Assignment of absolute configuration of a chiral phenyl-substituted dihydrofuroangelicin
Gennaro
Pescitelli†
a,
Nina
Berova
*a,
Tom L.
Xiao
b,
Roman V.
Rozhkov
b,
Richard C.
Larock
b and
Daniel W.
Armstrong
*b
aDept. of Chemistry, Columbia University, 3000 Broadway, New York, NY 10027, USA. E-mail: ndb1@columbia.edu
bDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: sec4dwa@iastate.edu
First published on 3rd December 2002
Abstract
A phenyl-substituted chiral dihydrofuroangelicin, 4-methyl-8-(2-E-phenylethenyl)-8,9-dihydro-2H-furo[2,3-h]-1-benzopyran-2-one, synthesized in racemic form, has been resolved by HPLC chiral separation, and its absolute configuration determined by the non-empirical exciton chirality method. The solution conformation has been investigated through NMR and molecular modeling methods: two minima found by molecular mechanics and DFT methods are in keeping with observed 1H–1H 3J coupling constants and NOE effects. The experimental CD spectrum for the second eluted enantiomer shows a positive couplet between 230 and 350 nm (amplitude A = + 15.7); by application of the exciton chirality method, the absolute configuration of this enantiomer at C8 is determined as (S). The experimental spectrum is in very good agreement with the one evaluated by means of DeVoe coupled-oscillator calculations, using the DFT calculated geometries.
Introduction
It is well known that many substituted furocoumarins are pharmacologically active. For example, they have been used for the treatment of skin diseases such as psoriasis and vitiligo.1 Warfarin is an anticoagulant that depresses formation of prothrombin and also increases the fragility of capillaries which can lead to hemorrhages.2 Recently, a variety of chiral, substituted dihydrofurocoumarins have been isolated as natural products and shown to have several useful pharmacological properties. For example, marmesin and columbionetin derivatives have been shown to exhibit cytotoxicity against KB cells,3 to inhibit c-AMP (which affects coronary vasodilation),4 and to mediate the action of acetylcholinesterase (which plays a role in Alzheimer's disease).5 A related dihydropsoralen, isolated from Dorstenia contrajerva, may moderate the adverse effects of rattlesnake venom.6Because of their importance, numerous syntheses of dihydrofurocoumarins have been published over the last 30 years.7,8 The earlier methods were characterized by numerous steps (8 to 10) and relatively low yields (2–20%).7 Later methods have required fewer steps and generally produce higher yields.8 One of the most recent and more general approaches involves the palladium catalyzed annulation of 1,3-dienes by o-iodoumbelliferones.9 This annulation proceeds in 70–85% yields with a variety of 1,3-dienes.9 Numerous substituted dihydrofuroangelicins 1 and dihydrofuropsoralens 2 (Chart 1) have been produced via this approach.
 | ||
| Chart 1 | ||
Although these compounds are usually chiral (when R1 ≠ R2), no efficient asymmetric synthesis has yet been reported. Also, the absolute configuration of some of the natural products has not been determined. In cases where one of the substituents (R1 or R2 above) contains an aromatic moiety, the possibility exists of using the exciton chirality method to determine the absolute configuration of these compounds. To test this possibility, compound 3 (Chart 2) was synthesized, resolved, and its absolute configuration investigated.
 | ||
| Chart 2 | ||
The exciton chirality method is a convenient and versatile approach for the structural investigation of chiral organic molecules when their absorption and circular dichroism spectra are dominated by intense electric dipole allowed transitions, such as the ones allied to strong aromatic chromophores.10 Any electric dipole allowed transition may be described in terms of a transition dipole moment. When two transition dipole moments are close in energy, lie near to each other in space and form a chiral array, their through-space interaction gives rise to distinctive spectral features: in the CD spectrum, a bisignate couplet is obtained whose sign is determined by the absolute sense of twist defined by the two dipoles; a positive twist corresponds to a positive couplet (i.e., with a positive long-wavelength branch), and vice versa. If the direction of transition dipoles within the molecular geometry is known, the absolute configuration may be derived.10 The presence of conformational ambiguity, or an unsuitable geometrical arrangement between dipoles, however, may preclude the straightforward application of this method.11
Full exciton-coupled CD spectra can be calculated with various methods; DeVoe coupled-oscillator calculations12,13 have been widely used for small organic molecules and polymers.14 The parameters necessary for the calculations are: (1) the molecular geometry, determined through experimental techniques and molecular modeling; (2) the spectral parameters (transition frequency, dipolar strength and bandwidth) extracted from the absorption spectra of the isolated chromophores; (3) position and direction of transition moments, usually determined by quantum mechanical calculation methods.
The inspection of the molecular structure of 3 (Chart 2) commends attention to the exciton chirality method as a most suitable approach for the determination of the absolute configuration in this case. This compound is in fact endowed with two strong aromatic chromophores (the coumarin and the styrene) located nearby in space, which give rise to intense electronic absorptions close in energy. Moreover, the spatial arrangement between the two chromophores, investigated through CD, will largely depend and be sensitive to the absolute configuration at the stereogenic center.
By taking into consideration these characteristic features of 3, we performed molecular mechanics and DFT modeling, as well as CD and NMR experimental studies, before applying DeVoe calculations of circular dichroic properties.
Results and discussion
Molecular modeling
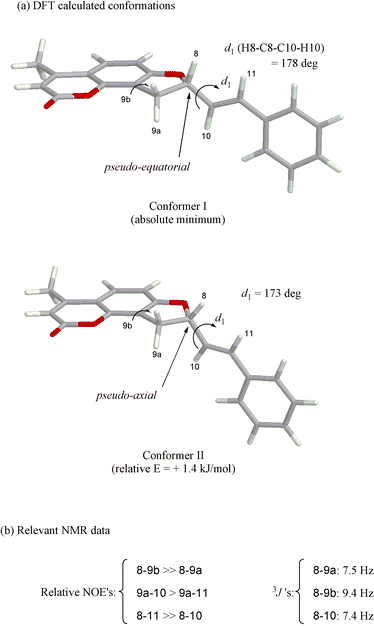
The molecular conformation of 3 was investigated through a combination of molecular mechanics and DFT calculations. The whole conformational space was sampled by means of Monte Carlo (MC) simulations15 with the molecular Merck force field (MMFFs), which is known to be very accurate for small organic molecules.16 MC/MMFFs calculations, run in CHCl3, reveal the presence of two main degrees of freedom, related to the five-membered ring conformation and the rotation around the C8–C10 bond, described through the H8–C8–C10–H10 dihedral angle d1 (Scheme 1a). Six minimum energy conformations were found overall by MC/MMFFs, which were further optimized by DFT calculations with B3LYP/6-31G** in CHCl3, resulting in the two structures shown in Scheme 1a as the lowest energy minima. In conformer I, the substituent occupies the pseudo-equatorial, and in conformer II the pseudo-axial position of the five-membered ring. In both structures the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C styrene double bond is syn to the C8–H8 bond, with d1
≈ 175°. The four other minima have energies higher than 6 kJ mol−1 with respect to conformer I and negligible Boltzmann populations at room temperature.
C styrene double bond is syn to the C8–H8 bond, with d1
≈ 175°. The four other minima have energies higher than 6 kJ mol−1 with respect to conformer I and negligible Boltzmann populations at room temperature.
 | ||
| Scheme 1 | ||
NMR spectra
1H NMR spectroscopy and, in particular, the chemical shifts, the 3J couplings and the relative NOE's between protons H8, H9, H10 and H11 (Chart 2 and Scheme 1), have been used to check the conformational picture arising from molecular modeling.Only one set of signals is apparent in the 1H NMR spectrum in CDCl3, proving that either only one conformer is present, or the interconversion between conformers is fast on the NMR timescale; this is expected for the five-membered ring flip and the rotamerism around the C8–C10 bond. Proton H9a is upfield shifted with respect to H9b by 0.4 ppm, and may be assigned a pseudo-axial position, considering both that axial protons generally resonate upfield of equatorial ones,17 and that the equatorial H9b is subjected to the deshielding effect of the phenyl ring current shift. Based on the relative NOE's (Scheme 1b), derived from a NOESY spectrum (not shown), proton H8 is closer and cis to H9b, and trans to H9a; this is also substantiated by the different values of 3J (JH8,H9a = 7.5 Hz, JH8,H9b = 9.4 Hz) analyzed through the Karplus equation.18 Thus, proton H8 lies in a pseudo-axial position, and the substituent at the stereogenic center occupies a preferred pseudo-equatorial position; this is the same situation occurring for the absolute minimum (Scheme 1a, conformer I) found by DFT.
The value of JH8,H10 = 7.42 Hz, analyzed through a Karplus-type equation for vinylic/allylic protons,19 leads to an estimated H8–C8–C10–H10 average dihedral angle d1 ≈ 140°; structures with large values of d1 are therefore dominant in solution, in agreement with the modeling results. This geometry around the C8–C10 bond is further confirmed by the strong NOE's between protons H8/H11 and H9a/H10 (Scheme 1b). It may be concluded that the DFT results faithfully depict the conformational situation in solution, and can be confidently used for the subsequent CD calculations.
Chromophore electronic structure
The UV absorption spectrum of compound 3 above 230 nm (Fig. 1, bottom) is dominated by the π–π* transitions of the aromatic chromophores. Two maxima are detected at 321 nm (ε = 12700 M−1 cm−1) and 252 nm (ε = 23000 M−1 cm−1) in acetonitrile, allied respectively to the conjugation or K bands of the coumarin and styrene chromophores.20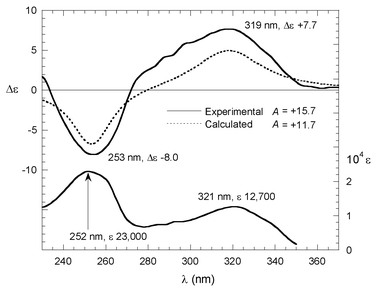 | ||
Fig. 1 UV (bottom) and CD (top) spectra of compound 3 in acetonitrile. Top, solid line: experimental CD spectrum for second eluted enatiomer 1.84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×10−5M in acetonitrile. Top, dotted line: CD calculated as Boltzmann average of DFT derived structures for the (S) configuration, with the DeVoe method and parameters as in Table 1 and Scheme 2. ×10−5M in acetonitrile. Top, dotted line: CD calculated as Boltzmann average of DFT derived structures for the (S) configuration, with the DeVoe method and parameters as in Table 1 and Scheme 2. | ||
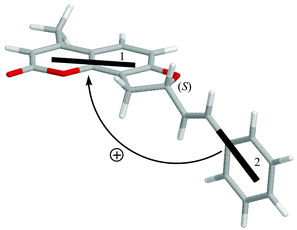
The relevant parameters for DeVoe calculations have been extracted from the UV spectra of 7-hydroxy-4,8-dimethylcoumarin in acetonitrile and styrene in hexane,21 and adapted to reproduce better the UV spectrum of 3, and are reported in Table 1. Both transitions have long-axis polarization in the respective ring systems. Due to the presence of substituents (in particular, the oxygen at position 7, Chart 2), the exact polarization of the coumarin transition needed to be calculated. By a CNDO-S/CI calculation on 7-hydroxy-4,8-dimethylcoumarin (geometry obtained with DFT, B3LYP/6-31G**), we found a small rotation (<5°) between the transition dipole and the coumarin ring long axis (as shown in Scheme 2).
 | ||
| Scheme 2 | ||
| Type | λ max/nm | D/D2 | ν max/cm−1 | Δν1/2/cm−1 | |
|---|---|---|---|---|---|
| a λ max wavelength maximum; D dipolar strength; νmax frequency maximum; Δν1/2 half-height width. b From the UV spectrum of 7-hydroxy-4,8-dimethylcoumarin in acetonitrile. c From the UV spectrum of styrene in hexane (ref. 21). | |||||
| 1 | Coumarinb | 318 | 20 | 31400 | 4000 |
| 2 | Styrenec | 254 | 30 | 39400 | 3600 |
CD spectrum, exciton chirality method and DeVoe calculation
The experimental CD spectrum of the second eluted enantiomer (Fig. 2; see Experimental Section for chiral HPLC conditions) of 3 in acetonitrile (Fig. 1, top, solid line) shows a positive couplet between 230 and 350 nm with a peak at 319 nm, Δε = +7.7, a trough at 253 nm, Δε = −8.0, and couplet amplitude A = +15.7. The symmetrical appearance of the couplet supports its interpretation as being due to non-degenerate exciton coupling between the two above discussed transitions, with small interference from higher energy ones. Integrated rotational strengths are +2.5×10−39 and −1.8×10−39 cgs units, respectively; the negative low wavelength branch is partially cancelled by superimposition with the positive Cotton effect appearing at higher energies.
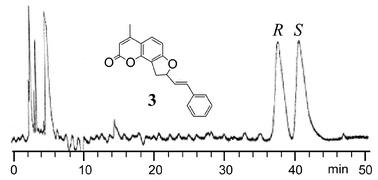 | ||
| Fig. 2 HPLC chromatogram of the enantiomeric separation of compound 3. The chromatogram was obtained using a Chirobiotic T column and a hexane plus 2% ethanol (by volume) mobile phase. The flow rate was 1.0 mL min−1. See the Experimental section for further details. The absolute configurations (S and R) of the first and the second eluted enantiomers for 3 were determined as outlined in the Results and discussion. | ||
Therefore, the absolute configuration of 3 can be assigned by the exciton chirality analysis of the experimental CD. By inspection of the molecular structure of the (S) enantiomer in the lowest energy conformation I (Scheme 2), it is apparent that the two above discussed transition dipoles define a positive chirality (that is, the transition moment 2 in front may be superimposed on the moment 1 in the back after a clockwise rotation). The same is true for the (S) enantiomer in the conformation II. Thus, the second eluted enantiomer, showing a positive CD couplet between 230 and 350 nm, may be assigned the (S) configuration.
Furthermore, this assignment is confirmed by quantitative CD theoretical calculations using the DeVoe method and the spectral and geometrical parameters discussed above, placing the dipoles in the middle of the styrene and coumarin chromophores (Scheme 2). The CD calculated as a Boltzmann-weighted average at room temperature for conformers I and II with an (S) configuration, shows a positive couplet with amplitude A = 11.7 (Fig. 1, top, dotted line), in good agreement with the experimental spectrum for the second eluted enantiomer. Other conformations with higher energies have calculated CD with intensities comparable to the ones for the first two, and after weighing with respective Boltzmann factors they give a negligible contribution. Small displacement (within 0.3 Å) and/or rotations (within 10°) of dipoles affected the calculated CD intensity only to a minor extent, and in no case was the sign reversed. Thus, DeVoe CD calculations can be reliably applied for the present configurational assignment.
Conclusion
The absolute configuration of a chiral, phenyl-substituted dihydrofuroangelicin (compound 3) has been assigned by means of the exciton chirality method and DeVoe CD calculations. The solution structure of 3 has been determined by NMR spectroscopy and molecular modeling with molecular mechanics and DFT methods. The spectroscopic parameters necessary for the CD calculations have been extracted from the UV spectra of isolated chromophores, and calculated by a CNDO method. The second eluted enantiomer of 3 (Fig. 2), with positive CD at 319 nm and negative CD at 253 nm, has an (S) absolute configuration.Experimental and computational section
General procedures
HRMS experiments were performed on the Kratos MS50TC double focusing magnetic sector mass spectrometer using EI at 70 eV. IR spectra were performed on the IR-Bomen Michelson MB-102 FT-IR spectrometer.1H and 13C spectra were recorded at 400 and 100.5 MHz respectively using a Varian 400 MHz instrument, and chemical shifts are reported in ppm relative to TMS (δ), with coupling constants (J) in Hz; proton numbering refers to Chart 2. 1H-NOESY spectrum was recorded at 600 MHz using a Varian Unity INOVA 600 with 1.5 s mixing time.
The UV spectrum for 3 was recorded on a Varian Cary 100 Bio UV-VIS spectrophotometer. The molar absorptivity was determined at three wavelengths (204 nm, 252 nm, and 321 nm). The concentration of all samples was adjusted so that their absorbance was in the range of 0.2 and 1.5 absorbance units. The molar absorptivities of 3 are: 59000, 23000, and 12700 at the aforementioned three wavelengths respectively.
CD spectra were measured in spectroscopy grade acetonitrile with a Jasco J-810 spectropolarimeter, using a 1 cm cell, and the following conditions: SBW 1 nm, 50 nm min−1, response 1 s, 8 scans.
MC/MMFFs and DFT calculations were run respectively with MacroModel 7.1 and Jaguar 4.1 (Schrödinger, Inc., Portland, OR).22
CNDO/S–CI calculations were run with a CNDO/M program, according to Del Bene and Jaffé's formulation,23 with Mataga approximation of two-electron repulsion integrals, including in the CI up to 648 singly excited states, with maximum energy values of 7.0 eV.
DeVoe calculations were run with a Fortran program due to Hug and co-workers.13
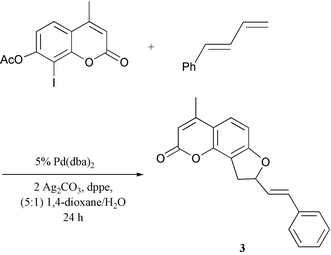
Synthesis of the dihydrofuroangelicin 4-methyl-8-(2-E-phenylethenyl)-8,9-dihydro-2H-furo[2,3-h]-1-benzopyran-2-one (3) was accomplished by annulation of (E)-1-phenylbuta-1,3-diene by the corresponding 7-acetoxy-8-iodo-4-methylcoumarin as reported elsewhere (Scheme 3).9
Mp 145–148 °C; 1H NMR (CDCl3)
δ/ppm 2.32 (3 H, d, JCH3,3
= 0.8 Hz, CH3), 3.16 (1 H, dd, JH9a,H9b
= 16.1 Hz, JH9a,H8
= 7.5 Hz, H9a), 3.55 (1 H, dd, JH9b,H9a
= 16.1 Hz, JH9b,H8
= 9.4 Hz, H9b), 5.49 (1 H, m, JH8,H9b
= 9.4 Hz, JH8,H9a
= 7.5 Hz, JH8,H10
= 7.4 Hz, JH8,H11
= 1.0 Hz, H8), 6.03 (1 H, d, J3,CH3
= 0.8 Hz, H3), 6.28 (1 H, dd, JH10,H11
= 15.8 Hz, JH10,H8
= 7.4 Hz, H10), 6.67 (1 H, dd, JH11,H10
= 15.8 Hz, JH11,H8
= 1.0 Hz, H11), 6.72 (1 H, d, JH6,H5
= 8.4 Hz, H6), 7.17–7.36 (5 H, m), 7.35 (1 H, d, JH5,H6
= 8.4 Hz, H5); 13C NMR (CDCl3)
δ 19.24, 33.28, 85.90, 106.77, 111.50, 113.69, 114.34, 125.76, 126.97, 127.33, 128.51, 128.85, 133.32, 136.02, 151.10, 153.26, 161.28, 163.44; IR (neat) 3050 (CH), 1727 (CO), 1615 (CC) cm−1; HRMS for C20H16O3 found: 304.1104, calc.: 304.1099.
 | ||
| Scheme 3 | ||
The enantiomeric separation of racemic 3 was performed on an HP 1050 HPLC system equipped with UV detector, auto-injector, and computer controlled Chem-station data processing software. Both preparative and analytical separations were carried out using a Chirobiotic T, 250 × 4.6 mm id, (Advanced Separation Technologies Inc., Whippany, NJ) column24 with baseline resolution and very good reproducibility (Fig. 2). The first eluted and second eluted peaks were collected manually. The normal phase mode was used for the enantiomeric separation with a mobile phase of hexane and 2% (by volume) ethanol. The injection volume was 2 µL. Separations were carried out isocratically at a flow rate of 1 mL min−1 at room temperature (22 °C). The mobile phase was premixed and degassed under vacuum conditions. All HPLC grade solvents were purchased from Fisher Chemical. Detection wavelengths were monitored at both 254 nm and 220 nm for confirmation that the same peak absorption ratio for the enantiomer pairs occurred.
Acknowledgements
Dr Lorenzo Di Bari, University of Pisa, is gratefully thanked for the help in NMR measurements. D.W.A. and T.L.X. acknowledge NIH grant RO1 GM53825–07 for partial support of this work. N.B. acknowledges NIH grants GM 34509 and GM 36564 for financial support. G.P. acknowledges the Italian C.N.R. grant (203.03.26) for financial support. R.V.R. and R.C.L. acknowledge the donors of the Petroleum Research Fund, administered by the American Chemical Society, for partial support of this research, and Kawaken Fine Chemicals Co., Ltd., and Johnson Matthey, Inc. for donations of Pd(OAc)2 and PPh3.References
- G. Rodigliero, J. Photochem. Photobiol., B, 1992, 14, 1–22 Search PubMed.
- R. G. Bell, J. A. Sadowski and J. T. Matschiner, Biochemistry, 1972, 11, 1959–1961 CrossRef CAS.
- D. Guilet, J.-J. Helesbeux, D. Seraphin, T. Sevenet, P. Richomme and J. Bruneton, J. Nat. Prod., 2001, 64, 563–567 CrossRef CAS.
- O. Thastrup, B. Fjalland and J. Lemmich, Acta Pharmacol. Toxicol., 1983, 52, 246–250 Search PubMed.
- S. Y. Kang, K. Y. Lee, S. H. Sung, M. J. Park and Y. C. Kim, J. Nat. Prod., 2001, 64, 683–685 CrossRef CAS.
- R. Tovar-Miranda, R. Cortes-Garcia, N. F. Santos-Sanchez and P. Joseph-Nathan, J. Nat. Prod., 1998, 61, 1216–1220 CrossRef CAS.
- (a) W. Steck, Can. J. Chem., 1971, 49, 1197–1201 CAS; (b) S. Yamaguchi, R. Miyakawa, S. Yonezawa and Y. Kawase, Bull. Chem. Soc. Jpn., 1989, 63, 3593–3597.
- (a) J. T. Trumble and J. G. Millar, J. Agric. Food. Chem., 1996, 44, 2859–2864 CrossRef CAS; (b) V. Stanjek, M. Milksch and W. Boland, Tetrahedron, 1997, 53, 17699–17710 CrossRef CAS.
- R. V. Rozhkov and R. C. Larock, Org. Lett., 2002, in press Search PubMed.
- (a) N. Harada and K. Nakanishi, Circular Dichroic Spectroscopy – Exciton Coupling in Organic Stereochemistry, University Science Books, Mill Valley, CA, 1983 Search PubMed; (b) N. Berova, K. Nakanishi, in Principles and Applications of Exciton Chirality Method, ed. N. Berova, K. Nakanishi, and R. Woody, Wiley-VCH, New York, 2000 Search PubMed.
- R. Person, K. Monde, H.-U. Humpf, N. Berova and K. Nakanishi, Chirality, 1995, 7, 128–135 CrossRef CAS.
- (a) H. DeVoe, J. Chem. Phys., 1964, 41, 393–400 CrossRef CAS; (b) H. DeVoe, J. Chem. Phys., 1965, 43, 3199–3208 CrossRef CAS; (c) C. Rosini, M. Zandomeneghi and P. Salvadori, Tetrahedron: Asymmetry, 1993, 4, 545–554 CrossRef CAS.
- C. L. Cech, W. Hug and I. Tinoco, Jr., Biopolymers, 1976, 15, 131–152 CrossRef CAS.
- (a) F. Castronovo, M. Clericuzio, L. Toma and G. Vidari, Tetrahedron, 2001, 57, 2791–2798 CrossRef CAS; (b) L. Di Bari, G. Pescitelli, G. Reginato and P. Salvadori, Chirality, 2001, 13, 548–555 CrossRef CAS; (c) C. Rosini, M. I. Donnoli and S. Superchi, Chem. Eur. J., 2001, 7, 72–79 CrossRef CAS; (d) L. Di Bari, S. Mannucci, G. Pescitelli and P. Salvadori, Chirality, 2002, 14, 611–617 CrossRef CAS; (e) A. Solladié-Cavallo, C. Marsol, G. Pescitelli, L. Di Bari, P. Salvadori, X. Huang, N. Fujioka, N. Berova, X. Cao, T. B. Freedman and L. A. Nafie, Eur. J. Org. Chem., 2002, 1788–1796 CrossRef CAS.
- G. Chang, W. C. Guida and W. C. Still, J. Am. Chem. Soc., 1989, 111, 4379–4386 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley-Interscience, New York, 1994 Search PubMed.
- From the Karplus equation 3J = 7 − cos ϕ + 5cos 2ϕ, where ϕ is either the H8–C8–C9a–H9a (ϕ8–9a) or the H8–C8–C9b–H9b (ϕ8–9b) dihedral angle, two possible sets of values are obtained: (1) ϕ8–9a = 23 and ϕ8–9b = 134°, or (2) ϕ8–9a = 144 and ϕ8–9b = 37°. Only the first set is consistent with the DFT molecular models..
- E. W. Garbisch, Jr., J. Am. Chem. Soc., 1964, 86, 5561–5564 CrossRef.
- (a) R. A. Friedel and M. Orchin, Ultraviolet Spectra of Aromatic Compounds, Wiley, New York, 1951 Search PubMed; (b) H. H. Jaffé and M. Orchin, Theory and Applications of Ultraviolet Spectroscopy, Wiley, New York, 1962 Search PubMed; (c) L. Salem, The Molecular Orbital Theory of Conjugated Systems, Benjamin, New York, 1966 Search PubMed.
- A. Kaito, A. Tajiri and M. Hatano, J. Am. Chem. Soc., 1976, 98, 384–388 CrossRef CAS.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS.
- J. Del Bene and H. H. Jaffe, J. Chem. Phys., 1968, 49, 1221–1229 CrossRef.
- D. W. Armstrong, Y. Liu and H. K. Ekborg-Ott, Chirality, 1995, 7, 474–497 CrossRef CAS.
Footnote |
| † Present address: Dipartimento di Chimica e Chimica Industriale, Università degli Studi di Pisa, via Risorgimento 35, I-56126 Pisa, Italy. |
| This journal is © The Royal Society of Chemistry 2003 |