Boron doped diamond electrode modified with iridium oxide for amperometic detection of ultra trace amounts of arsenic(III)
Abdollah Salimi†, Michael E. Hyde, Craig E. Banks and Richard G. Compton*
Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QZ
First published on 5th December 2003
Abstract
Boron doped diamond (BDD) electrodes modified by electrodeposition from hydrous iridium oxide (IrOx) have been developed for the detection of arsenic(III). Potential cycling is used to deposit films of hydrous iridium oxide onto boron doped diamond electrode from a saturated solution of alkaline iridium(III) solution. A stable reversible redox couple was observed at the surface of modified electrode in both acidic and basic solutions. The properties of iridium oxide films, stability and its electrochemical properties were investigated by atomic force microscopy (AFM) and cyclic voltammetry. The modified electrodes showed excellent electrocatalytic activity toward oxidation arsenic(III) over a wide pH range (2–8); also they showed an excellent analytical performance for the amperometric detection of arsenic(III). The detection limit, sensitivity, response time and linearity are 2 nM, 4.2 nA nM−1, 60 ms and 20 nM–50 µM. The precision for 10 replicate determinations of 40 µM arsenic was 0.80% (RSD). These analytical parameters compare favourably with those obtained with modern analytical techniques such as inductively coupled plasma mass spectrometry and hydride generation atomic fluorescence spectrometry. The advantageous properties of this modified electrode for arsenic determination are its inherent stability, excellent catalytic activity over a wide pH range, high sensitivity and simplicity.
Introduction
The detection of different forms of arsenic in industrial, biological and environmental samples has been the target of increasing attention in recent years. Many arsenic compounds are known to be toxic and the exposure of humans and animals especially, and ecosystems in general, to arsenic remain of international concern. The toxicity of arsenic depends strongly on its chemical form. The inorganic compounds are far more toxic than their organic metabolites. Four of the more toxic arsenic compounds are dimethylarsinate (DMA), arsenate (As(V)), monomethylarsonate (MMA)) and arsenite (As(III)) of which the later is most toxic.1 It is important to monitor even low concentrations of arsenite in aquatic systems.2For the determination of arsenic species in aquatic samples where the concentration of arsenic are usually below 1 µM L−1, highly sensitive techniques are required owing to the low concentration of the species. When the analytical techniques employed do not offer sufficient detection capability for such determination, preconcentration and separation steps are always necessary.
There are several options available to the analytical community for the detection of arsenic compounds including hydride generation atomic fluorescence spectrometry2–4 and inductively coupled plasma mass spectrometry.5,6 Unfortunately these embrace the most expensive instrumental methodologies. Recently electrochemical methods especially stripping voltammetry have been used for the detection of arsenic.7–11 Mercury electrodes have been used for these stripping methods but usually another metal such as copper is required to form an intermetallic compound with arsenic at the mercury electrode surface.9 Gold electrodes and also glassy carbon or pyrolytic graphite electrodes modified with gold films have been used successfully for the detection of arsenic with anodic stripping potentiometry and anodic stripping voltammetry.12–14 However, there are often problems associated with arsenic voltammetry at solid electrodes such as limited sensitivity, poor precision, low electron transfer reaction, high overvoltages at which the electron transfer process occurs, low stability over a wide range of solution composition and for optimum reproducibility the electrode surfaces were re-plated between each measurements, which makes this approach inconvenient for routine analysis. A rather disappointingly low sensitive method for the titration of As(III) with electrogenerated iodine in microelectrode arrays has been reported.15
The chemical modification of inert substrate electrodes with redox active thin films offers significant advantages in the design and development of electrochemical sensors. In operation, the redox active sites shuttle electrons between the substrate electrode and analytes with a significant reduction in activation overpotential and ideally, less surface fouling and oxide formation compared to inert substrate electrodes. A wide variety of compounds have been used as electron transfer mediator via modification of different electrode surfaces.
The use of boron doped diamond as a robust electrode substrate is well established due to its wide potential window in aqueous solutions,16 low background currents,17,18 long term stability19 and low sensitivity to dissolved oxygen.20 BDD has recently been utilized electroanalytically as a suitable electrode substrate for determination of several metals such as Ag,21 Pb,22 Cu23 and Mn.24 However As(III) is not oxidized inside the potential window of aqueous solutions at the surface of BDD electrodes. Accordingly for the detection of arsenic at the surface of BDD the surface should be modified with a thin film of electron transfer mediator. BDD25 and GC26 electrodes modified with iridium oxide have been used for the electrocatalytic oxidation of hydrogen peroxide and insulin. Also electrodeposited iridium oxides have been used as pH electrodes.27,28 Recently we reported the application of BDD electrodes modified with oxide layers of metals such as Ag,29,30 Sn31 and Pb.32
In the present study the BDD electrodes modified with electrodeposited iridium oxide are investigated by voltammetry and atomic force microscopy (AFM). The modified electrodes are used for the electrocatalytic oxidation of As(III), and finally the analytical performance of an iridium oxide modified BDD is described as an amperometric sensor for arsenic determination.
Experimental
Material
All reagents used were of analytical grade and used as received. Potassium hexachloroiridate(III) and sodium (meta)arsenite were obtained from Aldrich. All solutions and subsequent dilutions were prepared using deionized water from Vivendi Water Systems, UK with a resistivity of not less than 18 MΩ cm. The iridium containing deposition solution was prepared according to the method described by Baur.35 The first step in the preparation of this solution is the formation of the diaquaetetrachloroiridate(III) ion, Ir(H2O)2Cl4− from K3Ir(Cl)6.Accordingly a 1 mM solution of IrCl6−3 in 0.1 M HCl was aquated by heating at 80 °C for 2 h. The second step in the preparation of the deposition solution is the formation of iridium(III) oxide from Ir(H2O)2Cl4− with added base based according to following reaction.
 | (1) |
Prior to the addition of base, oxygen must be removed because the iridium(III) oxide is unstable in oxygen29. After removing the oxygen from the acidic solution of Ir(H2O)2Cl4−, the pH of solution was raised to 10.5 by adding anhydrous potassium carbonate, and this solution used for the deposition of iridium oxide onto the BDD electrode surfaces.
Instrumentation
Electrochemical analyses were carried out in a conventional three electrode cell using an μ-Autolab 2 PGSTAT computer controlled potentiostat (ECO-Chemie, The Netherlands). A boron doped diamond electrode (BDD, DeBeers Industry Diamond Division Ascot, supplied via Windsor Scientific, Slough, UK) was used as the working electrode; a platinum wire and saturated calomel electrode (SCE, Radiometer, Copenhagen) were used as the counter and reference electrodes.The boron doped diamond film electrode was housed in a Teflon mounting with an electrical connection to the rear graphite side made via a brass rod, attached using silver epoxy resin. The rotating disk electrode of BDD (5 × 5 mm BDD in Teflon) was made in the workshop of the Physical Chemistry Laboratory of Oxford University. The BDD electrodes modified with iridium oxide layers were characterized by atomic force microscopy (AFM). The AFM was a digital Instruments Multimode SPM., operating in ex situ Tapping Mode. A model J scanner was used, having a lateral range of 125 × 125 µm and a vertical range of 5 µm. Nanosensors NCH Tapping Mode tips having a spring constant of approximately 45 nm−1 and a resonant frequency of approximately 380 kHz were used.
Results and discussion
Electrodeposition of IrOx at BDD electrodes
Several methods have been developed for preparation of iridium oxide including thermal salt decomposition,33 reactive sputtering34 and electrochemical methods.25,26,35 Among these techniques the latter are very attractive, because they do not require high temperatures, they are comparable with a wide range of substrate materials, they favour the production of high effective surface areas and they allow large area film formation at room temperature. The cyclic voltametric,25,35 galvanostatic36 and potential pulsing27 methods have been used for electrodeposition of IrOx at the surface of different electrode materials. To our knowledge the simplest and fastest method for electodeposition of iridium oxide is cyclic voltammetry and in this study potential cycling was used to form iridium oxide layers at the surface of diamond electrodes.Boron doped diamond electrodes were carefully polished with diamond paste (0.1 µm), and then ultrasonically cleaned in distilled water. Then the clean electrode was immersed in the plating solution containing 0.5 mM iridium(III) complexes and cyclic voltammograms recorded with cycling of the potential from 0.2 to 1.2 V at scan rate 50 mV s−1. Fig. 1 shows that cyclic voltammograms resulting for the continuous cyclization of electrode potential (15 cycles). The currents on the consecutive scans increase indicating formation of an electroactive deposit on the electrode surface. The formation of iridium(IV) oxide layers is known to be initiated electrochemically at the surface of BDD electrodes based on the following reaction.35
| Ir2O3·xH2O (aq.) + 2OH− → 2IrO2·xH2O (s) + H2O + 2e− | (2) |
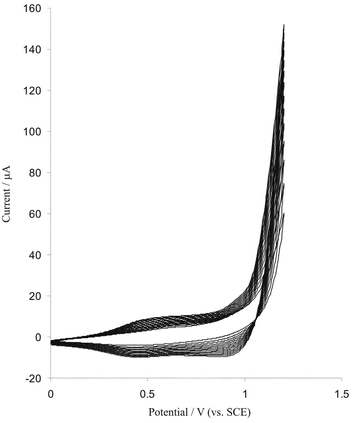 | ||
| Fig. 1 Cyclic voltammograms (15 cycles) at the bare BDD electrode (electrode surface area is 0.25 cm2) in a plating solution containing 0.5 mM Na3IrCl6 (pH = 10.5) scan rate 50 mV s−1. | ||
BDD electrodes modified with IrOx layers (prepared with 15 potential cycles in 0.5 mM iridium solution) were cleaned with distilled water and used for other applications as detailed below.
The electrochemical properties and stability of modified BDD electrodes
Cyclic voltammetry was used to study the stability and electrochemical properties of BDD electrodes modified with iridium oxide film layers. Fig. 2 shows typical cyclic voltammograms of modified and bare BDD electrodes at pH 2 recorded at a scan rate of 100 mV s−1. Nearly reversible voltammetric responses are observed whereas for an unmodified BDD electrode no signal was observed in the potential range −0.2 to 1.0 V. Similar results were observed at GC and BDD electrodes modified with iridium oxide.25–27,35 | ||
| Fig. 2 Cyclic voltammograms of bare and iridium oxide modified BDD electrodes at pH = 2, scan rate 100 mV s−1, electrode surface area is 0.25 cm2. | ||
Fig. 3 shows two atomic force microscopy images of polished BDD and BDD electrode modified with iridium oxide films (15 potential cycles in 0.5 mM iridium(III) solution). A thin film of iridium oxide is clearly deposited on the electrode surface.
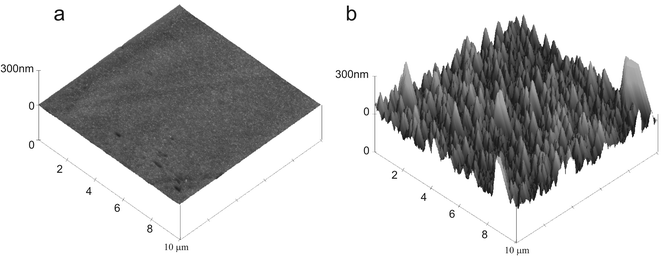 | ||
| Fig. 3 AFM images of (A) clean polished BDD surface and (B) iridium oxide modified BDD electrode. | ||
Fig. 4 shows the cyclic voltammograms of iridium oxide films at different scan rates in pH 7.4 phosphate buffer. A redox couple provides evidence for prolonged surface attachment: The ratio of the cathodic to anodic peak currents obtained at various scan rates (10–100 mV s−1) was almost unity, the anodic and cathodic peaks currents were proportional to scan rate and the peak to peak potential separations were small, about 20 mV at a scan rate of 10 mV s−1, as predicted for a diffusionless system. The apparent electrochemical (self-exchange) rate constant k0 for Ir(III)/Ir(IV) redox couple was calculated from Tafel diagrams according to the method described by Laviron.37 Anodic and cathodic transfer coefficients (αa, αc) and k0 were calculated from the slope and intercepts of plots Epversus log(v) for high scan rate. The slope of linear segment is equal to 2.303RT/αanF and the evaluated value for αa is 0.44. An electron transfer rate constant value of 2.4 s−1 was evaluated. Since the electron transfer rate constant for the iridium oxide film is high, it can be used as an excellent electron transfer mediator for electrocatalytic processes. The surface coverage (Γ) of iridium oxide films was evaluated from the following equation.
| Γ = Q/nFA | (3) |
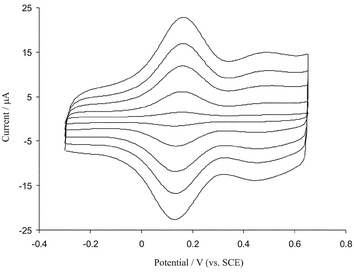 | ||
| Fig. 4 Cyclic voltammetric responses of BDD electrode (electrode surface area is 0.25 cm2) modified with iridium oxide in 0.1 M phosphates buffer (pH = 7.30) at scan rates (inner to outer) 20, 40, 60, 80 and 100 mV s−1. | ||
The stability of the IrOx films, electroplated from alkaline solutions of iridium, was investigated in background electrolyte solutions of different pH by continuous cycling of the electrode potential for over 500 cycles at a scan rate of 100 mV s−1. Cyclic voltammograms in Fig. 5A–C show that in phosphate buffer solutions with pH = 2, 7.4 and 11 after 500 cycles at scan rate 100 mV s−1 no significant decrease in peak currents and no potential shifts were observed suggesting no loss of the oxide from the electrode surfaces under potentiodynamic conditions. Also the stability of the modified electrode and the reproducibility of its electrochemical behaviour were investigated by cyclic voltammetry after storing it in a phosphate buffer solution for a long period of time (1–14 days) and then recording cyclic voltammograms. As shown in Fig. 5D after immersion of the modified electrodes in buffer solution (pH 7) for 2 weeks no recognizable change in the corresponding cyclic voltammograms was observed.
 | ||
| Fig. 5 The first and 500th cyclic voltammograms of iridium oxide film modified BDD electrodes in 0.1 M phosphate buffer solutions; (A) pH = 2,( B) pH = 7.4 and (C) pH = 11, scan rate 100 mV s−1. (D) Cyclic voltammograms of modified BDD electrode in pH = 7.3 (a), (b) same as (a) but after immersing the electrode for two weeks in phosphate buffer of pH = 7.3. Scan rate 50 mV s−1, electrode surface area is 0.25 cm2. | ||
These results indicate the excellent adherence of the iridium oxide layers on the surface of BDD electrodes. These materials can be potentially used as electrocatalytic oxide layers in electrochemical and sensor devices as is next described.
pH response of the iridium oxide layers on BDD electrodes
Iridium oxide electrodes exhibit high stability both in acidic and alkaline solutions but their pH response depends upon the conditions used during their preparation.32 To study the effect of pH on the electrochemical behaviour of the BDD electrodes modified with iridium oxide films the cyclic voltammograms of the modified electrode were recorded in electrolyte solutions over the pH range 1–12. Fig. 6 shows the results at a scan rate 40 mV s−1. To account for the electrochemical behaviour, the general reaction for the pH sensitivity of iridium oxide electrodes can be written as follows.35,36| Ir(IV) oxide + xH+ + ne− ⇆ Ir(III) oxide + yH2O | (4) |
 | ||
| Fig. 6 The cyclic voltammetry response of iridium oxide modified BDD electrode (electrode surface area is 0.25 cm2) at different pH 1–12 (from right to left); scan rate 40 mV s−1. Inset shows the variation of potential vs. pH. | ||
Electrocatalytic activity of modified BDD electrodes towards the oxidation of arsenic(III)
As mentioned above, the modified BDD with iridium oxide had a very stable electrochemical behaviour; it might therefore be usefully used as a sensor for analytical applications. One of the objectives of this work was to fabricate a modified electrode capable of the electrocatalytic oxidation of arsenic(III). In order to test the electrocatalytic activity of the modified electrodes, cyclic voltammograms were obtained in the absence and presence of 40 µM arsenic(III) in buffer solution (pH 4.3). Fig. 7 shows cyclic voltammograms for the electrooxidation of As(III) at a bare BDD and at modified surface electrodes. As is seen upon the addition 40 µM arsenic(III) there is a dramatic enhancement of the anodic peak current and the cathodic peak current disappeared (Fig. 8b), which indicate a strong catalytic effect. The anodic peak potential for the oxidation of arsenic is about 0.5 V while at the surface of bare BDD electrode arsenic(III) is not oxidized at the same potential range (Fig. 7c). Thus, a decrease in overall potential and enhancement peak current for arsenic oxidation is achieved with the modified electrode. In order to optimize the electrocatalytic response of the modified BDD electrode toward arsenic oxidation the effect of pH on the catalytic oxidation behaviour was investigated. The responses of iridium oxide film in 50 µM arsenic at different pH values were studied. With increasing pH the oxidation peak potential shifts to more positive values and is separated from the peak of iridium couple. The modified electrode shows the excellent electrocatalytic activity at pH range 1–10, but at pH > 6 the results are not repeatable and the activity of modified electrodes are decreased. Hence the pH value has an effect on the kinetics of the catalytic reaction; the optimum value for analysis is pH 4. Fig. 8 shows the dependence of the voltammetric response of modified BDD electrode on the addition of arsenic (10–60 µM); the anodic peak currents are increased. The dependence of peak current response on the concentration of arsenic was linear in the range 5–50 µM. Cyclic voltammograms for 50 µM arsenic(III) were recorded at different scan rates, the peak currents for the anodic oxidation of arsenic were proportional to the square root of the scan rate (not shown). These results indicate that at sufficiently positive potential the reaction is controlled by diffusion arsenic, which is the ideal case for quantitative applications. Also, it can be seen that the peak potential for the catalytic oxidation of arsenic shifts to positive values and the reverse scan peaks are observed to grow with increasing scan rate, suggesting a kinetic limitation in the reaction between the redox sites of iridium oxide and arsenic. Based on the results the following catalytic scheme (EC′) showed that IrO2 specifically catalysed arsenic oxidation in the following electrochemical catalytic pathway.| Ir (reduced form) → Ir (oxidized form) | (5) |
| Ir (oxidized form) + As(III) → Ir (reduced form) + As(V) | (6) |
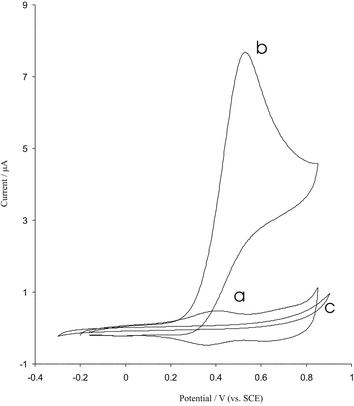 | ||
| Fig. 7 Cyclic voltammograms of modified BDD electrode in phosphate buffer pH = 4.3 at scan rate 10 mV s−1 in the absence (a) and presence (b) of 40 µM arsenic solution. (c) The cyclic voltammogram of the bare electrode in 40 µM arsenic solution, electrode surface area is 0.25 cm2. | ||
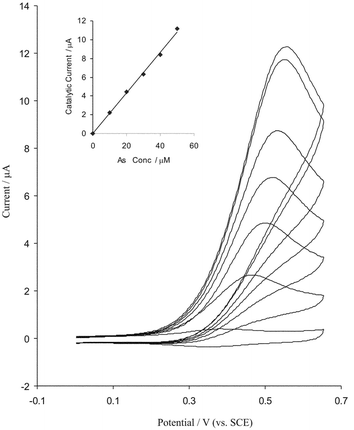 | ||
| Fig. 8 Cyclic voltammograms of modified BDD electrode in pH = 4.3 at scan rate 10 mV s−1 (electrode surface area is 0.25 cm2) with increasing the arsenic concentration (from inner to outer) 10, 20, 30, 40, 50, 60 and 70 µM arsenic. Inset, plot of peak current vs. arsenic concentrations. | ||
The stability of modified electrode and its catalytic activity for arsenic oxidation was investigated by recording the cyclic voltammograms of iridium oxide modified BDD electrodes before and after use in the presence of arsenic. As shown in Fig. 9 after cycling the potential for 90 cycles at a scan rate of 40 mV s−1 in a solution containing 50 µM arsenic, then recording cyclic voltammograms in the presence and absence of arsenic, the peak potentials are unchanged and the currents decreased by less than 2%. Also using the electrode again after leaving it unused for one week, the peak potential for arsenic oxidation were unchanged and the current signals showed only less than a 4% decrease of the initial response. In test of reproducibility, it was found that the relative standard deviation (RSD) of the cyclic voltammogram currents of 50 µM arsenic at pH 4.3 for ten replicate determination was <0.9%.
 | ||
| Fig. 9 Cyclic voltammograms of modified BDD electrode in the absence (a) and presence of 50 µM arsenic (c) at pH = 4.3, scan rate 10 mV s−1, electrode surface area is 0.25 cm2 (b and d as a and c) after the electrode was used for 90 cycles in buffer solution and arsenic containing solution. | ||
Amperometric detction of arsenic at modified BDD electrode
Since amperometry under stirred conditions or flow injection analysis with amperometric detection has much higher current sensitivity than cyclic voltammetry they can be used to estimate the lower limit of detection. As shown above the iridium oxide film modified BDD electrode has excellent and strong electrocatalytic properties and facilitates the low potential amperometric measurement of arsenic(III). Fig. 10 shows chronoamperograms recorded at a rotating modified BDD electrode (rotation speed 1200 rpm), under conditions where the potential was kept at 0.6 V in phosphate buffer solution with pH = 4.3. As shown during successive additions 500 and 20 nM a well defined response is observed. After each injection of arsenic at less than 1 s (response time) a sharp rise in the current was observed. The plot of current vs. arsenic concentration is shown in the inset of Fig. 10. The calibration plot was linear for a wide concentration range, 10 nM to 3 µM. Linear least squares calibration curve over the range 20–140 nM (7 points) had a slope of 4.2 nA nM−1 (sensitivity) and a correlation coefficient of 0.999. The detection limit was 2 nM when the signal to noise ratio was three and this value is comparable with modern instrumental techniques such as inductively coupled plasma mass spectrometry,5 hydride generation atomic fluorescence spectrometry3 and also with stripping voltammetric methods.9,39 The response time measured in amperometric measurements was 60 ms to 90% of full signal. Thus the modified iridium oxide film boron doped diamond was found to exhibit very high sensitivity and a fast response time for arsenic(III) detection. | ||
| Fig. 10 Amperometric response at rotating modified BDD electrode (rotation speed 1200 rpm) held at 0.6 V in phosphate buffer solution (pH = 4.30) for successive addition of (A) 0.5 µM and (B) 20 nM arsenic. C and D: Plot of chronoamperometric currents vs. arsenic concentrations. | ||
Conclusion
Cyclic voltammetry method was used to fabricate hydrous iridium oxide thin film modified boron doped diamond electrodes from a basic solution of iridium(III) oxide. The stability of the iridium oxide layers at the diamond substrate was also investigated by cyclic voltammetry, and it was found that the use of potential cycling ensures excellent mechanical stability and electrochemical reproducibility of the deposited electrocatalyst. The modified BDD electrodes were used for the electrocatalytic oxidation of arsenic(III) over a wide pH range. Electroplated nanometer thick films of iridium oxide, were used for the fast amperometric detection of arsenic(III) at nonomolar concentrations. The method is simple, fast and sensitive and is promising for routine analysis of ultra trace amounts of arsenic(III).Acknowledgements
A. Salimi thanks the Kurdistan University for financial support. CEB thanks the EPSRC for funding via a project studentship (Grant GR/R14392/01).References
- W. R. Cullen and M. Dodd, Appl. Organomet. Chem., 1988, 2, 1 CrossRef.
- X. P. Yan, X. B. Yin, X. W. He and Y. Jiang, Anal. Chem., 2002, 74, 2162 CrossRef CAS.
- Y. K. Lu, H. W. Sun, C. G. Yuan and X. P. Yan, Anal. Chem., 2002, 74, 1525 CrossRef.
- A. C. Lopez and M. D. L. de Castro, Anal. Chem., 2003, 75, 2011 CrossRef CAS.
- X. P. Yan, R. Kerich and M. J. Hendry, Anal. Chem., 1998, 70, 4736 CrossRef CAS.
- J. R. Pretty, E. A. Blubaugh and J. A. Caruso, Anal. Chem., 1993, 65, 3396 CrossRef CAS.
- H. Li and R. B. Smart, Anal. Chim. Acta, 1996, 325, 25 CrossRef CAS.
- C. M. Barra and M. M. C. Santoes, Electroanalysis, 2001, 13, 1098 CrossRef CAS.
- M. A. Ferreira and A. A. Barros, Anal. Chim. Acta, 2002, 459, 151 CrossRef CAS.
- U. Greulach and G. Henez, Anal. Chim. Acta, 1995, 306, 25 CrossRef CAS.
- J. Zima and C. M. G. Van den Berg, Anal. Chim. Acta, 1994, 289, 25 CrossRef CAS.
- S. B. Adeloju and T. M. Young, Anal. Lett., 1997, 30, 147 CAS.
- P. H. Davis, Anal. Chem., 1978, 50, 131 CrossRef CAS.
- Y. C. Sun, J. Mierzwa and M. H. Yang, Talanta, 1997, 44, 1379 CrossRef CAS.
- P. Tomcik, S. Jursa, Š. Mesároš and D. Bustin, J. Electroanal. Chem., 1997, 423, 115 CrossRef CAS.
- J. W. Strojek, M. C. Granger, T. Dallas, M. V. Holtz and G. M. Swain, Anal. Chem., 1996, 68, 2031 CrossRef CAS.
- S. Jolley, M. Koppang, T. Jakson and G. M. Swain, Anal. Chem., 1997, 69, 4099 CrossRef CAS.
- T. Yano, D. A. Tryk, K. Hashimoto and A. Fujishima, J. Electrochem. Soc., 1998, 145, 1870 CAS.
- E. Popa, H. Notsu, T. Miwa, D. A. Tryk and A. Fujishima, Electrochem. Solid State Lett., 1999, 2, 49 CrossRef CAS.
- T. N. Rao, I. Yagi, T. Miwa, D. A. Tryk and A. Fujishima, Anal. Chem., 1999, 71, 2506 CrossRef CAS.
- A. J. Saterlay, F. Marken, J. S. Foord and R. G. Compton, Talanta, 2000, 53, 403 CrossRef CAS.
- A. J. Saterlay, C. Agra-Gutierrez, M. P. Taylor, F. Marken and R. G. Compton, Electroanalysis, 1999, 11, 1083 CrossRef CAS.
- C. Prado, S. J. Wilkins, F. Marken and R. G. Compton, Electroanalysis, 2002, 14, 262 CrossRef CAS.
- A. J. Saterlay, J. S. Foord and R. G. Compton, Analyst, 1999, 124, 1791 RSC.
- C. Trashima, T. N. Rao, B. V. Sarada, N. Spataru and A. Fujishima, J. Electroanal. Chem., 2003, 544, 65 CrossRef.
- M. Pikulski and W. Gorski, Anal Chem., 2000, 72, 2696 CrossRef CAS.
- A. N. Bezbaruah and T. C. Zhang, Anal Chem., 2002, 74, 5726 CrossRef CAS.
- M. Wang, S. Yao and M. Madou, Sens. Actuators, B, 2002, 81, 313 CrossRef.
- A. J. Saterlay, S. J. Wilkins, C. H. Goeting, J. S. Foord, R. G. Compton and F. Marken, J. Solid State Electrochem., 2000, 4, 383 CrossRef CAS.
- A. J. Saterlay, F. Marken, J. S. Foord and R. G. Compton, J. Solid State Electrochem., 2000, 4, 383 CrossRef CAS.
- M. E. Hyde, A. J. Saterlay, S. J. Wilkins, J. S. Foord, R. G. Compton and F. Marken, J. Solid State Electrochem., 2002, 6, 183 Search PubMed.
- E. Hyde and R. G. Compton, J. Electroanal. Chem., 2002, 531, 19 CrossRef.
- S. Ardizzone, A. Carugati and S. Trasatti, J. Electroanal. Chem., 1981, 126, 287 CrossRef CAS.
- L. M. Schiavone, W. C. Dautremont-Smith, G. Beni and J. L. Shay, J. Electrochem., Soc., 1981, 128, 1339 CAS.
- J. E. Baur and T. W. Spaine, J. Electroanal. Chem., 1998, 443, 208 CrossRef CAS.
- S. Glab, A. Hulanicki, G. Edwall and F. Ingman, Crit. Rev. Anal. Chem., 1989, 21, 29 Search PubMed.
- E. Laviron, J. Electroanal. Chem, 1979, 101, 19 CrossRef CAS.
- D. O. Wipf, F. Ge, T. W. Spaine and J. E. Baur, Anal. Chem., 2000, 71, 4921 CrossRef CAS.
- C. Locatelli and G. Torsi, J. Electroanal. Chem., 2001, 509, 80 CrossRef CAS.
Footnote |
| † On sabbatical leave from: Department of Chemistry, Kurdistan University, P.O. Box, 416, Sanandaj, Iran. |
| This journal is © The Royal Society of Chemistry 2004 |