlations, and (d) Me–Me pair correlations. | ||
Fig. 1c shows the pair correlations between the methyl groups of DMSO and the water sites: Me–OW and Me–H. Once again, the increase in temperature produces a broadening of the peaks of the radial distribution functions. The similar positions of the first peaks reveal a nearly tangential arrangement of water molecules around the methyl groups, as has been reported earlier for the methyl groups of ethane in water.64 Such a geometric arrangement of water around non-polar groups is characteristic of hydrophobic hydration, allowing for hydrogen bonds to be maintained albeit at an entropic cost. Fig. 1d shows the pair correlation between the methyl groups of DMSO, i.e., the average tendency for aggregation of these non-polar groups in aqueous solutions. The peaks reveal a lack of any clear temperature trend, the main fingerprint of hydrophobic interactions,64–67 as the tendency for aggregation seems to decrease from 298 to 318 K before increasing significantly at 338 K. Similar simulation times but significantly smaller solute concentrations have been shown to be adequate enough to reveal the hydrophobic tendencies of small solutes in aqueous solution.65–67 Two inter-related factors make it difficult to judge whether hydrophobic effects should be expected in these solutions: the ratio of DMSO to water is nearly 1∶17 and there is very strong hydrogen bonding between water and the OS of DMSO. The well-defined strong aggregates mentioned earlier are thus likely to be the predominant interaction present in the solutions, and so truly temperature-dependent weak hydrophobic interactions might not be apparent except at very dilute DMSO concentrations. We can conclude that, along with the strong water–water interactions (responsible for the hydrophobic hydration of the non-polar Me groups of DMSO), the strong hydrophilic DMSO–water interactions dominate the behaviour of these solutions within the normal temperature range studied here.
3.3. Water structure
Since water molecules can establish an effective hydrogen-bonded network around the DMSO molecule, we decided to look at the orientational correlations between water molecules in the hydration shell of the Me and S (“hydrophobic” water) and OS (“hydrophilic” water) groups of DMSO. For this purpose, all water molecules within a 3.2 Å cut-off distance from the OS atom were classified as “hydrophilic water”, while all water molecules within a 5.5 Å cut-off distance from the Me group or within a 4.2 Å cut-off distance from the S atom were classified as “hydrophobic water”. These cut-off distances coincide with the positions of the first minimum in the OS–OW, Me–OW and S–OW pair correlation functions, respectively. A water molecule within the cut-offs of OS and either S or Me was classified as “hydrophilic” only, ensuring that “hydrophobic” water molecules were in the vicinity only of the non-polar groups of DMSO. The vector joining either the methyl or OS group of DMSO to the water oxygen (OW) subtends an angle α with the water dipole moment vector and an angle β with a vector perpendicular to the H–O–H molecular plane. Fig. 2 illustrates the definition of these angles. | ||
| Fig. 2 Definition of the water orientational α and β angles. A vector (labelled r) joining a DMSO group (such as Me or OS) with the OW of water subtends an α angle with the water dipole moment vector (labelled μ). The same r vector subtends a β angle with a vector (labelled τ) perpendicular to μ, which lies in the H–O–H plane. | ||
Fig. 3a shows the normalised (after division by the sine of the angle) distributions of the α and β angles for both hydrophilic and hydrophobic water, at 298 K. For hydrophobic water, the peaks near 70° for the α angle and the preferred nearly parallel/antiparallel orientations of the β angle correspond to a nearly tangential orientation of water molecules in the first hydration shell, as already mentioned in our analysis of the Me–water pair correlations. This arrangement in the vicinity of a non-polar group allows water molecules to straddle the surface of the group and maintain nearly tetrahedral hydrogen-bond coordination.63–68,69 There are no significant changes in these orientational distributions upon increasing the temperature, as can be see in Fig. 3b.
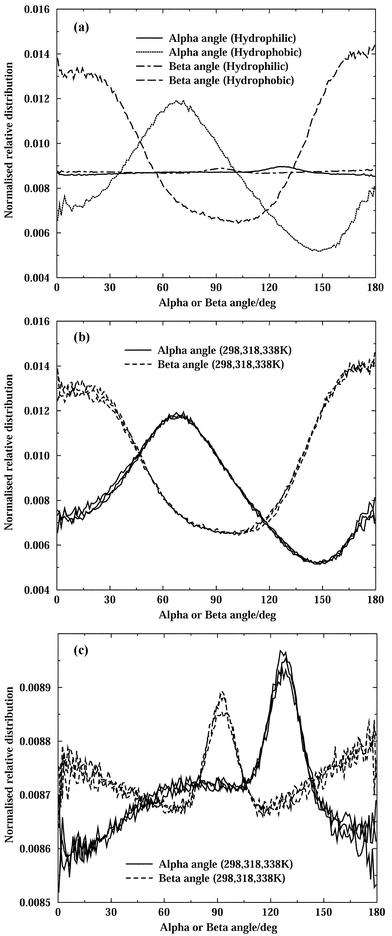 | ||
| Fig. 3 α- and β-angle distributions: (a) hydrophilic and hydrophobic water molecules at 298 K, (b) hydrophobic water molecules at all temperatures, and (c) hydrophilic water molecules at all temperatures. | ||
In the case of the hydrophilic water molecules, Fig. 3a shows that the α- and β-angle distributions are flat in relation to the distributions for hydrophobic water molecules. However, a rescaling of these distributions, as shown in Fig. 3c for all temperatures, reveals a clear solvation pattern around the sulfonyl group consistent with hydrogen bonding between this group and the neighbouring water molecules. In particular, the distribution is consistent with the oxygen of the sulfonyl group accepting nearly linear hydrogen bonds from neighbouring water molecules, as expected.49 All temperatures are plotted together, and although there seems to be a level of broadening of the distributions as the temperature increases, the noise level is too high. This reveals that the hydrogen-bonded DMSO–water aggregates are strongly held together and that the orientational preferences involved are not significantly disturbed by the rise of temperature.
3.4. Hydrogen-bonding structure
We carried out an analysis of the structural properties of the aqueous hydrogen-bonded network in the vicinity of the hydrophobic (Me and S) and hydrophilic (OS) groups of DMSO, as defined by the position of the first minimum in the previously calculated pair correlation functions. Bulk water was defined as all those water molecules not in the vicinity of these groups. As in earlier studies,63,68,70 water molecules were considered for possible hydrogen bonding if their oxygens were ≤3.5 Å apart. The hydrogen bond between two such water molecules is then chosen on a geometric criterion63 as the one having the minimum OW–HW distance (hydrogen-bond length) among the four possible combinations of intermolecular OW–HW distances. A hydrogen-bond angle is then the angle formed between the OW–HW bond vector of one water molecule and the intermolecular OW–HW hydrogen-bond vector with another water molecule. A hydrogen bond is defined to exist if it has a maximum length (H–O) of 2.5 Å and a hydrogen-bond angle between 130° and 180°.63 Within this definition, “strong” hydrogen bonds are shorter in length and closer to a linear geometry (an angle of 180°). All hydrogen bonds made between water molecules belonging to a different category (bulk, hydrophilic or hydrophobic) are taken into account twice: once for each water category.We have found that as the temperature increases, for all bulk, hydrophilic and hydrophobic water molecules, the average hydrogen-bond length decreases slightly, as can be seen in Figs. 4a to 4c. The most likely length of about 1.8 Å remains the same at all temperatures, as observed before in aqueous solutions of hydrocarbons.63,70 When comparing the different kinds of water molecules, hydrophobic water molecules show the sharpest distributions, followed by hydrophilic water and then closely by bulk water. This reveals that there is a slight enhancement of structure (more short hydrogen bonds). The same behaviour is observed at all temperatures.
 | ||
| Fig. 4 Hydrogen-bond length distributions at all temperatures: (a) bulk water, (b) hydrophilic water, and (c) hydrophobic water. | ||
We have also observed that, as the temperature increases, for all bulk, hydrophilic and hydrophobic water molecules, the average hydrogen-bond angle decreases, as can be seen in Figs. 5a to 5c. The most likely hydrogen-bond angle goes from 165° at 298 K to 162/163° at 338 K, which again resembles the behaviour seen in aqueous solutions of hydrocarbons.63,70 When comparing the different kinds of water molecules, there is an enhancement (more linear hydrogen bonds) going from bulk to hydrophilic to hydrophobic water. The same behaviour is observed at all temperatures.
 | ||
| Fig. 5 Hydrogen-bond angle distributions for all kinds of water molecules: (a) 298 K, (b) 318 K, and (c) 338 K. | ||
The behaviour of the hydrogen-bond lengths and angles in the various regions of the solution reveals that the polar OS group of DMSO is a strong hydrogen-bonding group that promotes an enhancement in the water structure. This is revealed by the presence of shorter and more linear hydrogen bonds between water molecules in the vicinity of this group. A further enhancement is seen around the non-polar Me and S groups of DMSO, where hydrogen bonds between water molecules are seen to be even shorter and more linear than in the bulk of the solution, in resemblance to the behaviour that has been observed in the hydration shell of purely non-polar substances in aqueous solution.70 Consequently, both the hydrophilic and hydrophobic groups of DMSO are seen to enhance the structure of water. It is possible that a further enhancement of the hydration structure around the polar OS group of DMSO could be observed if a polarisable potential of water were used. However, the pairwise TIP4P and P2 potentials of water and DMSO used in this work seem to capture the main structuring features that exist around such a polar group in aqueous solution.
The hydrophobic hydration of the Me and S groups of DMSO seems to be even further stabilised by the strong hydrophilic hydration of the OS group. This is suggested by the above water structure analysis, which reveals that the orientational preferences of water molecules around the Me and S groups (as well as the OS group) are rather temperature-independent. However, in this case, an enhanced hydrophobic hydration shell around the Me groups of DMSO does not seem to lead to an enhanced hydrophobic interaction between these groups, as shown earlier. This is likely to be due to the strength of the hydrophilic DMSO–water interactions around the polar OS group of DMSO, which give rise to 1(DMSO):2(H2O) aggregates that dominate the overall interaction between DMSO molecules.
In earlier studies of non-polar substances in water,70–72 an analysis of the average number of hydrogen bonds and, in particular, of the fraction of broken hydrogen bonds has proven extremely useful for rationalising the thermodynamics of hydrophobic hydration. We have followed the same method as in previous studies.70,72 The average number of hydrogen bonds (Nhb) was calculated for bulk, hydrophilic and hydrophobic water molecules. The fraction of broken hydrogen bonds was then obtained as
| f=(4−Nhb)/2 | (1) |
The results at each temperature for the different aqueous regions can be seen in Table 3. A plot of the fraction of broken hydrogen bonds in each region is shown in Fig. 6. We can see that as the temperature increases, the average number of hydrogen bonds decreases for all kinds of water molecules, indicating that more hydrogen bonds are broken as the temperature rises. This is confirmed by the increase in the fraction of broken hydrogen bonds, again showing that as the temperature increases the fraction of broken hydrogen bonds increases for all water types. It can also be seen that bulk water has the smallest fraction of broken hydrogen bonds (and hence the largest number of hydrogen bonds), which increases when going to hydrophilic water and being the largest for hydrophobic water molecules (which hence have the lowest number of hydrogen bonds). As has been reported in the past, this shows that there are fewer hydrogen bonds in the vicinity of a non-polar group compared to the bulk and a hydrophilic region of a solute (as shown here), although water molecules near a non-polar group will have a more ordered or enhanced structure (revealed by its orientational properties). There is a slight tendency for the difference in the fraction of broken H-bonds between either bulk and hydrophobic water or hydrophilic and hydrophobic water to increase with increasing temperature. This behaviour had already been predicted theoretically73 and observed in computer simulations of non-polar substances in water.70–72
| T/K | Nhb/f (bulk) | Nhb/f (hydrophilic) | Nhb/f (hydrophobic) |
|---|---|---|---|
|
a
T=temperature. Nhb=average number of hydrogen bonds (with standard deviations in brackets). f=fraction of broken hydrogen bonds.
|
|||
| 298.1 | 3.783 (0.180)/0.109 | 3.547 (0.041)/0.227 | 3.249 (0.142)/0.375 |
| 318.1 | 3.714 (0.199)/0.143 | 3.483 (0.044)/0.258 | 3.190 (0.136)/0.405 |
| 338.0 | 3.637 (0.193)/0.181 | 3.422 (0.047)/0.289 | 3.095 (0.149)/0.452 |
 | ||
| Fig. 6 Temperature dependence of the fraction of unbroken hydrogen bonds for all kinds of water molecules. | ||
At all temperatures, when comparing the different kinds of water molecules, it can be seen that bulk water molecules have the highest average number of hydrogen bonds, followed by hydrophilic water molecules and then hydrophobic water molecules. More importantly, the above results show that there is a small hydrogen-bonding penalty experienced by the water molecules in the vicinity of the OS group of DMSO, although this loss of water–water hydrogen-bonding is clearly compensated by the formation of strong DMSO–water hydrogen bonds.46,49 These results provide further evidence that the hydrophilic hydration of the OS group of DMSO is the predominant interaction.
3.5. Hydrogen-bonding dynamics
We also investigated the time-dependent behaviour of the hydrogen-bonding network in the various regions of the DMSO solutions. For this purpose, we calculated hydrogen-bond breaking functions by obtaining the fraction of unbroken hydrogen bonds in time from a histogram containing the number of hydrogen bonds that break after a given time,74 as implemented in the study of hydrophobic hydration.63Figs. 7a to 7c show the time decay functions thus obtained. As far as the effect of temperature is concerned, for all bulk, hydrophilic and hydrophobic water molecules, the fraction of unbroken hydrogen bonds decays more rapidly with time, showing a decrease in hydrogen-bond lifetimes as temperature rises, as expected.63 | ||
| Fig. 7 Hydrogen-bond breaking functions for all kinds of water molecules: (a) 298 K, (b) 318 K, and (c) 338 K. | ||
When comparing the different kinds of water molecules, the fraction of unbroken hydrogen bonds decays progressively more rapidly with time along the series: hydrophilic, hydrophobic and bulk water, showing that the lifetime of hydrogen bonds between water molecules in the vicinity of the OS group of DMSO is the longest, followed by hydrogen bonds between water molecules in the vicinity of the Me and S groups, and then by hydrogen bonds between bulk water molecules. The same behaviour is seen at all temperatures.
This complements earlier reports,46 which established a correlation between the stronger OS–HW hydrogen bonds (in relation to water–water hydrogen bonds) and the longer lifetimes of such hydrogen bonds. The slower hydrogen-bond breaking dynamics in aqueous DMSO solutions compared to pure water have been interpreted in terms of a reduced likelihood of fluctuations of the hydrogen-bond network in relation to the presence of free hydrogen-bonding sites, as hydrogen bonds most frequently break during a process of switching allegiances when a newly formed hydrogen bond replaces a broken one.75 However, the actual mechanism of hydrogen bond breaking does not seem to involve exclusively large amplitude librations.76
We have seen that hydrogen bonds between water molecules near the OS group of DMSO are long-lived, while those hydrogen bonds between water molecules hydrating the non-polar Me and S groups of DMSO break up faster, while the hydrogen bonds between bulk water molecules are short-lived. This is again likely to be the result of the strong hydrophilic DMSO–water correlations, in particular those arising from the hydrogen bonding of water with the OS group. It is important to mention here that the dynamics of hydrogen bonds are expected to be slowed down with respect to pure water on thermodynamic grounds: in the case of hydrophilic water there is an energetic factor involved in the stronger DMSO–water hydrogen bonds, which in the case of hydrophobic water there is an entropic factor involved in the reduction of the number of possible ways that water can form hydrogen bonds in the vicinity of the non-polar Me and S groups.76
4. Conclusions
We have carried out the first series of MD simulations of DMSO in water at different temperatures to investigate the temperature dependence of the hydrophilic and hydrophobic behaviour of water around DMSO at a concentration of 0.055 mole fraction. Our simulations indicate the existence of an enhancement in the structure of water around both the hydrophilic and hydrophobic portions of DMSO with respect to the bulk of the solution. This is revealed by the existence of a well-defined hydration structure around the OS group of DMSO, with clear directional linear hydrogen-bonding with water molecules. Water molecules in the vicinity of this group lose some hydrogen bonds with other water molecules, but this is compensated by the formation of stronger hydrogen bonds with DMSO. Such interactions increase the lifetime of the shorter and more linear water–water hydrogen bonds in the vicinity of the OS group. The effect of temperature on all these effects is what would be expected from the increased thermal energy of the system.There is clear evidence of hydrophobic hydration around the Me and S groups of DMSO, as shown by the formation of an even more ordered hydration structure around these groups, with strong water–water hydrogen bonds that have longer lifetimes than in the bulk of the solution. This region has the largest fraction of broken water–water hydrogen bonds, followed by the hydrophilic region around the OS group of DMSO and then the bulk of the solution. On the other hand, no evidence for a temperature-dependent hydrophobic interaction between the Me groups was observed.
These results reveal that the balance between hydrophilic and hydrophobic interactions is clearly shifted in favour of the former. The increase in temperature did not produce an enhancement of the hydrophobic tendency for aggregation between the Me groups of DMSO, while the orientational preferences of water molecules in the vicinity of the whole of the DMSO molecules remained rather temperature independent. The overall picture that has emerged is that the hydrophilic interactions between water and the OS group of DMSO dominate the structural properties of these solutions within the temperature range studied and, while seemingly favouring the hydrophobic hydration of the Me and S groups of DMSO, do not allow a for truly temperature-dependent hydrophobic interaction to be observed between the Me groups of the DMSO molecules.
Acknowledgements
RLM is also a Research Fellow of Hughes Hall, Cambridge, United Kingdom. Part of this work (MC, JS) was carried out within the project No 70/4/6485AU of the University of Athens, Greece.References
- Dimethyl Sulfoxide, ed. S. W. Jacobs, E. E. Rosenbaum and D. C. Wood, Marcel Dekker, New York, 1971 Search PubMed.
- D. Martin and H. G. Hauthal, Dimethyl Sulfoxide, Wiley, New York, 1975 Search PubMed.
- C. M. Kinart, W. J. Kinart and L. Skulski, Pol. J. Chem., 1986, 60, 879 CAS.
- G. V. Roshkovskii, R. A. Ovchinnikova and N. V. Penkina, Zh. Prikl. Khim., 1982, 55, 1858 Search PubMed.
- J. M. G. Cowie and P. M. Toporowski, Can. J. Chem., 1964, 39, 2240.
- J. Mazurkiewicz and P. Tomasik, J. Phys. Org. Chem., 1990, 3, 493 CrossRef.
- U. Kaatze, M. Brai, F.-D. Sholle and R. Pottel, J. Mol. Liq., 1990, 44, 197 CrossRef CAS.
- M. Y. Doucet, F. Calmes-Perault and M. T. Durand, C. R. Hebd. Seances Acad. Sci., 1965, 260, 1878 Search PubMed.
- E. Tommila and A. Pajunen, Suom. Kemistil., 1969, B41, 172 Search PubMed.
- A. Luzar, J. Chem. Phys., 1989, 91, 3603 CrossRef CAS.
- H. L. Clever and S. P. Pigott, J. Chem. Thermodyn., 1971, 3, 221 CAS.
- M. F. Fox and K. P. Whittingham, J. Chem. Soc., Faraday Trans. 1, 1975, 71, 1407 RSC.
- J. Kenttammaa and J. J. Lindberg, Suom. Kemistil., 1960, B33, 32 Search PubMed.
- D. H. Rasmussen and A. P. Mackenzie, Nature, 1968, 220, 1315 CAS.
- A. Luzar, J. Mol. Liq., 1990, 46, 221 CrossRef CAS.
- Q. F. Ahkong, D. Fischer, W. Tampion and J. A. Lucy, Nature, 1975, 253, 194 CAS.
- T. J. Anchordoguy, J. F. Carpenter, J. H. Crowe and L. M. Crowe, Biochim. Biophys. Acta, 1992, 117, 1104.
- J. E. Lovelock and M. W. H. Bishop, Nature, 1959, 183, 1394 CAS.
- T. J. Anchordoguy, C. A. Ceccini, J. N. Crowe and L. M. Crowe, Cryobiology, 1991, 28, 467 CAS.
- S. W. Jacob and R. Herschler, Cryobiology, 1986, 23, 14 CAS.
- J. R. Willigan and J. F. Ward, Radiat. Res., 1994, 137, 295.
- H. Bertagnolli, E. Schultz and P. Chieux, Ber. Bunsenges. Phys. Chem., 1989, 93, 88 Search PubMed.
- G. J. Safford, P. C. Schaffer, P. S. Leung, G. F. Doebbler, G. W. Brady and E. F. X. Lyden, J. Chem. Phys., 1969, 50, 2140 CAS.
- G. Brink and M. Falk, J. Mol. Struct., 1970, 5, 27 CrossRef CAS.
- A. Burneau, J. Mol. Liq., 1990, 46, 99 CrossRef CAS.
- Y. Higashigaki, D. H. Christensen and C. H. Wang, J. Phys. Chem., 1981, 85, 2531 CrossRef CAS.
- H. Kelm, J. Klowoski and E. Steger, J. Mol. Struct., 1975, 28, 1 CrossRef CAS.
- A. Bertulozza, S. Bonora, M. A. Battaglia and P. Monti, J. Raman Spectrosc., 1979, 8, 231.
- A. Allerhand and P. von R. Schleyer, J. Am. Chem. Soc., 1963, 85, 175.
- D. E. Bowen, M. A. Priesand and M. P. Eastman, J. Phys. Chem., 1974, 78, 2611 CrossRef CAS.
- W. M. Madigsky and R. W. Warfield, J. Chem. Phys., 1983, 78, 1912 Search PubMed.
- T. Kondo, L. L. Kirschenbaum, J. Kim and P. Riesz, J. Phys. Chem., 1993, 97, 522 CrossRef CAS.
- T. Tokuhiro, L. Menafra and H. H. Szmant, J. Chem. Phys., 1974, 61, 2275 CrossRef CAS.
- J. A. Glasel, J. Am. Chem. Soc., 1970, 92, 372 CrossRef CAS.
- K. J. Packer and D. J. Tomlinson, Trans. Faraday Soc., 1971, 67, 1302 RSC.
- T. Freech and H. G. Hertz, Z. Phys. Chem. (Munich), 1984, 142, 43.
- B. C. Gordalla and M. D. Zeidler, Mol. Phys., 1986, 59, 817 CAS.
- B. C. Gordalla and M. D. Zeidler, Mol. Phys., 1991, 74, 975 CAS.
- E. S. Barker and J. Jonas, J. Phys. Chem., 1985, 89, 1730 CrossRef.
- W. Feder, H. Dreizler, H. D. Rudolph and V. Trypke, Z. Naturforsch. A, 1969, 24A, 266.
- E. S. Verstakov, P. S. Yastremskii, Y. M. Kessler, V. Goncharov and V. V. Kokovin, Zh. Strukt. Khim., 1980, 21, 91 Search PubMed.
- U. Kaatze, R. Pottel and M. Schafer, J. Phys. Chem., 1989, 93, 5623 CrossRef CAS.
- T. A. Novskova, V. I. Gaiduk, V. A. Kudryashova and Y. I. Khurgin, Sov. J. Chem. Phys., 1991, 8, 1636 Search PubMed.
- B. G. Rao and U. C. Singh, J. Am. Chem. Soc., 1990, 112, 3803 CrossRef CAS.
- I. I. Vaisman and M. L. Berkowitz, J. Am. Chem. Soc., 1992, 114, 7889 CrossRef CAS.
- A. Luzar and D. Chandler, J. Chem. Phys., 1993, 98, 8160 CrossRef CAS.
- A. K. Soper and A. Luzar, J. Chem. Phys., 1992, 97, 1320 CrossRef CAS.
- A. Luzar, A. K. Soper and D. Chandler, J. Chem. Phys., 1993, 99, 6836 CrossRef CAS.
- A. K. Soper and A. Luzar, J. Phys. Chem., 1996, 100, 1357 CrossRef CAS.
- I. A. Borin and M. S. Skaf, J. Chem. Phys., 1999, 110, 6412 CrossRef CAS.
- A. Vishnayakov, A. P. Lyubartsev and A. Laaksonen, J. Phys. Chem., 2001, 105, 1702 Search PubMed.
- M. Chalaris and J. Samios, J. Mol. Liq., 2002, 98–99, 399 CAS.
- B. Kirchner and J. Hutter, Chem. Phys. Lett., 2002, 364, 497 CrossRef CAS.
- K. Refson, Comput. Phys. Commun., 2000, 126, 310 CrossRef CAS.
- A. Dullweber, B. Leimkuhler and R. McLachlan, J. Chem. Phys., 1997, 107, 5840 CrossRef CAS.
- K. Refson, manuscript in preparation.
- S. D. Bond, B. J. Leimkuhler and B. B. Laird, J. Comput. Phys., 1999, 151, 114 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Clarendon Press, Oxford, 1993 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- M. Ferrario, M. Haughley, I. R. McDonald and M. L. Klein, J. Chem. Phys., 1990, 93, 5156 CrossRef.
- M. L. Strader and S. E. Feller, J. Phys. Chem. A, 2002, 106, 1074 CrossRef CAS.
- R. L. Mancera, POLYatomic Monte Carlo simulation code, POLYMC, version 1.0 Search PubMed.
- R. L. Mancera and A. D. Buckingham, J. Phys. Chem., 1995, 99, 14632 CAS.
- N. T. Skipper, Chem. Phys. Lett., 1993, 207, 424 CrossRef CAS.
- R. L. Mancera and A. D. Buckingham, Chem. Phys. Lett., 1995, 234, 296 CrossRef CAS.
- N. T. Skipper, C. H. Bridgeman, A. D. Buckingham and R. L. Mancera, Faraday Discuss., 1996, 103, 141 RSC.
- R. L. Mancera, A. D. Buckingham and N. T. Skipper, J. Chem. Soc., Faraday Trans., 1997, 93, 2263 RSC.
- P.-L. Chau and R. L. Mancera, Mol. Phys., 1999, 96, 109 CrossRef CAS.
- I. I. Vaisman, F. K. Brown and A. Tropsha, J. Phys. Chem., 1994, 98, 5559 CrossRef CAS.
- R. L. Mancera, J. Chem. Soc., Faraday Trans., 1996, 92, 2547 RSC.
- K. E. Laidig and V. Daggett, J. Phys. Chem., 1996, 100, 5616 CrossRef CAS.
- R. L. Mancera, J. Phys. Chem. B., 1999, 103, 3774 CrossRef CAS.
- N. Muller, Acc. Chem. Res., 1990, 23, 23 CrossRef CAS.
- M. Haughney, M. Ferrario and I. R. McDonald, J. Phys. Chem., 1987, 91, 4934 CrossRef CAS.
- A. Luzar, Faraday Discuss., 1996, 103, 29 RSC.
- J. T. Cabral, A. Luzar, J. Teixeira and M-C. Bellissent-Funel, J. Chem. Phys., 2000, 113, 8736 CrossRef CAS.
Footnote |
| † Present address: Hellenic Fire Corps Headquarters, Mourouzi Str.10172, Athens, Greece. |
| This journal is © the Owner Societies 2004 |