Liquid chromatography and tandem mass spectrometry: a powerful approach for the sensitive and rapid multiclass determination of pesticides and transformation products in water†
Juan V.
Sancho
,
Oscar J.
Pozo
and
Félix
Hernández
*
Analytical Chemistry, Experimental Sciences Dept., University Jaume I, P.O. Box 224, E-12080 Castellón, Spain. E-mail: hernandf@exp.uji.es; Fax: 964 728066; Tel: 964 728100
First published on 11th December 2003
Abstract
In this paper we will show the results of our research on the direct simultaneous determination of multi-class pesticides and transformation products with different polarities and acid–base properties by applying an on-line trace enrichment coupled to the chromatographic system supplied with electrospray interface (SPE-LC-MS/MS method). The specific chromatographic separation allows the correct determination of almost fifty compounds (37 pesticides and 10 transformation products) using very low sample volume and very little sample handling. Recoveries between 70–120% were obtained for all compounds in drinking and groundwater, meanwhile in surface water 44 compounds were correctly quantified. Relative standard deviations lower than 15% were obtained for all compounds. Even at the lowest concentration level tested (25 ng L−1) 40 compounds presented satisfactory recoveries and repeatability. The use of methanol as organic modifier and the increase of injection volume are also studied. The applicability of the developed method to a monitoring programme is demonstrated by applying it to the analysis of hundreds of samples.
Introduction
Recent years have witnessed the growing use of pesticides, chiefly in agriculture, but also in public works (road and rail maintenance) and home products.1 This development has undeniable repercussions on the environment, and the quality control of natural waters, both groundwater and surface water, has become a major issue. To address the subject, several national and international regulations now exist, including the European regulation on drinking water quality, which sets a maximum concentration of 0.1 µg L−1 for an individual pesticide and 0.5 µg L−1 for total pesticides present in the sample. The variety of substances determined and their degradation products entail the use of wide-spectrum, reliable and sensitive multi-residue methods to produce analysis to meet the regulations.The potential of liquid chromatography coupled with mass spectrometry (LC-MS) for the determination of pesticide residues in water samples was recognized early.2 Since then LC-MS has became an alternative to the common gas chromatography-mass spectrometry (GC-MS) methods in this area.3–4 Sample pre-treatment is normally more tedious by using GC-MS, besides most pesticide residues found in ground and surface waters tend to be polar compounds and/or transformation products (TPs) of the parent compounds applied.5 Therefore, LC-MS is becoming even more attractive in this type of analysis.
In the last years, due to the introduction of ion-trap mass spectrometers and because triple quadrupoles became commercially available, applications in the determination of pesticide residues in water samples by using tandem mass spectrometry have drastically increased.6 With these instruments the method selectivity has been notably increased due to the inherent selectivity of the technique. Furthermore, detection limits are decreased by using selected reaction monitoring (SRM) instead of selected ion monitoring (SIM) simplifying in this way sample pre-treatment and minimizing analytical errors associated to sample handling.
Although some papers described the use of quadrupole instruments in order to identify unknown pesticides,7 most of the developed methods with these instruments consist of the determination of a limited number of compounds. Thus, LC-MS methods for the determination of phenylureas,8,9 phenolic and acidic herbicides,10 chlorinated compounds11 or imidazolinone herbicides12 have been described and some organophosphorus,13 phenylureas,14 triazines14 and quaternary ammonium pesticides15 have been determined by LC-MS/MS methods. Normally these methods involved a screening of preselected compounds as only one transition for each was used in order to determine the analyte. Although sometimes it is difficult to obtain more than one transition for one compound due to the soft ionisation in API interfaces, the main reason for the choice of screening instead confirmatory methods has been the loss of sensitivity when more transitions are added. For quadrupole instruments an interrelationship exists between the sensitivity of detection and the time spent recording one transition.16 A high sensitivity requires long collection time for each ion to be detected, and this time decreases when more transitions are recorded at the same time. Therefore, the higher the number of compounds, the lower the collection time and the lower the sensitivity of the method.
On the other hand, a different problem appears in the use of LC-MS/MS for quantitative purposes. The presence of matrix coeluting organic compounds can interfere with the ionisation of the target analyte severely affecting the quantification.17 Although this matrix effect could be compensated by using labelled isotope standards or by using matrix-matched calibration, both solutions are not suitable in the environmental analysis as it is difficult to obtain commercially available isotope labelled standards for all analytes, and normally the use of only one internal standard in order to correct several compounds does not generate satisfactory results.18 Furthermore, the variability of the matrix between series of samples misadvises the matrix-matched calibration. A suitable clean-up step is usually performed in order to reduce this matrix effect. The use of off-line mode or on-line trace enrichment by solid-phase extraction7–9,19,20 (SPE-LC) is the most common approach for sample pre-treatment in the environmental field although some papers described liquid–liquid extraction7 or dual precolumn extraction.21 Obviously, this clean-up step is also used to concentrate the sample, however coeluting matrix interferences are usually concentrated and, although sensitivity is improved, the matrix effect is not reduced.7,19,22 This problem can be solved by reducing the sample volume23 or by improving the chromatographic separation using LC-LC coupling.24
All considerations indicated above have to be kept in mind when developing realistic methods to be applied at the sub-ppb concentration levels.
This paper describes a rapid method for the determination of 47 selected pesticides and transformation products in water. The method, based on SPE in combination with LC and tandem mass spectrometry, employs only 1.3 mL of sample and reaches detection limits lower than 100 ng L−1 for all analytes, as recommended by the European guideline for drinking water. The method has been validated and implemented for routine analysis of surface and groundwater samples in the Mediterranean coast of Spain.
Experimental
Reagents and chemicals
Pesticides and transformation products were chosen on the basis of information available on pesticides used in citrus production, which is the most important crop in our area (Western Mediterranean Coast of Spain), and considering the commercial availability of standards as well (mainly for TPs).Pesticides and metabolite reference standards were purchased from Dr Ehrenstorfer (Augsburg, Germany), Riedel de Haën (Seelze, Germany) and Sigma (St. Louis, MO, USA). HPLC grade acetonitrile and methanol were purchased from ScharLab (Barcelona, Spain). LC grade water was obtained by purifying demineralized water in a Nanopure II system (Barnstead, Newton, MA, USA). Formic acid (HCOOH, content > 98%) were supplied by Fluka (Buchs, Switzerland).
Stock standard solutions were prepared dissolving 25 mg, accurately weighted, in 50 ml of HPLC grade acetonitrile obtaining a final concentration of 500 µg mL−1. For LC-MS analysis, the stock solutions were mixed and diluted with acetonitrile or HPLC grade water.
Liquid chromatography
A triple quadrupole mass spectrometer was interfaced to an HPLC system based on a 233XL autosampler with a loop of 1330 µL (Gilson, Villiers-le-Bel, France) and 2 pumps: an Agilent 1100 (Agilent, Waldbron, Germany) binary pump used to condition and wash the cartridge (P-1) and a Waters Alliance 2690 (Waters, Milford, MA, USA) quaternary pump used for the chromatographic separation (P-2). The experimental setup can be found elsewhere.23 The SPE preconcentration was performed with a polymeric phase Hamilton PRP-1 using a small cartridge, 10 × 2 mm (Teknokroma, Barcelona, Spain) as C-1. For the LC separation a column 80 × 2 mm packed with Nucleosil C18 5 µm from ScharLab was used as C-2. Mobile phases consisted in water and acetonitrile in P-1 and mixtures of 0.01% HCOOH in acetonitrile–0.01% HCOOH in water in P-2.Mass spectrometry
A Quattro LC (quadrupole-hexapole-quadrupole) mass spectrometer with an orthogonal Z-spray-electrospray interface (Micromass, Manchester, UK) was used. Drying gas as well as nebulising gas was nitrogen generated from pressurized air in a NG-7 nitrogen generator (Aquilo, Etten-Leur, NL). The nebuliser gas flow was set to approximately 80 L h−1 and the desolvation gas flow to 800–900 L h−1. Infusion experiments were performed using a Model 11 single syringe pump (Harvard Instruments, Holliston, MA, USA), directly connected to the interface.For operation in MS/MS mode, collision gas was Argon 99.995% (Carburos Metalicos, Valencia, Spain) with a pressure of 5 × 10−4 mbar in the collision cell. Capillary voltages of −3 kV and 3.5 kV were used in negative and positive ionization mode respectively. The interface temperature was set to 350 °C and the source temperature to 120 °C. Dwell times of 0.1 s scan−1 were chosen.
Masslynx NT v 3.4 (Micromass, Manchester, UK) software was used to process the quantitative data obtained from calibration standards and from water samples.
Recommended procedure
Water samples were centrifuged at 3500 rpm for 10 min (only in the case of suspended particulate matter) and acidified with formic acid at 1%. This acidified sample was loaded in the loop and directly injected into the SPE-LC-(ESI)-MS/MS system.The conditioning of the PRP-1 cartridge was achieved with acetonitrile at a flow rate of 2 mL min−1 for 2 min, following by 2 min more with water. An aliquot of the acidified water sample (1330 µL) was preconcentrated into the cartridge and washed with 4 mL of water at 2 mL min−1. After been washed, the sample was transferred in backflush mode to the C-2 column and a gradient in P-2 started.23 Pesticide standard solutions used for quantification were also preconcentrated by SPE using the same procedure as for samples.
To perform the chromatographic separation a 0.01% HCOOH in acetonitrile:0.01% HCOOH in water gradient was optimised. The percentage of organic modifier was changed linearly as follows: 0 min, 5%; 6 min, 70%; 9 min, 70%; 10 min, 90%; 14 min, 90%; 15 min, 5%, 18 min 5% at a flow rate of 0.3 mL min−1
Finally, the determination was carried out by MS/MS under optimised conditions that are shown in Table 1.
| Compound | Na | Polarity | Precursor ion (m/z) | Cone/V | Collision/eV | Product ion (m/z) | Group |
|---|---|---|---|---|---|---|---|
| a N: Peak number. b Transformation products. c DIA: Desisopropylatrazine. d Carbofuran 3-OH: 3-hydroxy-carbofuran. e TCPY: 2,4,6-trichloro-3-pyridinol. f MCPA: 2-methyl-4-chlorophenoxyacetic acid. | |||||||
| Oxamyl | 1 | ES+ | 342 | 30 | 18 | 72 | A |
| Methomyl | 2 | ES+ | 185 | 25 | 10 | 128 | A |
| 2-Aminobenzimidazolb | 3 | ES+ | 134 | 45 | 25 | 92 | A |
| DIAbc | 4 | ES+ | 174 | 45 | 20 | 96 | A |
| 6-Chloronicotinic acidb | 5 | ES− | 156 | 25 | 10 | 112 | A |
| Carbendazim | 6 | ES+ | 192 | 35 | 20 | 160 | A |
| Carbofuran-3-OHbd | 7 | ES+ | 238 | 20 | 15 | 163 | A |
| Imidacloprid | 8 | ES+ | 256 | 35 | 15 | 209 | A |
| Dimethoate | 9 | ES+ | 230 | 30 | 10 | 199 | A |
| Carbofuran-7-phenol-3-ketob | 10 | ES− | 177 | 45 | 15 | 177 | A |
| Bromacil | 11 | ES− | 259 | 40 | 20 | 203 | B |
| Pyrimicarbe | 12 | ES+ | 239 | 30 | 20 | 72 | B |
| Fluroxypyr | 13 | ES− | 253 | 20 | 10 | 195 | B |
| Terbacil | 14 | ES− | 215 | 35 | 18 | 159 | B |
| 4-Chloroanilineb | 15 | ES+ | 128 | 40 | 20 | 93 | B |
| TCPYbe | 16 | ES− | 196 | 30 | 5 | 196 | B |
| Simazine | 17 | ES+ | 202 | 35 | 20 | 132 | B |
| 3-Methyl-p-nitrophenolb | 18 | ES− | 152 | 35 | 17 | 122 | B |
| Carbofuranb | 19 | ES+ | 222 | 30 | 15 | 165 | B |
| MCPAf | 20 | ES− | 199 | 25 | 15 | 141 | B |
| Ethiofencarb | 21 | ES+ | 226 | 20 | 15 | 107 | B |
| Diuron | 22 | ES+ | 233 | 30 | 16 | 72 | B |
| 3,4-Dichloroanilineb | 23 | ES+ | 162 | 40 | 20 | 127 | B |
| Fluazifop | 24 | ES− | 326 | 30 | 15 | 254 | B |
| Methiocarb | 25 | ES+ | 226 | 20 | 20 | 121 | B |
| Methidathion | 26 | ES+ | 303 | 20 | 8 | 145 | C |
| Terbuthylazine | 27 | ES+ | 230 | 30 | 20 | 174 | C |
| Azinphos-methyl | 28 | ES+ | 160 | 25 | 15 | 132 | C |
| Fomesafen | 29 | ES− | 437 | 50 | 15 | 437 | C |
| Pyridaphenthion | 30 | ES+ | 341 | 40 | 25 | 189 | C |
| Molinate | 31 | ES+ | 188 | 30 | 15 | 126 | C |
| Malathion | 32 | ES+ | 331 | 30 | 10 | 127 | C |
| Terbumeton | 33 | ES+ | 226 | 35 | 18 | 170 | C |
| Mecarbam | 34 | ES+ | 330 | 22 | 12 | 227 | C |
| Quinalphos | 35 | ES+ | 299 | 35 | 20 | 147 | C |
| Terbutryne | 36 | ES+ | 242 | 30 | 20 | 186 | C |
| Diazinon | 37 | ES+ | 305 | 30 | 15 | 169 | C |
| Thiobencarb | 38 | ES+ | 258 | 30 | 18 | 125 | D |
| Chlorpyrifos-methyl | 39 | ES+ | 322 | 30 | 18 | 125 | D |
| Pirimiphos-methyl | 40 | ES+ | 306 | 35 | 22 | 164 | D |
| Tebufenpyrad | 41 | ES+ | 334 | 50 | 30 | 145 | D |
| Pyriproxyfen | 42 | ES+ | 322 | 30 | 15 | 96 | D |
| Hexythiazox | 43 | ES+ | 353 | 25 | 16 | 228 | D |
| Chlorpyrifos | 44 | ES+ | 352 | 30 | 20 | 200 | D |
| Pendimethalin | 45 | ES+ | 282 | 20 | 10 | 212 | D |
| Buprofezin | 46 | ES+ | 306 | 25 | 12 | 201 | D |
| Pyridaben | 47 | ES+ | 365 | 35 | 25 | 147 | D |
Validation study
The precision (expressed as relative standard deviation, in %) was evaluated within-day by determining all pesticides in three standard solutions prepared at 25, 100 and 500 ng L−1 (n = 8 for each concentration level). The calibration curve was obtained by analysing standard solutions in triplicate at eight concentrations between 0 and 500 ng L−1.The recoveries were obtained by analysing drinking, ground and surface water samples spiked at three concentration levels each (25, 100 and 500 ng L−1). These experiments were performed in quintuplicate (n = 5).
The limit of quantification (LOQ) was taken as the lowest concentration level validated, for which adequate recoveries (between 70 and 120%) and precisions (RSD < 15%) were obtained. The limit of detection (LOD), defined as the lowest concentration that the analytical process can reliably differentiate from background levels, was estimated when the signal was three times the background noise from the chromatograms at the lowest analyte concentration assayed.
Results and discussion
MS optimisation
The full-scan mass spectra and the MS/MS spectra were obtained from infusion of 5 µg mL−1 50:50 acetonitrile:water solutions of each compound at a flow rate of 10 µL min−1. The multi-class nature of the selected pesticides as well as their different acid–base properties made that 10 out of 47 compounds presented only negative ionisation meanwhile the rest showed preferably positive ionisation.Most of compounds presented a [M + H]+ or [M − H]− ion, which led to a main product ion by fragmentation in the collision cell. However, carbofuran-7-phenol-3-keto (N = 10, see Table 1), trichloropirydinol (TCPY) (N = 16) and fomesafen (N = 29) showed MS/MS spectra without any important fragment. In these cases, the measured fragment ion was the selected precursor ion, but using a collision energy high enough for breaking interferences in the collision cell without affecting to an important degree sensitivity; therefore cleaner chromatograms were obtained.25 As an example, for the determination of carbofuran-7-phenol-3-keto (N = 10) case, both the precursor ion and product ion were selected at m/z 177, and a collision energy of 15 eV was set in order to break interferences preserving this molecule.
Moreover, azinphos-methyl (N = 28) was not measured by fragmentation of its protonated molecule [M + H]+ ion of m/z 318. A more sensitive transition was obtained under in-source fragmentation, as azinphos-methyl undergoes entrance-cone fragmentation and produces a fragment at m/z 160 which generates a product ion at m/z 132 in the collision cell.
The mass spectrometry parameters selected, as precursor and product ions, cone voltage and collision cell energy, for selected compounds are shown in Table 1.
LC optimization
The preconcentration cartridge selected was filled with polymeric material rather than C18-modified silica in order to increase the retention for the most polar transformation products and pesticides. 4 mL of water was selected as washing volume as it is shown elsewhere.23Moreover, the number of pesticides and transformation products has been increased in relation to our previous work, including mainly acidic analytes. This fact implies a previous acidification of the water sample before on-line trace enrichment as well as the use of different analytical column as the polar embedded stationary phase used before23 did not produce acceptable chromatographic peaks for acidic pesticides. The water sample was acidified with formic acid up to 1% and a standard reversed phase microbore separation column was used instead of the Supelco ABZ+ column.
Another result of including acidic analytes is that mobile phase has to be also acidified. Acidic analytes are detected under electrospray process as deprotonated molecules in negative mode. Thus, the acidification of mobile phase leads to a decrease in the MS sensitivity due to the ionisation suppression. On the other hand, acidified mobile phase allowed us to enhance the sensitivity for the compounds analysed in positive electrospray mode. Therefore, a compromise between correct chromatographic peaks and sensitivity should be appraised by optimising the content of formic acid in the mobile phase. A 0.01% formic acid in both water and acetonitrile was finally selected and a gradient was optimised to perform the group separation. Optimised gradient is shown in experimental section.
The chromatographic separation between groups of analytes is required in order to ensure enough points for defining the chromatographic peak, as detection is performed sequentially. The smaller the group, the higher the number of points available for defining the peak, thus improving the quality of data. Under optimised conditions, chromatographic groups of less than 16 compounds that eluted in the same group (16 transitions) were obtained and good peak shapes were achieved for all compounds. These chromatographic groups are shown in Table 1. The total chromatographic run was only 14 min for 47 analytes, ranging from very polar transformation products as 2-aminobenzimidazol (N = 3) to nonpolar pesticides as pyridaben (N = 47), and the overall analytical process, including the on-line trace enrichment was around 27 min. However, the LC-MS equipment used allowed us to overlap the cartridge conditioning and the next sample loading with the chromatographic separation of the current sample rendering a real analysis time per sample of only 18 min.
In this work, we only selected one transition for both quantitation and qualification as the use of another transition for confirmation implied the use of at least 94 channels. This high number of channels would require longer chromatographic runs in order to increase the data points available per peak, decreasing the sample throughput of the developed procedure. As our main objectives were multi-residuality and short run-time period, we kept only one channel per analyte. Moreover, for some analytes is difficult to find more product ions in the spectra that can be used for confirmation.
Analytical characteristics
Standard curves showed excellent linearity, with correlation coefficients greater than 0.994 for all the 47 compounds in the range from 0 to 1000 ng L−1. The linear range was lower in few cases as 2-aminobenzimidazol (N = 3) where a quadratic curve was applied. The method showed good precision (RSD < 15%) for all compounds with very few exceptions, the most notable being diazinon (N = 37) with a RSD up to 27%. Even at a concentration as low as 25 ng L−1, 39 out of 47 compounds still presented good repeatability.Limits of detection (LOD) were calculated from the most diluted standard analysed and ranged from 0.1–0.5 ng L−1 for some compounds like carbendazim (N = 6), carbofuran (N = 19) and pyrimicarb (N = 12) to around 50 ng L−1 for other as chlorpyrifos (N = 44), tebufenpyrad (N = 41) and hexythiazox (N = 43), the poorest values being for pyridaben (N = 47) (LOD 210 ng L−1). For 33 out of 47 compounds the LOD were lower than 10 ng L−1, using as low as 1.3 mL of sample, and only 7 compounds presented detection limits above 25 ng L−1, the LOD objective in this work. The majority of these 7 compounds were late eluting analytes.
In order to improve this situation two alternatives were tested: the use of a ternary acetonitrile/methanol/water gradient or the increase of sample volume to be preconcentrated in the PRP-1 cartridge.
As regards the use of methanol in the mobile phase, some improvement in the sensitivity by using this organic modifier has been described.18,26 However, methanol is not strong enough to desorb the analytes from the polymeric cartridge,18 therefore a ternary gradient was used. The gradient started with a mixture acetonitrile/water and after 6 min the mobile phase was changed to methanol/water so late-eluting analytes entered the electrospray interface together with a more protic solvent as methanol that may enhance the signal in positive mode. This effect was found to be compound-dependent and varied from no effect to two-three fold signal increase. Anyway, the signal improvement did not allow reaching detection limits below 25 ng L−1 for several of the late-eluting analytes.
The effect of increasing the sample volume to be preconcentrated on the polymeric cartridge was also investigated. Keeping in mind a short overall analysis time and considering the presence of very polar metabolites within the group of selected analytes, the loop size was increased from 1.3 mL up to only 4.4 mL, in spite of the fact that a polymeric cartridge was employed. Using an acetonitrile/water gradient and the same loading scheme, the sensitivity was increased enough for the late-eluting analytes, achieving limits of detection below 25 ng L−1 for all the 47 studied compounds. However, sample breakthrough started to occur for early-eluting analytes, and similar or even worse limits of detection were obtained for some polar pesticides and metabolites, although LOD were in all cases below 25 ng L−1. Recoveries were satisfactory for all compounds in drinking and groundwater matrices showing the non existence of matrix effect. However, in the surface water tested the higher amount of interferences preconcentrated on the polymeric cartridge produced signal suppression in the electrospray source for some analytes, leading to recoveries below 70%, or signal enhancement in others giving recovery values above 120%.
Consequently, the method was not modified, and 1.3 mL of sample with an acetonitrile: water gradient were used for the sample analysis.
Method validation
Precision and accuracy of the overall analytical procedure were evaluated for surface, ground and drinking water samples spiked at 25, 100 and 500 ng L−1 level. The high number of data (3 matrices at 3 concentration levels and 47 analytes) makes difficult their presentation in a table as a whole (see Electronic supplementary information (ESI)).†The method was found to be precise (RSD < 15%) for all the compounds studied at 100 and 500 ng L−1 spiking level. Besides, recoveries were satisfactory (between 70% and 120%) in drinking and groundwater for all analytes. As regards surface water, the presence of more interferences resulted in lower recoveries (60–70%) for only three compounds (methomyl (N = 2), oxamyl (N = 1) and the metabolite 6-chloronicotinic acid (N = 5)), although with satisfactory precision (RSD < 15%).
At the 25 ng L−1 spiking level, 40 compounds could still be satisfactorily quantified, and only three analytes (imidacloprid (N = 8), 4-chloroaniline (N = 15) and TCPY (N = 16)) presented unacceptable precision with RSD > 15%. All average recoveries at this low concentration level in the three water matrices tested were in the 70–120% range. The method could not be validated at 25 ng L−1 for three insecticides (pyriproxyfen (N = 42), chlorpyrifos methyl (N = 39) and chlorpyrifos (N = 44)), 3 acaricides (tebufenpyrad (N = 41), hexythiazox (N = 43) and pyridaben (N = 47)) and 1 TP (carbofuran-7-phenol-3-keto (N = 10)), but all of them (except for pyridaben (N = 47)) were determined satisfactorily at the 100 ng L−1 level.
In conclusion, the majority of pesticides and metabolites could be satisfactorily determined at sub-ppb levels in drinking, ground and surface water using only 1.3 mL of sample. The poorest results were obtained for pyridaben, as it was the last analyte eluting at the end of the gradient, surely co-eluting with interferent compounds that would suppress its ionisation. In the developed method the different matrix components of surface water in relation to drinking and groundwater seemed to not affect robustness, as only 1.3 mL of sample were preconcentrated. As an example, Fig. 1 shows representative chromatograms for groundwater spiked at 25 ng L−1.
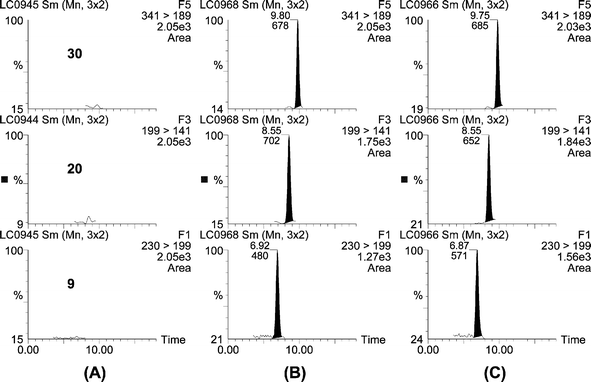 | ||
| Fig. 1 SPE-LC-ESI-MS/MS chromatograms from a groundwater sample (A) blank. (B) spiked at 25 ng L−1 and (C) 25 ng L−1 standards of dimethoate (N = 9), MCPA (N = 20) and pyridaphenthion (N = 30). | ||
The method developed in this paper presents some advantages over those previously reported. The most important one is the low injection volume used which is around 10 times lower than previously reported methods, this reduction allows the correct quantification of almost all the analytes at the regulatory level by using electrospray interface without any matrix effect observed. This result can be compared with other methods where the high amount of matrix introduced in the source generated poor recoveries for some compounds,19,21,22 as it was also observed in this paper when increasing the sample volume for surface waters. On the other hand, this small volume allows the easy automatisation of the analysis and reduces considerably the total run time, as one of the most time consuming tasks in the determination of pesticide residues in water is the preconcentration step.
Monitoring of pesticides in environmental waters
More than 100 samples of ground and surface water were collected in selected sites from Spanish Mediterranean area (Castellon, Valencia, Alicante and Murcia provinces). This is an important citrus-growing area, where many pesticides from different chemical families are applied, mainly insecticides and herbicides. Several horticultural crops are also frequent, and additionally exists an important area for rice crop, which the most representative site is the Albufera Lagoon of Valencia.After collection of samples, they were transported to the laboratory and stored at −18 °C until required for residue analysis.
Several pesticides and metabolites were detected at relatively high concentrations (µg L−1 level) in both ground and surface water.27 The high sensitivity as well as the multiresiduality of the method applied allowed us to detect a variety of pesticides and TP at very low levels. As an example, Fig. 2 shows chromatograms for groundwater samples containing several pesticides at the sub-ppb level.
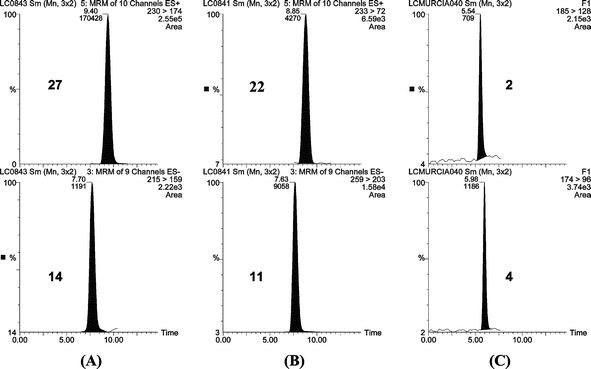 | ||
| Fig. 2 Typical SPE-LC-ESI-MS/MS chromatograms from positive real groundwater samples. (A) sample containing 0.47 µg L−1 of terbuthylazine (N = 27) and 0.13 µg L−1 of terbacil (N = 14). (B) sample containing 0.11 µg L−1 of diuron (N = 22) and 2.5 µg L−1 of bromacil (N = 11). (C) sample containing 0.04 µg L−1 of methomyl (N = 2) and 0.12 µg L−1 of DIA (N = 4). | ||
Some of the areas monitored presented severe pollution by pesticides in surface water. As an example, in a surface water from a wetland surrounded by citrus orchards sited at Burriana fourteen compounds were detected, in several cases at concentrations higher than 1 µg L−1 (Fig. 3).
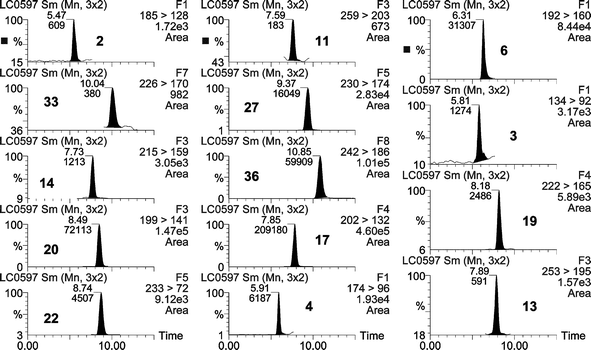 | ||
| Fig. 3 SPE-LC-ESI-MS/MS chromatogram from a surface water sample collected from the Clot de la Mare de Deu in Burriana containing 0.70 µg L−1 of diuron (N = 22), 3.96 µg L−1 of MCPA (N = 20), 0.13 µg L−1 of terbacil (N = 14), 0.010 µg L−1 of terbumeton (N = 33), 0.05µg L−1 of methomyl (N = 2), 1.33 µg L−1 of DIA (N = 4), 4.10 µg L−1 of simazine (N = 17), 1.89 µg L−1 of terbutryne (N = 36), 0.37 µg L−1 of terbuthylazine (N = 27), 0.07 µg L−1 of bromacil (N = 11), 0.11 µg L−1 of fluroxypyr (N = 13), 0.26 µg L−1 of carbofuran (N = 19), 0.17 µg L−1 of 2-aminobenzimidazol (N = 3) and 0.81 µg L−1 of carbendazim (N = 6). | ||
For several compounds, the good peak obtained (i.e. terbumeton, N = 33) allowed us the quantification of the analyte at concentrations lower than 25 ng L−1 (the LOQ of the method), although the method was not validated at such a low levels. For example, a concentration of ca. 10 ng L−1 was estimated for terbumeton from the chromatogram shown in Fig. 3.
Summarising, herbicides were the most commonly detected pesticides in groundwater. Triazines were found in around 79% of samples, as an example, 15% of samples contained more than 0.1 µg L−1 of terbuthylazine. Other herbicides as bromacil, terbacil and diuron were detected in around 54% of groundwater samples (e.g. 34% of samples contained more than 0.1 µg L−1 of bromacil). Molinate was also frequently detected. In relation to insecticides, pyrimicarb, carbofuran and ethiofencarb were detected in around 25% of samples (always below 0.1 µg L−1). None of the acaricides investigated were detected in the groundwater samples. However, the fungicide carbendazim was largely detected (51%) in the samples. As regards transformation products, 2-aminobenzimadol and carbofuran were the most commonly detected in the samples.
Regarding surface water samples, herbicides were again the most frequently detected, their concentrations levels being notably higher than in groundwater. As an example, 36% of surface waters presented concentrations of triazines higher than 0.1 µg L−1 with a mean of 1.7 µg L−1 and a maximum of 7 µg L−1. Moreover, organophosphorous pesticides were often detected, and 34% of samples contained methidathion, chlorpyrifos or dimethoathe.
The robustness of the method was proved by more than 4 months of performing the analysis of surface and groundwater samples. Some variations in the sensitivity of the instrument were occasionally observed, making the quantification at the lowest validated level (0.025 µg L−1) difficult for some compounds, although no relevant problems were observed at the 0.1 µg L−1 concentration level. In these situations, a simple cleaning of the Z-Spray source was enough to recover sensitivity up to satisfactory levels.
Conclusion
The developed procedure allows the simultaneous determination of 37 pesticides and 10 transformation products with different polarities and acid–base properties in drinking, ground and surface water with a global analysis time of 18 min (sample throughput of ca. 80 samples day−1) using only 1.3 mL of sample. All compounds are correctly quantified at the 0.1 µg L−1 concentration level, which is the restrictive regulatory level for drinking water in the E.U. Besides, most of them (40 out of 47) can also be correctly quantified at 25 ng L−1. Detection limits are for most compounds (33 out of 47) even lower than 10 ng L−1. Satisfactory precision and recoveries are obtained and sample preparation is minimal, rendering a fast and robust assay. The high automation of the method as well as the reduced sample pre-treatment make affordable an easy confirmation of the positive samples by reanalysing them using a confirmatory transition, wherever available.In order to optimise the selection of transformation products to be included in future analytical methods, the degradation of the most used pesticides in our area is being studied in our laboratory by using a hybrid quadrupole-time of flight (QTOF) instrument, with exact mass capabilities, in combination with dedicated software tools, as Metabolynx.
Acknowledgements
The authors are very grateful to the Serveis Centrals d'Instrumentació Científica (SCIC) of University Jaume I for the use of the Quattro LC triple quadruple mass spectrometer. This work is included within the research project (ref: REN2002-01818) developed under financial support of the Ministerio de Ciencia y Tecnología, Spain.References
- V. Pignon, R. Jeannot and E. Sauvard, Int. J. Environ. Anal. Chem., 1999, 75(4), 345–366 CAS.
- R. D. Voyksner and C. A. Haney, Anal. Chem., 1985, 57, 991–996 CAS.
- W. M. A. Niessen, J. Chromatogr., A, 1999, 856, 179–197 CrossRef CAS.
- R. B. Geerdink, W. M. A. Niessen and U. A. Th. Brinkman, J. Chromatogr., A, 2002, 970, 65–93 CrossRef CAS.
- D. W. Kolpin, E. M. Thurman and S. M. Linhart, Sci. Total Environ., 2000, 248, 115–122 CrossRef CAS.
- T. Reemtsma, Trends Anal. Chem., 2001, 20, 500–517 CrossRef CAS.
- H. Sabik and R. Jeannot, J. Chromatogr., A, 1998, 818, 197–207 CrossRef CAS.
- Di Corcia, A. Costantino, C. Crescezi and R. Samperi, J. Chromatogr., A, 1999, 852, 465-474.
- E. Baltussen, H. Snijders, H. G. Janssen, P. Sandra and C. A. Cramers, J. Chromatogr., A, 1998, 802, 285–295 CrossRef CAS.
- A. C. Hogenboom, I. Jagt, R. J. J. Vreuls and U. A. Th. Brinkman, Analyst, 1997, 122, 1371–1377 RSC.
- A. J. H. Louter, A. C. Hogenboom, J. Slobodnik, R. J. J. Vreuls and U. A. Th. Brinkman, Analyst, 1997, 122, 1497–1503 RSC.
- G. D'Ascenzo, A. Gentili, S. Marchese and D. Perret, J. Chromatogr., A, 1998, 800, 109–119 CrossRef CAS.
- A. Ingelse, R. C. J. van Dam, R. J. Vreeken, H. G. J. Mol and O. M. Steujger, J. Chromatogr., A, 2001, 918, 67–78 CrossRef CAS.
- C. Hogenboom, W. M. A. Niessen and U. A. Th. Brinkman, J. Chromatogr. A, 1998, 794, 201–210 CrossRef CAS.
- R. Castro, E. Moyano and M. T. Galceran, J. Chromatogr., A, 2001, 914, 11–121 CrossRef CAS.
- T. Reemtsma, Trends Anal. Chem., 2001, 20, 533–542 CrossRef CAS.
- K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 1998, 70, 882–889 CrossRef CAS.
- P. G. M. Kienhuis and R. B. Geerdink, Trends Anal. Chem., 2000, 19, 460–474 CrossRef CAS.
- Koppen and N. H. Spliid, J. Chromatogr., A., 1998, 803, 157–168 CrossRef CAS.
- R. Curini, A. Gentili, S. Marchese, A. Marino and D. Perret, J. Chromatogr., A, 2000, 874, 187–198 CrossRef CAS.
- R. J. C. A. Steen, A. C. Hogenboom, P. E. G. Leonards, R. A. L. Peerboom, W. P. Cofino and U. A. Th. Brinkman, J. Chromatogr., A, 1999, 857, 157–166 CrossRef CAS.
- A. C. Hogenboom, M. P. Hofman, D. A. Jolly, W. M. A. Niessen and U. A. Th. Brinkman, J. Chromatogr., A, 2000, 885, 377–388 CrossRef CAS.
- F. Hernández, J. V. Sancho, O. Pozo, A. Lara and E. Pitarch, J. Chromatogr. A, 2001, 939, 1–11 CrossRef CAS.
- E. Dijkman, D. Mooibroek, R. Hoogerbrugge, E. Hogendoorn, J. V. Sancho, O. Pozo and F. Hernandez., J. Chromatogr., A, 2001, 926, 113–125 CrossRef CAS.
- J. V. Sancho, O. Pozo and F. Hernández, Rapid Commun. Mass Spectrom., 2000, 14, 1485–1490 CrossRef CAS.
- R. B. Geerdink, A. Kooistra-Sijpersma, J. Tiesnitsch, P. G. M. Kienhuis and U. A. Th. Brinkman, J. Chromatogr., A, 1999, 863, 147–155 CrossRef CAS.
- I. Morell, R. Aragón, J. L. García, O. Pozo, S. Grimalt, J. V. Sancho and F. Hernández, Environ. Sci. Technol. Search PubMed , submitted.
Footnote |
| † Electronic supplementary information (ESI) available: Method validation data. See http://www.rsc.org/suppdata/an/b3/b312236k/ |
| This journal is © The Royal Society of Chemistry 2004 |