Selective 1,3-complexation of p-tBu-calix[4]arene by [TiCp2Me2]
Antonella J.
Petrella
a,
Nicholas K.
Roberts
a,
Donald C.
Craig
a,
Colin L.
Raston
b and
Robert N.
Lamb
a
aSchool of Chemical Sciences, University of New South Wales, Sydney, Australia. E-mail: r.lamb@unsw.edu.au
bSchool of Biomedical and Chemical Sciences, University of Western Australia, Crawley, Perth, Australia. E-mail: clraston@chem.uwa.edu.au
First published on 25th November 2003
Abstract
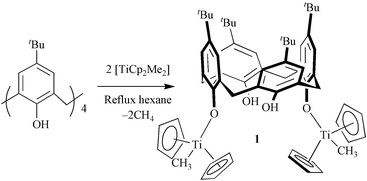
Reaction of p-tBu-calix[4]arene with dimethyl titanocene results in high yield selective 1,3-dimetallation of the calixarene in the cone conformation by selective cleavage of one methyl group.
Calix[4]arenes represent a unique class of partially constrained polyalkoxide ligand, and their variable conformations permit a variety of metal binding environments, and even the simultaneous binding of several metal centres.1 In the ubiquitous cone conformation, a calix[4]arene presents a preorganised set of four pendant oxygen atoms for metal ion complexation, which generally leads to the formation of molecular assemblies where the metal centres bridge two calix[4]arene molecules through phenolate moieties.1 Mono-calix[4]arene metal complexes have been made using partially O-alkylated derivatives.2 In contrast, mono-nuclear complexes of underivatised calixarene acting as one quadridentate ligand to one metal centre are much less common, since the quasi-planar set of four phenoxy groups can only accommodate one side of the metal coordination sphere. Moreover, the metal precursors generally used to form metal complexes (homoleptic metal amides, chlorides, and alkyls) typically undergo complete displacement of all their ligands, thereby opening up sites for bridging metal centres. Notwithstanding this generality, there are some cases where the selective displacement of ligands from metal precursors allows the formation of several mononuclear species.3
Our interest in forming titanium/calix[4]arene complexes devoid of bridging phenolate moieties prompted us to investigate the reaction of [TiCp2Cl2] (Cp = η-C5H4) with alkali metal dianions formed by deprotonation of p-tBu-calix[4]arene, [tBu–L(OH)4]. The incorporation of Cp groups was intended to circumvent formation of metallo-bridged species through steric buttressing. This proves to be the case, as manifested in the formation of a novel mono-calixarene/tetra-nuclear complex [tBu–L(O)4Ti4O4Cp4],4 albeit with the cleavage of one Cp ligand per titanium centre. We were unable to form a complex incorporating the titanocene fragment intact, under a variety of conditions involving [TiCp2Cl2]. In general metallocene bis-alkoxides are not easily accessible through salt metathesis reactions involving [TiCp2Cl2] due to competing Cp displacement and disproportionation reactions, typically affording a complex mixture of products, complicating the isolation of the dialkoxides.5 In contrast p-tBu-calix[6]arene readily reacts selectively with one or two equivalents of [TiCp2Cl2] with cleavage of both chloro ligands and one Cp.6 A clean reaction of [tBu–L(OH)4] could, however, be obtained by replacing the metallocene dichloride by the corresponding dimethyl metallocene, [TiCp2Me2], affording a complex bearing two metallocene units attached to a single calixarene. The expectation was that the more reactive di-alkyl would react under mild conditions, in a solvent of low polarity, through selective protonolysis of the alkyl groups. Surprisingly only one methyl group is displaced from each titanium centre, the resulting product, 1,† comprising the calix[4]arene bearing two TiCp2Me groups in the 1,3 arrangement, eqn. (1).
 | (1) |
The reaction of [TiCp2Me2] with tBu–L(OH)4 was investigated under a range of conditions. A 1 : 1 product could not be isolated from reactions involving a 1 : 1 ratio of [TiCp2Me2] to tBu–L(OH)4. 1H NMR spectroscopy revealed that two equivalents of [TiCp2Me2] react with one equivalent of calix[4]arene, affording a mixture of 1 and native calixarene. This result was insensitive to both the choice of solvent (toluene, hexane, ether) and the temperature of the reaction. Optimum conditions for the formation of 1 were under reflux in hexane, with solid [TiCp2Me2] being added to a suspension of tBu–L(OH)4 in hexane. Complex 1 deposited as a pale yellow solid which was isolated easily by filtration in air (81% yield). Remarkably, the complex is stable in air, and its existence as a hemihydrate is testimony of its stability towards hydrolysis.
The finding that two equivalents of [TiCp2Me2] react selectively with the calix[4]arene is reminiscent of the known regio-selective 1,3-di-O-alkylation of the same calix[4]arene.7 To understand the provenance of the present 1,3-dimetallated complex it is tempting to draw parallels with the known alkylation process. The regiospecificity in the latter process is believed to arise by the initial mono-alkylation augmenting the acidity of the proton in the 3-position of the calix[4]arene. While this is also possible in the present study, another explanation is that the initial mono-titanocene complex is more soluble in hexane than the parent calix[4]arene, rendering it available for a second metallation. In this scenario, steric hindrance, which is readily perceptible in the X-ray structure of 1, could account for the high regioselectivity.
Despite the higher reactivity of [ZrCp2Me2] relative to the titanium analogue towards protic reagents, it gave no analogous reaction with p-But-calix[4]arene in refluxing hexane, toluene or mesitylene, even until the onset of decomposition of [ZrCp2Me2] in the latter. The reactivity of the dimethyl titanocene reagent may relate to its lower thermal stability relative to the dimethyl zirconocene. Whereas [TiCp2Me2] is thermally (and light) sensitive,8 [ZrCp2Me2] is thermally (and light) stable, being readily purified by sublimation at 110 °C.9 Although the thermal decomposition of [TiCp2Me2] has been the subject of numerous investigations,10 the exact mechanism of decomposition remains unclear. EPR studies11 have shown the presence of transitory reduced titanium species, suggesting that the reaction of [TiCp2Me2] with the calixarene could occur through a reduced coordinatively unsaturated decomposition intermediate such as TiCp2CH3.
In contrast, both [TiCp2Me2] and [ZrCp2Me2] were found to react with p-tBu-calix[6]arene in refluxing hexane, although affording a complex mixture (1H NMR). That [ZrCp2Me2] reacts with a calix[6]arene and not with a calix[4]arene may relate to the greater conformational flexibility of calix[6]arene as compared to calix[4]arene which may make the calix[6]arene phenols more accessible for reaction. Complex 1 crystallises from toluene in the space group Aba2,‡ with the asymmetric unit being two half molecules, the other half generated by a two fold rotation axis, Fig. 1.12 The calix[4]arene assumes the pinched cone conformation, with the 1,3-phenoxide groups coordinated to titanium having approximately parallel orientation. The pitch angles of the two complexed and uncomplexed phenyl planes are 85.9(5), 86.0(5) and 44.1(5), 44.0(5) relative to the plane of the four methylene carbon atoms of calixarene. The pinching of the calixarene cone precludes the incorporation of toluene in the calixarene cavity, although toluene is found only in interstitial sites in the structure. Coordination around each Ti(IV) centre is comprised of two perhapto-cyclopentadienyl ligands, and a phenolate and methyl groups. In respect to the Cp centroid, the O–Ti–Cp bonds angles in 1 range from 104.10(5)–115.0(5) ° and the Ti–Cp bond lengths from 2.07(5) and 2.13(5) Å, which are consistent with previously reported values for such complexes.4,6
 | ||
| Fig. 1 (a) (b) Molecular projections of complex 1. Key bond distances (Å) and angles (°): Ti1A–(O1A, C1M1, Cp1, Cp2) 1.902(6), 2.044(21), 2.073, 2.130, Ti1B–(O1B, C1M2, Cp3, Cp4) 1.899(5), 2.157(22), 2.076, 2.112, O1A–Ti1A–(C1M1, Cp1, Cp2) 103.8(8), 105.318, 110.744, O1B–Ti1B–(C1M2, Cp1, Cp2) 100.7(8), 115.01, 104.996, Cp represents the centroids of the C5H5 rings. | ||
Access to complex 1 in high yield is a significant advance in preparing metal calixarene complexes. Essentially the molecule is functionalised as a strong Lewis base (alkyl groups) and a weak Lewis acid (phenol groups), with scope for cleavage of one or more of the Cp moieties. These offer potential for using the complex to build up more sophisticated structures.
Notes and references
- Calixarenes 2001, eds. Z. Asfari, V. Bohmer, J. Harrowfield and J. Vicens, Kluwer, Dordrecht, 2001 Search PubMed; C. Wieser, C. B. Dieleman and D. Matt, Coord. Chem. Rev., 1997, 165, 93–136 Search PubMed.
- A. Zanotti-Gerosa, E. Solari, L. Giannini, C. Floriani, N. Re. Chiesi-Villa and C. Rizzoli, Inorg. Chim. Acta, 1998, 270, 298–311 CrossRef CAS.
- J. A. Acho, H. L. Doerrer and S. J. Lippard, Inorg. Chem., 1995, 34(10), 2542–56 CrossRef CAS; F. Corazza, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1991, 30, 4465 CrossRef CAS.
- A. J. Petrella, N. K. Roberts, M. Thornton-Pett, C. L. Raston and R. N. Lamb, Chem. Commun., 2003, 1238 RSC.
- M. Bottrill, P. D. Gavens, J. W. Kelland, J. McMeeking, Comprehensive Organometallic Chemistry, eds. G. Wilkinson, F. G. A. Stone and E. W. Abel, Pergamon Press, Oxford, 1982, 33 Search PubMed.
- A. J. Petrella, N. K. Roberts, D. C. Craig, C. L. Raston and R. N. Lamb, Chem. Commun., 2003, 1014 RSC.
- L. C. Groenen, B. H. M. Ruel, A. Casnati, W. Verboom, A. Pochini, R. Ungaro and D. N. Reinhoudt, Tetrahedron, 1991, 47(39), 8379–84 CrossRef CAS; K. Iwamoto, A. Yanagi, T. Arimura, T. Matsuda and S. Shinkai, Chem. Lett., 1990, 10, 1901–4; K. Iwamoto, A. Yanagi, K. Araki and S. Shinkai, Chem. Lett., 1991, 10, 473–6; C. D. Gutsche, B. Dhawan, J. A. Levine, H. No Kwang and L. J. Bauer, Tetrahedron, 1983, 39(3), 409–26 CrossRef CAS.
- K. Clauss and H. Bestian, Liebigs Ann. Chem., 1962, 654, 8–19 Search PubMed.
- P. C. Wailes, H. Weigold and A. P. Bell, J. Organomet. Chem., 1972, 34, 155 CrossRef CAS.
- G. J. Erskine, D. A. Wilson and J. D. McCowan, J. Organomet. Chem., 1976, 114(2), 119–25 CrossRef CAS; H. G. Alt, F. P. Di Sanzo, M. D. Rausch and P. C. Uden, J. Organomet. Chem., 1976, 107(2), 257–63 CrossRef CAS; G. A. Razuvauev, V. P. Mar'in and Yu. A. Andrianov, J. Organomet. Chem., 1979, 174(1), 67–75 CrossRef.
- G. A. Razuvauev, V. N. Latyaeva, L. I. Vyshinskaya and G. A. Kilyakova, Zh. Obshch. Khim., 1966, 36, 1491.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14.1, 33–38 CrossRef ; http://www.povray.org..
Footnotes |
| † Synthesis:. 1 To a suspension of tBu–L[OH]4 (1.0 g, 1.54 mmol) in hexane (50 ml) was added [TiCp2Me2] [0.70 g, 3.36 mmol]. The mixture was refluxed in hexane for 2 hours whereupon a bright yellow solid formed (1.3 g, 81%). 1H NMR (CDCl3, 300 MHz, 298 K): δ 7.04 (s, 4 H, aryl), 6.45 (s, 4 H, aryl), 6.11 (s, 20 H, Cp), 3.87 (d, 4 H, J = 12 Hz, CH2), 3.07 (d, 4 H, J = 12 Hz, CH2), 1.34 (s, 18 H, tBu), 0.96 (s, 6 H, CH3), 0.81 (s, 18 H, tBu). Crystals suitable for X-ray diffraction studies were grown from a concentrated toluene solution; data were collected at 150(2) K on a Bruker-AXS SMART 1000 CCD diffractometer with Mo–Kα radiation and the structure solved by direct methods (SIR92) and refined with a full matrix least-squares refinement on F (RAELS); (C66H78O4Ti2)·3(C7H8)·0.5(H2O), M = 1316.6, orthorhombic, a = 21.0123(4), b = 21.0224(3) c = 38.6495(6) Å, U = 17072.6(5) Å3, T = 150(2) K, space group Aba2, Z = 8, μ(Mo–Kα) = 0.226 mm−1, 105317 reflections measured, 9685 unique (Rint = 0.047). The final R = 0.119, and wR = 0.159 (observed data). |
| ‡ CCDC 217096. See http://www.rsc.org/suppdata/cc/b3/b309455c/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2004 |