Intercalating nucleic acids (INAs) with insertion of N-(pyren-1-ylmethyl)-(3R,4R)-4-(hydroxymethyl)pyrrolidin-3-ol. DNA (RNA) duplex and DNA three-way junction stabilities
Vyacheslav V.
Filichev
and
Erik B.
Pedersen
*
Nucleic Acid Center†, Department of Chemistry, University of Southern Denmark, DK-5230 Odense M, Denmark. E-mail: EBP@chem.sdu.dk; Fax: +45 6615 8780; Tel: +45 6550 2555
First published on 29th November 2002
Abstract
N-(Pyren-1-ylmethyl)-(3R,4R)-4-(hydroxymethyl)pyrrolidin-3-ol was synthesised from (3R,4R)-4-(hydroxymethyl)pyrrolidin-3-ol and (3R,4S)-4-[(1S)-1,2-dihydroxyethyl]pyrrolidin-3-ol using alkylation with 1-(chloromethyl)pyrene or reductive amination with pyrene-1-carbaldehyde and NaCNBH3. The incorporation of N-(pyren-1-ylmethyl)azasugar moiety into oligodeoxynucleotides (ODN) as a bulge to form an intercalating nucleic acid (INA) induced a slight destabilization of INA–DNA duplex, whereas the INA–RNA duplex was strongly destabilized and 9 °C difference per modification in thermal stability between INA–DNA over INA–RNA duplexes was observed. The stabilization of a DNA three way junction (TWJ) was improved when the intercalator moiety was inserted into the junction region as a bulge.
1. Introduction
The stabilized formation of the secondary structures of nucleic acids with chemically modified nucleosidic units is the main goal at the transcriptional (antigene) and translational (antisense) level of the gene expression.1 The increased affinity and sequence selectivity are desirable properties to achieve a good recognition for therapeutic targets. It is known that there are some differences in the three-dimensional structures of DNA–DNA and DNA–RNA duplexes. By the chemical modification of nucleosidic glycons some oligonucleotide analogues having better increased affinity towards single stranded RNA (ssRNA) and towards single stranded DNA (ssDNA) were discovered, for example locked nucleic acids (LNA),2 hexitol nucleic acids (HNA)3 and others.4 On the other hand all nucleic acids are stabilized both by hydrogen bonding interactions and by base stacking.1 Therefore polyaromatic units with hydrophobicity in conjunction with a large surface area can be used in place of the natural nucleic bases for mimicking base stacking in Watson–Crick/Hoogsteen base pairing.5 The design and the synthesis of various oligonucleotides containing planar polycyclic aromatic chromophores such as pyrene, anthracene, dansyl and others, which have a long fluorescence lifetime and possibilities to form π-stacking interactions in aqueous solutions, have been the subject of active research in recent years.6 However there are only a few articles focused on the possible discrimination between ssDNA and ssRNA with the modification or substitution of nucleic bases in the complementary sequences. Bleczinski et al. observed the DNA/RNA discrimination by insertion of cholic acid and its deoxy derivatives into the 5′-termini of short oligonucleotides.7 Recently we have found that intercalating nucleic acids (INAs) containing 1-O-(pyrenylmethyl)glycerol insertions have a strong preference for hybridization with DNA over RNA,8 but little is known about the requirements of the backbone structure for the selectivity between DNA and RNA.Single stranded oligonucleotides are often written as single stretches, but because of self pairing, branched structures (hairpins, internal loops, junctions) have been observed.9 For this reason, the number of possible targets is lower than it is often believed. It has actually been shown that recognising the structure of nucleic acids could be achieved by forming a three way junction (TWJ) without disruption of the stem.10 Formation of the TWJ has been confirmed in NMR studies on complexes with two unpaired bases at the branch point and a preferred coaxial base stacking interaction was indicated.11 Based on these findings we started out investigations devoted to the synthesis of pyren-1-yl units linked to different sugar-mimicking fragments which were used for insertions into ODNs for hybridization studies.12
Recently we synthesised the enantiomerically pure 1-aza analogue of 2-deoxy-D-ribofuranose having a nitrogen instead of the anomeric carbon and a methylene group instead of the ring oxygen.13 The corresponding 1′-aza-C-pseudonucleosides in which a carbon in the heterocyclic base is linked to the nitrogen of (3R,4R)-4-(hydroxymethyl)pyrrolidin-3-ol were also obtained.14 In continuation of this we now investigate the synthesis of N-pyrenemethyl type pseudonucleoside of the enantiomerically pure 1-aza analogue of 2-deoxy-D-ribofuranose and its insertion into several ODNs to form INAs in order to investigate their thermal stability when forming duplexes with complementary ssDNA and ssRNA or when forming DNA TWJs.
2. Results and discussion
The synthesis of 1′-aza pyrenemethyl pseudonucleoside 4 started from the previously published enantiomerically pure 1-aza analogues of 2-deoxy-D-hexofuranose 1 or 2-deoxy-D-ribofuranose 2.13 To find the most efficient synthesis of the target compound 4, we used pyrene substrates having chloromethyl and carbaldehyde functionalities that could be coupled with the secondary amines 1 and 2. We found that alkylation by 1-(chloromethyl)pyrene in the presence of Et3N led to a lower yield (57% and 46% for 3 and 4, respectively) compared to reductive amination with pyrene-1-carbaldehyde and NaCNBH3 (63% and 57%, respectively, Scheme 1). Surprisingly low overall yield 40% was observed in the synthesis of 4 using oxidative cleavage of the diol 3 followed by reduction of the obtained aldehyde. This may be due to low stability of the pyren-1-ylmethyl azasugars 3 and 4, which should be stored in a refrigerator under nitrogen or used immediately to avoid low yields in subsequent reactions.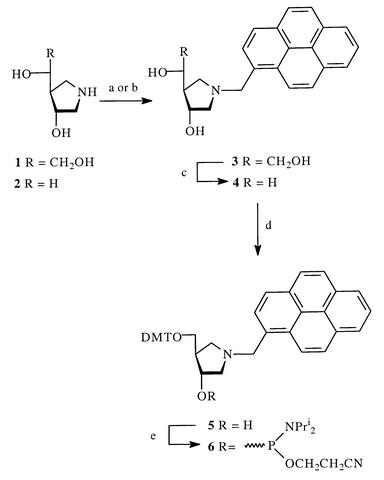 | ||
| Scheme 1 (a) 1-(chloromethyl)pyrene, Et3N, DMF; (b) pyrene-1-carbaldehyde, NaCNBH3, DMF–EtOH (3 ∶ 1); (c) 1) NaIO4, 2) NaBH4; (d) DMTCl, pyridine; (e) NC(CH2)2OP(NPri2)2, N,N-diisopropylammonium tetrazolide, CH2Cl2. | ||
The 4,4′-dimethoxytriphenylmethyl (DMT) protected phosphoramidite 6 is required for the oligonucleotide synthesis. The primary alcohol 4 was treated with an excess of DMTCl in pyridine with further purification on a silica gel column to give compound 5 in 61% yield. The synthesis of the final phosphoramidite by treatment with 2-cyanoethyl-N,N-isopropylchlorophosphoramidite in the presence of the excess of Hunig's base12 failed. To obtain the required phosphoramidite 6 we used an alternative method with 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphorodiamidite and N,N-diisopropylammonium tetrazolide.15 The compound 6 was obtained in 57% yield.
The phosphoramidite 6 was incorporated into different oligonucleotide sequences to give INAs on an automated solid phase DNA synthesizer using an increased coupling time (24 min) and repeating the cycle twice. The coupling efficiencies for the pyrene azasugar derivative 6 were approximately 80–85% compared to approximately 99% for commercial phosphoramidites (2 min coupling).
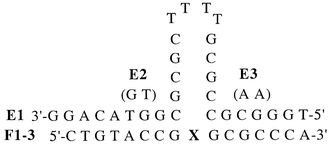
The synthesised INAs were used in the hybridization studies of INA–DNA and INA–RNA duplexes (Table 1) and INA–DNA three way junction (TWJ) (Table 2).
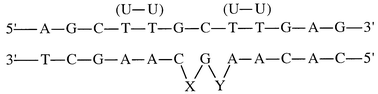
| No. | X | Y | T m/°C (DNA–DNA) | ΔTm/°C | T m/°C (DNA–RNA) | ΔTm/°C | ΔΔTmDNA–RNA |
|---|---|---|---|---|---|---|---|
| I = inserted nucleoside analogue 4, ΔTm = decrease in Tm per modification, ΔΔTmDNA–RNA = discrimination in Tm between DNA–DNA and DNA–RNA duplexes per modification | |||||||
| A | — | — | 43.0 | 42.2 | |||
| B | G | — | 32.2 | −10.8 | 32.6 | −9.6 | 1.2 |
| C | I | — | 41.8 | −1.2 | 32.2 | −10.0 | 8.8 |
| D | I | I | 39.4 | −1.2 | 21.8 | −10.2 | 9.0 |
|
F1
(X = 0) |
F2
(X = A) |
F3
(X = I) |
|
|---|---|---|---|
| I = inserted nucleoside analogue 4 | |||
| E1 | 38.6 | 39.4 | 48.6 |
| E2 | <18.0 | 20.2 | 24.2 |
| E3 | <18.0 | 19.4 | <18.0 |
INA with incorporation of the N-(pyren-1-ylmethyl)azasugar as the bulge resulted in lowering of the melting temperature with 1.2 °C per modification towards ssDNA (Table 1). The corresponding reference duplex in entry B containing a bulging deoxynucleotide (dG) had a considerably lower Tm = 32.2 °C (Tm = −10.8 °C). For the INA–RNA duplexes the pyrene containing sequence C and the reference B decreased the stability of the INA–RNA duplex with 10 °C and 9.6 °C, respectively, compared to the perfectly matched duplex (entry A). Consequently, INA with pyrene azasugar incorporated as the bulge has better hybridization affinity towards the complementary ssDNA than towards ssRNA. The differences in melting temperatures for ssDNA and ssRNA seems to be additive with respect to the number of pyrene moieties in the targeting ODN. These results are also in agreement with other investigations where 1-O-(pyren-1-ylmethyl)glycerol was inserted twice as bulges. A larger discrimination up to 25.8 °C between INA–DNA and INA–RNA was then observed.8 In that case the INA–DNA structure is stabilized compared to the wild type duplex in contrast to our case where a slight decrease is observed for Tm. The flexibility of the bulge may be an important factor to obtain both duplex stabilization and discrimination. In order to understand the nature of the difference in the thermal stability between DNA and RNA duplexes observed, further experiments are in progress using NMR spectroscopy for structural examination of the duplexes. The synthesis of different linkers and planar aromatic moieties is also in progress. The RNA/DNA discrimination displayed may be applied for purification or detection of DNA targets in a mixture with the very same sequences of RNA.
As a second target in our investigation we chose a DNA three way junction (TWJ) composed of two arms linked to a stem (Table 2). Here we observed a considerable stabilization when the pyrene azasugar intercalator was inserted in the INA (F3) compared to the ODN having dA at the same position (F2) or without an insertion in the ODN (entry F1). To be sure that hybridization in the arms is important for the stability of the complex; we prepared ODNs with mismatches in either arm of TWJ (entry E2 and E3). In both cases it resulted in a large lowering of the hybridization affinity.
3. Conclusion
Thus we prepared and used N-(pyren-1-ylmethyl)-(3R,4R)-4-(hydroxymethyl)pyrrolidin-3-ol (4) in the synthesis of several INAs and investigated the hybridization affinity of INA–DNA, INA–RNA duplexes and DNA TWJ region. When the N-(pyren-1-ylmethyl)azasugar was inserted as a bulge we observed good discrimination between stabilities of INA–DNA and INA–RNA duplexes and the increased stability of a DNA three-way junction. We believe that these findings will be helpful in molecular design of targets and probes for DNA diagnostics.4. Experimental
4.1 General
NMR spectra were recorded on a Bruker AC-300 FT NMR spectrometer at 300 MHz for 1H NMR and at 75.5 MHz for 13C NMR. Internal standards used in 1H NMR spectra were TMS (δ: 0.00) for CDCl3, CD3OD; in 13C NMR were CDCl3 (δ: 77.0), CD3OD (δ: 49.0). Accurate ion mass determination was performed on a Kraton MS-50-RF equipped with FAB source. The [M + H]+ ions were peakmatched using ions derived from the glycerol matrix. Thin layer chromatography (TLC) analyses were carried out with use of TLC plates 60 F254 purchased from Merck and were visualized in UV light (254 and/or 343 nm) and/or with a ninhydrin spray reagent (0.3 g ninhydrin in 100 cm3 butan-1-ol and 3 cm3 HOAc) for azasugars and their derivatives. The silica gel (0.063–0.200) used for column chromatography was purchased from Merck. ODNs were synthesised on an Assembler Gene Special DNA-Synthesizer (Pharmacia Biotech). Purification of 5′-O-DMT-on and 5′-O-DMT-off ODNs were accomplished using a Waters Delta Prep 4000 Preparative Chromatography System. The modified ODNs were confirmed by MALDI-TOF analysis on a Voyager Elite Biospectrometry Research Station from PerSeptive Biosystems. All solvents were distilled before use. The reagents used were purchased from Aldrich, Sigma or Fluka. The reagents for Gene Assembler were purchased from Cruachem (UK).Method A. Azasugar 1 (50 mg, 0.34 mmol) was dissolved in DMF (5 cm3); 1-(chloromethyl)pyrene (103 mg, 0.41 mmol) and Et3N (0.057 cm3, 0.41 mmol) were added. The reaction mixture was stirred at room temp. under nitrogen overnight. The solvent was evaporated under reduced pressure and co-evaporated with toluene (2 × 5 cm3). The residue was chromatographed on a silica gel column with CH2Cl2–MeOH (0–20%, v/v) as eluent affording the pure product 3 (70 mg, 57%): Rf 0.20 (10% MeOH–CH2Cl2); δH(CD3OD) 2.36 (1 H, m, H-4), 2.95 (1 H, m, H-5), 3.08 (1 H, dd, J 2.8 and 10.5, H-2), 3.22 (1 H, dd, J 5.4 and 12.0, H-5), 3.34 (1 H, m, H-2), 3.50–3.65 (3 H, m, CH[OH]CH2OH), 4.42 (1 H, m, H-3), 4.65 (2 H, s, CH2pyren-1-yl), 4.88 (3 H, br s, 3 × OH), 7.90–8.40 (9 H, m, Harom); δC(CD3OD) 50.7 (C-4), 56.7 (C-5), 57.2 (C-2), 62.7 (CH2pyren-1-yl), 65.7 (CH2OH), 71.8 (CH[OH]), 72.7 (C-3), 123.8, 125.5, 125.8, 125.9, 126.6, 126.8, 127.3, 128.0, 128.2, 129.1, 129.4, 129.9, 131.2, 131.9, 132.5, 133.2 (pyren-1-yl); m/z (FAB) 362.1748 [M + H]+, C23H24NO3 requires 362.1756.
Method B. Azasugar 1 (70 mg, 0.48 mmol) was dissolved in DMF–EtOH (3 ∶ 1, 10 cm3) and pyrene-1-carbaldehyde (270 mg, 1.18 mmol) and NaCNBH3 (74 mg, 1.18 mmol) were added. The reaction mixture was stirred at room temp. under nitrogen overnight. Concentrated HCl was added until pH < 2. Solvent was evaporated under reduced pressure and co-evaporated with toluene (2 × 5 cm3). The residue was purified using silica gel column chromatography with CH2Cl2–MeOH (0–20%, v/v) affording the compound 3 (110 mg, 63%).
Method A. Azasugar 2 (100 mg, 0.86 mmol) was dissolved in DMF (10 cm3) and 1-(chloromethyl)pyrene (257 mg, 1.03 mmol) and Et3N (0.140 cm3, 1.03 mmol) were added. The reaction mixture was stirred at room temp. under nitrogen overnight. The solvent was evaporated under reduced pressure and co-evaporated with toluene (2 × 5 cm3). The residue was dissolved in H2O–CH2Cl2 (1 ∶ 1, 40 cm3) and the water layer was extracted with CH2Cl2. The combined organic fractions were dried (Na2SO4), evaporated in vacuo and chromatographed on a silica gel column with CH2Cl2–MeOH (0–20%, v/v) affording the title compound 4 (130 mg, 46%): Rf 0.17 (10% MeOH–CH2Cl2); δH (CDCl3) 2.08 (1 H, m, H-4), 2.29 (1 H, m, H-5), 2.57 (1 H, dd, J 2.8 and 10.2, H-2), 2.85 (2 H, m, H-2 and H-5), 3.43 (2 H, s, CH2OH), 3.47 (2 H, br s, 2(OH)), 4.08 (1 H, m, H-3), 4.19 (2 H, s, CH2pyren-1-yl), 7.90–8.40 (9 H, m, Harom); δC (CDCl3) 49.8 (C-4), 55.9 (C-5), 57.5 (C-2), 62.3 (CH2pyren-1-yl), 64.2 (CH2OH), 73.9 (C-3), 123.3, 124.4, 124.6, 124.8, 125.1, 125.9, 127.3, 127.6, 127.8, 129.5, 130.7, 130.9, 131.1 (pyren-1-yl); m/z (FAB) 332.1631 [M + H]+, C23H24NO3 requires 332.1651.
Method B. Azasugar 2 (1.18 g, 10.1 mmol) was dissolved in DMF–EtOH (3 ∶ 1, 150 cm3) and pyrene-1-carbaldehyde (3.47 g, 15.1 mmol) and NaCNBH3 (950 mg, 15.1 mmol) were added. The reaction mixture was stirred at room temp. under nitrogen overnight. Concentrated HCl was added until pH<2. The solvent was evaporated under reduced pressure and co-evaporated with toluene (2 × 5 cm3). The residue was dissolved in H2O–CH2Cl2 (1 ∶ 1, v/v, 150 cm3) and the water layer was extracted with CH2Cl2 (3 × 75 cm3). The combined organic fractions were dried (Na2SO4) and evaporated under diminished pressure. The residue was purified using silica gel column chromatography with CH2Cl2–MeOH (0–20%, v/v) affording the compound 4 as an oil which crystallised on standing (1.9 g, 57%), mp 104–105 °C.
Method C. A cooled solution of compound 3 (110 mg, 0.304 mmol) in EtOH (4 cm3) was added to a solution of NaIO4 (71.6 mg, 0.335 mmol) in H2O (1.5 cm3) under stirring. After 30 min NaBH4 (12.3 mg, 0.335 mmol) was added. After 30 min the resulting solution was acidified with 2 M HCl until pH<2 under vigorous stirring. The solvent was removed in vacuo. The residue was dissolved in H2O–CH2Cl2 (1 ∶ 1, v/v, 20 cm3) and extracted with CH2Cl2 (4 × 15 cm3). The combined organic layers were dried (Na2SO4), evaporated under diminished pressure to dryness affording compound 4 (40 mg, 40%).
4.2 Synthesis and purification of modified and unmodified oligodeoxynucleotides
The oligodeoxynucleotides were synthesised on a Pharmacia Gene Assembler® Special synthesizer in 0.2 µmol-scale (7.5 µmol embedded per cycle, Pharmacia primer supportTM) using commercially available 2-cyanoethylphosphoramidites and 6. The synthesis followed the regular protocol for the DNA synthesizer. The coupling time for 6 was increased from 2 to 24 min and the cycle was repeated twice. The 5′-O-DMT-on ODNs were removed from the solid support and deprotected with 32% aqueous NH3 (1 cm3) at 55 °C for 24 h and then purified on preparative HPLC using a Hamilton PRP-1 column. The solvent systems were buffer A [950 cm3 0.1 M NH4HCO3 and 50 cm3 CH3CN (pH = 9.0)] and buffer B [250 cm3 0.1 M NH4HCO3 and 750 cm3 CH3CN (pH = 9.0)] which were used in the following order: 5 min A, 30 min linear gradient of 0–70% B in A, 5 min liner gradient of 70–100% B in A. Flow rate was 1 cm3 min−1. The purified 5′-O-DMT-on ODNs eluted as one peak after approximately 30 min [UV control 254 nm and 343 nm (for pyrene containing ODNs)]. The fractions were concentrated in vacuo followed by treatment with 10% aqueous HOAc (1 cm3) for 20 min and further purification on HPLC under the same conditions to afford detritylated ODNs which eluted at 23–28 min. The purity of oligos synthesised was 99–100% according to the preparative HPLC. The resulted solutions were evaporated in vacuo and co-evaporated twice with water to remove volatile salts to afford ODNs, which were used in melting temperature measurements. All oligonucleotides containing pyrenylmethylazasugar derivative 6 were confirmed by MALDI-TOF analysis (entry C: found 4005.65, calcd. 4005.76; entry D: 4398.02, calcd. 4398.87; entry F3: found 4903.05, calcd. 4904.89).4.3 Melting experiments
Melting temperature measurements were performed on a Perkin-Elmer UV/VIS spectrometer fitted with a PTP-6 Peltier temperature-programming element. The absorbance at 260 nm was measured from 18 °C to 85 °C in 1 cm cells. The melting temperature was determined as the maximum of the derivative plots of the melting curve. The oligodeoxynucleotides were dissolved in a medium salt buffer (pH = 7.0, 1 mM EDTA, 10 mM Na2HPO4 × 2H2O, 140 mM NaCl) to a concentration of 1.0 µM for each strand.Acknowledgements
We thank Mr U. B. Christensen for the great help in the synthesis and purification of ODNs.References
- (a) E. Uhlmann, Curr. Opin. Drug Discovery Dev., 2000, 3, 203 Search PubMed; (b) A. De Mesmaeker, R. Häner, P. Martin and H. E. Moser, Acc. Chem. Res., 1995, 28, 366–374 CrossRef CAS; (c) M. Egli, Angew. Chem., Int. Ed., 1996, 35, 1894–1910 CrossRef; (d) S. Verma and F. Eckstein, Annu. Rev. Biochem., 1998, 67, 99–134 CrossRef CAS; (e) D. Praseuth, A. L. Guieysse and C. Helene, Biochim. Biophys. Acta, 1999, 1489, 181–206 CrossRef CAS; (f) J. Micklefield, Curr. Med. Chem., 2001, 8, 1157–1179 Search PubMed; (g) E. T. Kool, Chem. Rev., 1997, 97, 1473–1487 CrossRef CAS.
- J. Wengel, Acc. Chem. Res., 1999, 32, 301–310 CrossRef CAS.
- (a) A. Van Aerschot, I. Verheggen, C. Hendrix and P. Herdewijn, Angew. Chem., Int. Ed. Engl., 1995, 34, 1338–1339 CrossRef CAS; (b) C. Hendrix, H. Rosemeyer, I. Verheggen, F. Seela, A. Van Aerschot and P. Herdewijn, Chem. Eur. J., 1997, 3, 110–120 CrossRef CAS.
- C. J. Leumann, Bioorg. Med. Chem., 2002, 10, 841–854 Search PubMed.
- (a) I. Luyten and P. Herdewijn, Eur. J. Med. Chem., 1998, 33, 515–576 CrossRef CAS; (b) P. Herdewijn, Antisense Nucleic Acid Drug Dev., 2000, 10, 293–310 CrossRef; (c) K. M. Guckian, B. A. Schweitzer, R. X.-F. Ren, C. J. Sheils, D. C. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 2000, 122, 2213–2222 CrossRef CAS.
- M. J. Davies, A. Shah and I. J. Bruce, Chem. Soc. Rev., 2000, 29, 97–107 RSC.
- C. F. Bleczinski and C. Richert, J. Am. Chem. Soc., 1999, 121, 10889–10894 CrossRef CAS.
- U. B. Christensen and E. B. Pedersen, Nucleic Acids Res., 2002, 30, 4918–4925 CrossRef CAS.
- (a) R. T. Batey, R. P. Rambo and J. A. Doudna, Angew. Chem., Int. Ed., 1999, 38, 2326–2343 CrossRef; (b) F. Belmont, J.-F. Constant and M. Demeunyck, Chem. Soc. Rev., 2001, 30, 70–81 RSC.
- (a) S. T. Cload and A. Schepartz, J. Am. Chem. Soc., 1994, 116, 437–442 CrossRef CAS; (b) M. Zhong, M. S. Rashes, N. B. Leontis and N. R. Kallenbach, Biochemistry, 1994, 33, 3660–3667 CrossRef CAS; (c) Q. Guo, M. Lu, M. E. A. Churchill, T. P. Tullius and N. R. Kallenbach, Biochemistry, 1990, 29, 10927–10934 CrossRef CAS; (d) F. Stuhneier, J. B. Welch, A. I. H. Murchie, D. M. J. Lilley and R. M. Clegg, Biochemistry, 1997, 36, 13530–13538 CrossRef.
- (a) M. A. Rosen and D. J. Patel, Biochemistry, 1993, 32, 6563–6575 CrossRef CAS; (b) M. A. Rosen and D. J. Patel, Biochemistry, 1993, 32, 6576–6587 CrossRef CAS.
- (a) O. M. Ali, T. Franch, K. Gerdes and E. B. Pedersen, Nucleic Acids Res., 1998, 26, 4919–4924 CrossRef CAS; (b) C. S. Poulsen, E. B. Pedersen and C. Nielsen, Acta Chem. Scand., 1999, 53, 425–431 Search PubMed; (c) A. A.-H. Abdel-Rahman, O. M. Ali and E. B. Pedersen, Tetrahedron, 1996, 52, 15311–15324 CrossRef CAS; (d) A. F. Khattab and E. B. Pedersen, Nucleosides, Nucleotides, 1998, 17, 2351–2365 Search PubMed; (e) S. A. El-Kafrawy, M. A. Zahran, E. B. Pedersen and C. Nielsen, Helv. Chim. Acta, 2000, 83, 1408–1416 CrossRef CAS.
- V. V. Filichev, M. Brandt and E. B. Pedersen, Carbohydr. Res., 2001, 333, 115–122 CrossRef CAS.
- V. V. Filichev and E. B. Pedersen, Tetrahedron, 2001, 57, 9163–9168 CrossRef CAS.
- J. Burmeister, A. Azzawi and G. Von Kiedrowski, Tetrahedron Lett., 1995, 36, 3667–3668 CrossRef CAS.
Footnote |
| † A research center funded by The Danish National Research Foundation for studies on nucleic acid chemical biology |
| This journal is © The Royal Society of Chemistry 2003 |