Möbius bis and tris-spiroaromatic systems†
David
Hall
and
Henry S.
Rzepa
*
Department of Chemistry, Imperial College London, UK SW7 2AY
First published on 2nd December 2002
Abstract
We propose that a general class of bis and tris-spiro 7-membered ring systems with a common atom X, of which there are a number of examples characterised crystallographically, can in fact be considered as spiroaromatic molecules in which each ring exhibits some degree of Möbius 4nπ-electron aromaticity. The aromaticity is probed as a function of the spiro-atom using ab initio calculations of the NICS(0) values, which indicate that the Möbius-aromaticity increases as the spiro-atom is changed e.g. from Al to P, and from e.g. P to As.
Heilbronner was the first to suggest1 that a 180° phase shift in the pπ–pπ overlap of a cyclic array of such orbitals, delocalised over the entire ring, would form a so-called Möbius aromatic system if the resulting closed shell π molecular orbitals were occupied by 4n electrons. Following the proposal by Schleyer and co-workers2 34 years later that the conjugated 4n pπ C9H9+ cation exhibits such aromaticity, an increasing number of diverse systems exhibiting in variable degree Möbius aromaticity have been identified.3–7 We recently suggested8 potential metal ligands of the general formula 1 (Y = NR, O; X = C−, Si−) which as 4n pπ systems exhibit predicted geometrical distortions towards C2 symmetry, driven by concomitant reduction of Hückel anti-aromaticity and increase in Möbius aromaticity. We subsequently argued7 that if the atom X is capable of 4- or higher coordination, then conjugated ring systems joined at the single atom X become possible. We used the term spiroaromatic to indicate those cases where each ring was itself capable of sustaining aromaticity, and suggested several examples where the spiro atom comprised a group V element such as phosphorus9 or arsenic. The analogy to the Möbius aromaticity present in the co-called coarctate transition states10,11 was also noted. In the present article, we extend the motif of 1 to the series 1–3, where both bis and tris Möbius spiroaromaticity might be revealed. Hexacoordinated systems of type 3 in particular constitute an important class of complex involved in asymmetric two-center catalysis12 for which Möbius aromaticity has not hitherto been previously studied or recognised.
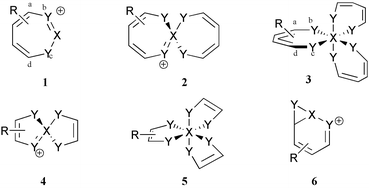
Computational procedure
Molecular structures were normally optimised without symmetry constraints using Gaussian9813 at the closed shell B3LYP/6-31G(d) level. The aromaticity of these systems was quantified using a NICS14 probe placed at the ring centroid. Coordinates are available via the electronic supplementary data (ESI).†Results and discussion
A search of the Cambridge crystal database15 reveals a number of examples of the structural motifs of either 2 and 3, or the 6π (4n+2) analogues 4 and 5. The diversity includes examples of the heteroatoms Y = O, S and N and the spiro atom X spans the main group IIIB–VIB elements and the analogous transition series group IIIA–VIA. Typical (but not exhaustive) examples are shown in Table 1, for which we note that for the main group elements, the oxidation state implied by XYn(z−n) (z = group number) tends to be followed. In the transition series, the oxidation state of X can be lower, e.g. Cr(III) rather than Cr(VI). Amongst the closest analogues are those for 2 and particularly 3 (for which X = Al, In, P, Y, Cr(III); R=dinaphthyl). Where examples are missing we suspect it may be because of lack of synthetic incentive rather than instability. We note that in general these types of compound, and in particular the series 2 and 3 have not hitherto been analysed in terms of their potential Möbius aromaticities, which is the purpose of the current article. Here we report spiro-systems derived from main group elements, which are adequately described by single reference wavefunctions. We found that calculations on the group IIIA–VIA transition metal analogues require multi-reference wavefunctions due to the ground states having several electronic configurations of similar energy. The MCSCF convergence for such systems tend to be problematic, and currently no MCSCF-NICS procedure14 is implemented.13 For these reasons, calculations for the transition series are not included here.| a Cambridge reference code and oxidation state of X as determined from the charge on the counter ions (not shown). 3D models of each structure are available via the ESI. | |||
|---|---|---|---|
| yetreu/B(III)16 | perjeb/Al(III)17 | zohvib/Al(III)18 | ubiqur/In(III)19 |
| kezqah/Ga(III)20 | gukdiz/Si(IV)21 | gukdul/Ge(IV)21 | gajfus/Sn(IV)22 |
| mspaps/P(V)23 | paqwuz/P(V)24 | eapdop10/P(V)25 | doxlor/As(V)26 |
| hegday/Sb(V)27 | yegduj/Bi(III)28 | tpcate/Te(IV)29 | tectcl/Te(IV)30 |
| qirpuc/La(III)31 | qazreo/Y(III)32 | govsoz/Ti(IV)33 | cigtuh/Ti(IV)34 |
| bzdtzr10/Zr(IV)35 | HOVTUH/V(IV)36 | rilhoj/Nb(V)37 | bzdtnb10/Nb(V)38 |
| cuqgea/Cr(III)39 | doxqai/Cr(V)40 | sixwif/Mo(V)40 | gefkor/W(IV)41 |
The dicoordinate series 1
We previously reported3 that for the hetero-rings 1 with X = C−, Si− a C2 distortion from planarity is predicted to occur, accompanied by a reduction in the planar anti-aromaticity. Here we tabulate the analogous properties of the X = N and P series (Y = O, NH, NF, R = H, F, Table 2), which we include for comparison with the four and six coordinated analogues 2 and 3. As was previously noted for R = H, the C2 distorted geometry is a true minimum in the potential surface, revealing no negative roots for the force constant matrix. The only exceptions are when Y = NF, for which the C2 geometry exhibits a small imaginary normal mode corresponding to a distorsion to a lower energy bicyclic form 6.| Substituentsb | Energy, Y = O | NICS (angle)a | Energy, Y = NR | NICS (angle)a |
|---|---|---|---|---|
| a NICS(0) value, ppm (dihedral angle a–b–c–d). b The designation RR/RS or RRR/RRS indicates relative rather than absolute chiral configurations for the 7-membered rings. c Wavenumber for imaginary normal mode corresponding to distortion to 6. d Dissociates to C4F4O2 and C4F4NO2+. e Energy (kcal mol−1) relative to chiral diastereomer. | ||||
| 1, X = N, R = H | −359.53314 | 17.4 (45.7) | −319.93038 | 26.7 (34.9) |
| 1, X = N, R = F | −756.39305 (365.7i)c | 11.1 (54.0) | −915.07525 (189.9i)c | −1.0 (59.0) |
| 1, X = P, R = H | −646.31012 | 18.6 (23.7) | −606.60521 | 19.4 (38.0) |
| 1, X = P, R = F | −1043.17777 (167.6i)c | 14.3 (41.2) | −1201.76006 (112.6i)c | −2.0 (54.6) |
| 2, RR, X = N, R = H | −664.72355 | −1.3 (76.6) | −585.37168 | −2.8 (73.8) |
| 2, RR, X = N, R = F | d | d | −1775.73438 | −9.0 (84.3) |
| 2, RR, X = P, R = H | −951.62957 | −0.2 (56.6) | −872.17703 | −0.7 (55.4) |
| 2, RS, X = P, R = H | 0.2e | −0.3 (57.4) | 4.8e | −0.7 (54.2) |
| 2, RR, X = P, R = F | −1745.37896 | −4.5 (63.2) | −2062.50669 | −9.3 (64.7) |
| 2, RS, X = P, R = F | 0.9e | −4.5 (62.9) | −0.6e | −8.8 (60.0) |
| 4, X = P, R = H | −796.78568 | −7.4 (0.0) | −717.36303 | −9.8 (0.0) |
| 3, RRS, X = Al, R = H | −1158.15341 | 0.2, 0.3 (33.5, 34.4) | −1038.79166 | 0.0, 0.0 (39.0, 39.1) |
| 3, RRS, X = Si, R = H | −1205.36111 | −1.0, −0.4 (43.0, 53.0) | −1086.03770 | −1.4, −1.6 (42.5, 42.3) |
| 3, RRS, X = Ge, R = H | −2990.82401 | −2.5, −2.3 (57.7, 58.9) | −2871.52125 | −2.2, −2.9 (44.5, 53.9) |
| 3, RRS, X = P, R = H | −1257.16780 | −2.6, −2.7 (67.2, 66.9) | −1137.88650 | −3.4, −3.4 (42.0, 53.9) |
| 3, RRR, X = P, R = H | 1.7e | −2.2 (66.5) | −4.7e | −4.2 (55.0) |
| 3, RRS, X = P, R = F | −2447.98997 | −6.7, −6.9 (69.2, 69.0) | −2923.62738 | −9.4, −10.0 (69.2, 69.0) |
| 3, RRR, X = P, R = F | 0.8e | −6.6 (69.3) | 5.0e | −10.2 (62.8) |
| 3, RRS, X = As, R = H | −3149.58333 | −3.7, −3.4 (67.3, 68.3) | −3030.31362 | −4.3, −4.4 (56.7, 58.8) |
| 5, X = P, R = H | −1024.96531 | −6.3 (0.0) | −905.65126 | −3.0 (0.0) |
The computed (positive) NICS(0) values reveal the basic ring system 1, X = N or P to be strongly anti-aromatic, despite a moderate degree of Möbius-aromatising C2 twisting. This anti-aromaticity is reduced (or the aromaticity increased) by replacing the ring hydrogens with fluorine, and the degree of ring twist is also increased (Table 2) by this substitution. This “Möbius-fluorine” effect appears to be stronger when Y = NF than when Y = O, but the effects of changing from X = N to X = P are small. The Möbius character can be qualitatively illustrated in the form of the highest occupied molecular orbitals (Fig. 1a and b). For both X = N and X = P, the HOMO takes the form of a catenane or rotaxane-like interlinking of the two phases of the wavefunction, characteristic4 of Möbius aromatic rings.
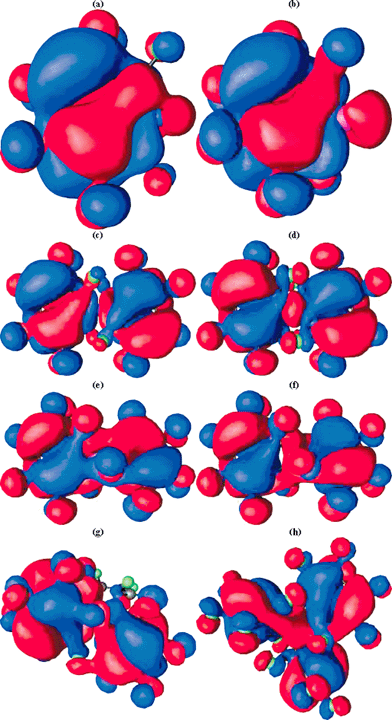 | ||
| Fig. 1 RHF/6-31G* computed molecular orbitals, contoured at 0.01 Hartree for (a) HOMO of 1, X = N, Y = NF, (b) HOMO of 1, X = P, Y = NF, (c) HOMO of 2, X = N, Y = NF, (d) HOMO-1 of 2, X = N, Y = NF, (e) HOMO of 2, X = P, Y = N, (f) HOMO-1 of 2, X = P, Y = NF, (g) HOMO of 3, X = P, Y = NF, (h) HOMO-1 of 3, X = P, Y = NF. | ||
The next issue to resolve is whether the reduction of Hückel anti-aromaticity can be sustained when two heterocyclic rings 1 are spiro-fused via the central atom X, and whether the greater ability of the 2nd period elements (X = Al–P) to involve in some degree the 3d orbital shell could promote the ability of the ring to sustain Möbius aromaticity. Each individual ring again has local optimised C2 symmetry; attempts to find other ring conformations all converged to this form. The modestly negative NICS(0) values for Y = O, NH and the high degree of ring twist indicate that even for X = N, each ring has lost the anti-aromaticity associated with planar 1. One possibility is that X = N is acting as an insulator, inhibiting π conjugation across it, and hence creating a formally non (or homo) aromatic system. The form of the molecular orbitals suggests otherwise (Fig. 1). The HOMO for 2, X = N, Y = NF contains nodes for each ring essentially identical that for the single ring in 1. Using rotaxane terminology, the two orbital phases are associated respectively with the “eye” and “thread” of the needle for one ring and become reversed for the other ring (i.e. what was the eye becomes the thread, and vice versa). The HOMO reveals a node at the X = N atom, indicating little cross-ring conjugation. With X = P however an increased contribution from the P-3d orbitals indicates the HOMO and HOMO-1 for this system (Fig. 1e and f) to be more strongly coupled across the rings; the HOMO shows an eye/thread:thread/eye motif for the two rings, whilst the HOMO-1 reveals a eye/thread:eye/thread nodal pattern. When fluoro substituted, each ring becomes modestly aromatic (compared to e.g. benzene which has a NICS(0) value of approximately −10 ppm) with a concomitant increase in the ring twisting.
The other noteworthy feature is that the twist of each ring can have either the same or opposite chirality, resulting in the possibility of chiral diastereomers for the overall molecule. The calculations indicate that these differ insignificantly in energy (Table 2).
Changing the 7-membered 8π (4n) ring to a five membered 6π (4n+2) system as in 4 reveals the latter ring to be entirely planar and untwisted, with NICS(0) values which indicate significant (Hückel-like) aromaticity. A similar observation of ring aromaticity was made previously by Wang and Schleyer9 for tetracoordinate phosphabenzene systems.
The hexacoordinate series 3 and 5
In 3, the central atom is octahedrally coordinated, and with each of the 7-membered rings being capable of a C2 chiral distortion, two chiral diastereoisomers become possible. With X = P− and Y = O, the parent ring appears to favour the RRS/SSR chiral isomer; the combination X = P− and Y = NH in contrast favours the RRR/SSS form. Compared with 2, the NICS(0) value is slightly more negative (aromatic) and the degree of ring twist slightly greater, both suggesting a slight increase in Möbius character. As before, ring fluorination increases the NICS(0) value to a similar magnitude to that for benzene (∼−10ppm) and again increases the degree of ring twist. By these criteria at least, these rings are now amongst the most Möbius-aromatic systems yet identified. The form of the degenerate HOMO for 3, X = P, Y = NF and HOMO-2 reveal a similar nodal pattern as previously described for 2.We also explored variation of X to see how this might influence any Möbius characteristics. The series X = Al, Si, P reveals an increase in the negative NICS(0) values and this is also true in moving down a column, i.e. from Si to Ge, and from P to As; thus the latter system is predicted to carry the greatest degree of Möbius aromaticity of the systems studied. Finally in the hexacoordinate series, we note that the five membered 6π (4n+2) system as in 5 appears to sustain lower (Hückel) aromaticity compared to the four-coordinate analogue.
Conclusions
We have studied a series of 4- and 6-coordinate systems containing a main group atom (X) which can act as a spiroaromatic bridge across two aromatic ring systems. When these rings correspond to 8π systems, a C2 distortion can occur to alleviate the formal planar anti-aromaticity. As components of the bis-spiro rings 2, the effect is to create mildly Möbius-aromatic rings, and the aromaticity is enhanced by ring fluorination, and by the nature of X, with lower row or higher main group elements favouring aromaticity. The form of the computed molecular orbitals shows a significant degree of cross-conjugation between the spiro-rings. Examples of this type of compound are also known to occur in the transition metal series (e.g., V, Cr, etc.) for which variable oxidation states add a further level of subtlty. Calculations on these systems currently present technical challenges which we anticipate will find solution in the future.References
- E. Heilbronner, Tetrahedron Lett., 1964, 29, 1923 CrossRef.
- M. Mauksch, V. Gogonea, H. Jiao and P. von R. Schleyer, Angew. Chem. Int. Ed., 1998, 37, 2395 CrossRef CAS.
- (a) C. Kastrup, S. Oldfield and H. S. Rzepa, Chem. Commun., 2002, 642–643 RSC; (b) W. L. Karney, C. J. Kastrup, S. P. Oldfield and H. S. Rzepa, J. Chem. Soc., Perkin Trans 2, 2002, 388–392 RSC.
- (a) S. Martin-Santamaria and H. S. Rzepa, Chem. Commun., 2000, 1089 RSC; (b) S. Martin-Santamaria and H. S. Rzepa, J. Chem. Soc., Perkin Trans. 2, 2000, 2372–2377 RSC.
- C. Castro, C. M. Isborn, W. L. Karney, M. Mauksch and P. von R. Schleyer, Org. Lett., 2002, 4, 3431–3434 CrossRef CAS.
- (a) R. P. Johnson and K. J. Daoust, J. Am. Chem. Soc., 1996, 118, 7381–7385 CrossRef CAS; (b) R. W. A. Havenith, J. H. Van Lenthe and L. W. Jenneskens, Int. J. Quantum Chem, 2001, 85, 52–60 CrossRef CAS.
- H. S. Rzepa and K. Taylor, J. Chem. Soc., Perkin Trans 2, 2002, 1499 RSC.
- C. Kastrup, H. S. Rzepa and S. V. Oldfield, J. Chem. Soc., Dalton Trans., 2002, 2421 RSC.
- Z.-X. Wang and P. von R. Schleyer, Helv. Chim. Acta, 2001, 84, 1578–1600 CrossRef CAS.
- (a) R. Herges, Angew. Chem. Int. Ed. Engl., 1994, 33, 255 CrossRef; (b) C. Berger, C. Bresler, U. Dilger, D. Geunenich, R. Herges, H. Rottele and G. Schroder, Angew. Chem. Int. Ed. Engl., 1998, 37, 1850 CrossRef CAS.
- F. Hernandez Blanco, D. C. Braddock, S. Martin-Santamaria and H. S. Rzepa, Internet J. Chem., 2001, 4, article 10 Search PubMed.
- M. Shibasaki, M. Kanai and K. Funubashi, Chem. Commun, 2002, 1989–1999 RSC.
- Gaussian 98 (Revision A.11), M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 1998.
- (a) P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS; (b) H. Jiao and P. von R. Schleyer, J. Phys. Org. Chem., 1998, 11, 655 CrossRef CAS.
- F. H. Allen and O. Kennard, Chemical Design Automation News, 1993, vol. 8, pp 1 & 31–37 Search PubMed.
- K. Ishihara, M. Miyata, K. Hattori, T. Tada and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 10520 CrossRef CAS.
- T. Arai, H. Sasai, K. Yamaguchi and M. S. Hibasaki, J. Am. Chem. Soc., 1998, 120, 441 CrossRef CAS.
- T. Arai, H. Sasai, K. Aoe, K. Okamura, T. Date and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1996, 35, 104 CrossRef CAS.
- S. Chitsaz and B. Neumuller, Organometallics, 2001, 20, 2338 CrossRef CAS.
- S. Matsunaga, J. Das, J. Roels, E. M. Vogl, N. Amamoto, T. Iida, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2000, 122, 2252 CrossRef CAS.
- R. Tacke, A. Stewart, J. Becht, C. Burschka and I. Richter, Can. J. Chem., 2000, 78, 1380 CrossRef CAS.
- R. R. Holmes, S. Shafieezad, V. Chandrasekhar, A. C. Sau, J. M. Holmes and R. A. Day, J. Am. Chem. Soc, 1998, 110, 1168.
- H. Wunderlich, Acta Crystallogr., Sect.B, 1981, 37, 995 CrossRef.
- D. J. Sherlock, A. Chandrasekaran, T. K. Prakasha, R. O. Day and R. R. Holmes, Inorg. Chem., 1998, 37, 93 CrossRef CAS.
- H. R. Allcock and E. C. Bissell, J. Am. Chem. Soc., 1973, 95, 3154 CrossRef CAS.
- B. A. Borgias, G. G. Hardin and K. N. Raymond, Inorg. Chem., 1986, 25, 1057 CrossRef CAS.
- J. Wegener, K. Kirschbaum and D. M. Giolando, J. Chem. Soc., Dalton Trans., 1994, 1213 RSC.
- G. Smith, A. N. Reddy, K. A. Byriel and C. H. L. Kennard, Aust. J. Chem., 1994, 47, 1413 CAS.
- K. von Deuten, W. Schnabel and G. Klar, Cryst. Struct. Commun., 1980, 9, 161 Search PubMed.
- O. Lindqvist, Acta Chem. Scand., 1967, 21, 1473 Search PubMed.
- T. Nemoto, T. Ohshima, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 2725 CrossRef CAS.
- H. C. Aspinall, J. F. Bickley, J. L. M. Dwyer, N. Greeves, R. V. Kelly and A. Steiner, Organometallics, 2000, 19, 5416 CrossRef CAS.
- T.-B. Wen, B.-S. Kang, C.-Y. Su, D.-X. Wu, L.-G. Wang, S. Liao and H.-Q. Liu, Bull. Chem. Soc. Jpn., 1998, 71, 2339 CAS.
- B. A. Borgias, S. R. Cooper, Y. B. Koh and K. N. Raymond, Inorg. Chem., 1984, 23, 1009 CrossRef CAS.
- M. Cowie and M. J. Bennett, Inorg. Chem., 1976, 15, 1595 CrossRef CAS.
- B.-S. Kang, X.-J. Wang, C.-Y. Su, H.-Q. Liu, T.-B. Wen and Q.-T. Liu, Transition Met. Chem., 1999, 24, 712 Search PubMed.
- P. R. Challen, D. H. Peapus and K. A. Magnus, Polyhedron, 1997, 16, 1447 CrossRef CAS.
- M. Cowie and M. J. Bennett, Inorg. Chem., 1976, 15, 1589 CrossRef CAS.
- R. J. Cross, L. J. Farrugia, D. R. McArthur, R. D. Peacock and D. S. C. Taylor, Inorg. Chem., 1999, 38, 5698 CrossRef CAS.
- H.-C. Chang, S. Kitagawa, M. Kondo and T. Ishii, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1999, 335, 183 Search PubMed.
- C. Lorber, J. P. Donahue, C. A. Goddard, E. Nordlander and R. H. Holm, J. Am. Chem. Soc., 1998, 120, 8102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Coordinates for the molecular structures. Seehttp://www.rsc.org/suppdata/ob/b2/b210415f/ |
| This journal is © The Royal Society of Chemistry 2003 |