A formal total synthesis of (+)-apicularen A: base-induced conversion of apicularen-derived intermediates into salicylihalamide-like products
Arwel
Lewis
a,
Ian
Stefanuti
a,
Simon A.
Swain
a,
Stephen A.
Smith
b and
Richard J. K.
Taylor
*a
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD
bGlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage, UK SG1 2NY
First published on 5th December 2002
Abstract
A synthesis of apicularen precursor (−)-6 in 18 steps from D-glucal is reported. As (+)-6 has been converted into the potent, naturally occurring salicylate anti-cancer agent, (−)-apicularen A in 8 steps, this study constitutes a formal total synthesis of (+)-apicularen A. Key steps in the synthetic route include: (i) useful D-glucal elaboration processes, (ii) organometallic displacements at carbohydrate C-6 triflates using Knochel-type and related functionalised, aromatic Grignard reagents, (iii) stereoselective allyltrimethylsilane–acetal reactions generating C-allyl systems, (iv) stereocontrolled aldehyde allylation processes from both substrate and reagent, and (v) a novel Keck-type macrolactonisation. In addition, preliminary studies are reported in which a procedure has been devised to convert apicularen-derived intermediates into salicylihalamide-like products.
Introduction
Apicularen A (1) (Fig. 1) was originally isolated from the myxobacterial genus Chondromyces by Jansen et al. in 1998.1 Apicularen B (2), a glycosylated co-metabolite of apicularen A was isolated from the same source. Biological screening indicated that apicularen B showed weak antimicrobial activity against some Gram-positive bacteria, e.g.Micrococcus luteus (MIC 12.5 µg ml¬1), and moderate cytotoxic activity with IC50 values ranging between 0.2 and 1.2 µg ml¬1. Apicularen A did not show any antibiotic activity but exhibited remarkably high cytostatic activity (IC50 values between 0.3 and 3 ng ml¬1 against nine different human cancer cell lines). Furthermore, Sasse2 observed several abnormal effects in tumour cells suggesting a novel mode of action.Other related salicylate anti-tumour natural products are known,3 including salicylihalamide A (3),3a lobatamide C (4),3b CJ-129503c and oximidine I (5).3d These families of compounds were tested in the NCI 60 cell-line tumour screen and it was found that as well as acting via a novel mode of action they all share the same (or similar) molecular target. Recent studies have established that these compounds are selective inhibitors of mammalian vacuolar (H+)-ATPase.4
The potent biological activity, structural novelty and limited availability of these salicylate anti-tumour natural products has resulted in the development of numerous synthetic approaches5 culminating in recent total syntheses of salicylihalamide A by De Brabander et al.,6a Fürstner et al.,6b Labrecque et al.,6c Smith and Zheng6d and Snider and Song,6e of lobatamide C by Porco et al.,6f and of apicularen A by De Brabander's group,6g together with a formal total synthesis of the latter compound.7
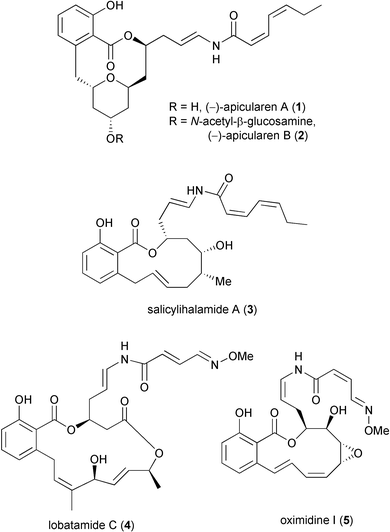 | ||
| Fig. 1 | ||
This paper details the formal total synthesis of apicularen A using a carbohydrate based route.7D-Glucal is used to prepare the protected, side-chain truncated macrolide (−)-6 (Scheme 1); (+)-6 has been utilised by De Brabander et al. during their total synthesis of apicularen A.5d,6g We also report our findings concerning the base-induced ring opening of the tetrahydropyran ring in an apicularen intermediate. This kind of process may be involved in the biosynthetic conversion of apicularens into other members of the salicylate family (or vice versa).5p,r
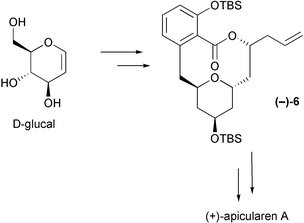 | ||
| Scheme 1 | ||
(i) Formal total synthesis of (+)-apicularen A
The absolute stereochemistry of apicularen A was mis-assigned in the original publication1a and was subsequently revised to the enantiomeric form.1c As we commenced our study before the publication of the structural revision, we concentrated our efforts on the synthesis of the enantiomer of the natural product.The unsaturated enamide side chains are characteristic structural features of the salicylate anti-tumour natural products, and provide a significant synthetic challenge. We decided to construct these side chains via vinyl organometallic addition to unsaturated isocyanates, and in model studies successfully developed this methodology for the preparation of enamide side chains of the apicularen/salicylihalamide (hydrocarbon terminated),5g and lobatamide/oximidine (oxime terminated)5h types.
Retrosynthetic analysis of (+)-apicularen A 1 therefore suggested disconnection of the Z,Z-enamide to give isocyanate 7 and organometallic reagent 8 (Scheme 2). We have previously described the preparation of reagent 8 (M = Cu) by a double acetylene carbocupration process,8 and indeed this methodology has been successfully employed in the total syntheses of salicylihalamide A reported by the groups of Smith6d and Snider.6e Isocyanate 7 should be available by Curtius rearrangement of the acyl azide derived from carboxylic acid 9, and again we carried out model studies5g,h to establish the viability of this approach. Horner–Wadsworth–Emmons alkene formation and functional group interconversion lead back to the allylic intermediate 10, itself obtained by macrolactonisation of hydroxy-acid 11. Alternatively, functionalised intermediates 12 and 13 were also considered viable at this stage.
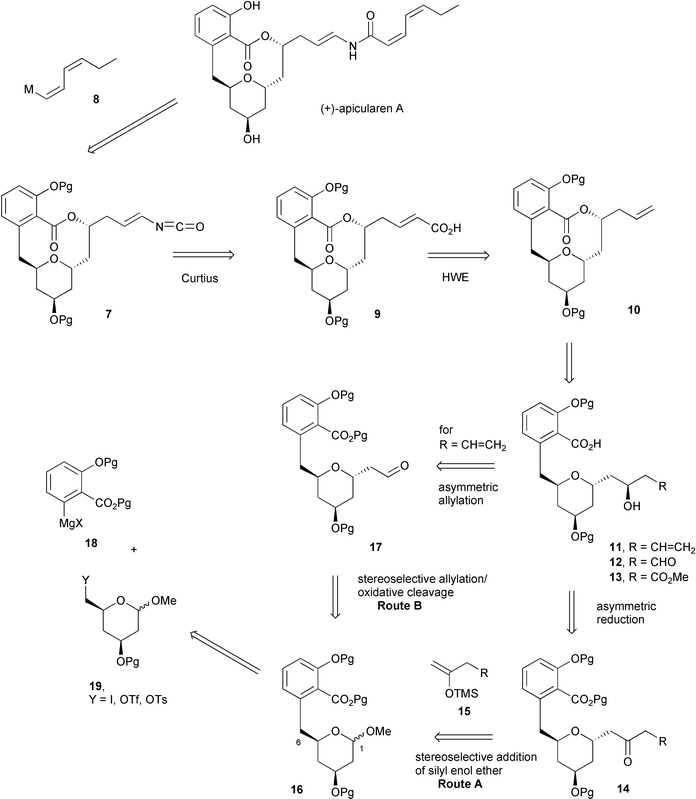 | ||
| Scheme 2 | ||
We envisaged two possible strategies for incorporation of the appendage at C-1 of the tetrahydropyran (Routes A and B, Scheme 2). The chiral alcohol could be produced by β-alkoxy- or reagent-directed asymmetric reduction of a ketone 14. Side-chains containing ketone functionalities have been introduced at C-1 of sugar derivatives with good α-selectivity by reaction of silyl enol ethers 15 with acetals of type 169 and we were keen to establish whether functionalised silyl enol ethers were applicable to this type of reaction (Route A). An alternative methodology involved the use of an allyl group as a masked aldehyde (Route B). In this second route, the asymmetric allylation of aldehyde 17 would be employed to install the secondary alcohol in the side chain with the appropriate stereocontrol. Lewis acid-promoted reactions of acetals of type 16 with allyltrimethylsilane are well-established for the generation of C-glycosides possessing the required α-stereochemistry.10
Construction of the sp2–sp3 bond of 16 at C-6 was considered possible by reaction of a functionalised organometallic of type 18 with a suitable electrophile 19 (Scheme 2). Although Grignard or organolithium reagents incorporating esters are generally considered to be synthetically non-viable, recent work by Knochel11 and Oshima12 and their co-workers indicates that reagents 18 should be accessible. We were aware, however, that β-oxygenated electrophiles such as 19 are known to possess reduced electrophilicity.13 We therefore set out to explore the synthesis of system 16 from precursors such as 18 and 19 as the first part of this study.
Knochel et al. have shown that a range of functionalised aryl iodides can be converted into the corresponding Grignard reagents at low temperature by use of an iodine–magnesium exchange reaction.11 As a model system, we initially examined whether Grignard reagent 21 could be derived from ethyl o-iodobenzoate 20 and then coupled with triflate 22 to produce adduct 23 (Scheme 3).
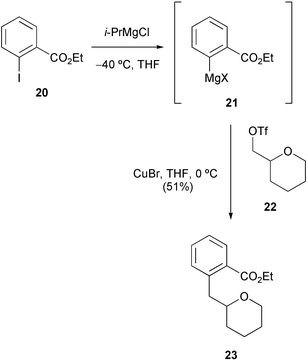 | ||
| Scheme 3 | ||
After generation of the Grignard reagent 21 using Knochel's conditions (iPrMgCl, THF, −40 °C), copper(I)-catalysed coupling with triflate 22 at the same temperature failed to produce adduct 23. However, by allowing the reaction temperature to warm to 0 °C, the required coupling proceeded in reasonable yield (51%) to produce 23. Encouraged by this result, we embarked upon construction of the core tetrahydropyran-ring system 28 required in our synthesis (Scheme 4). Derivatisation of each hydroxy of D-glucal 24 in a one-pot procedure using a three reagent sequence (successively TBSCl, TPSCl then thiocarbonyldiimidazole) gave the protected derivative 25 in 60% overall yield after chromatography. The tri-protected D-glucal 25 then underwent Bu3SnH-mediated deoxygenation via a Barton–McCombie type process14 to give dihydropyran 26. Acid-catalysed addition of methanol to the enol ether moiety,15 together with concomitant deprotection of the primary TBS group, was then achieved using triphenylphosphine hydrobromide in dichloromethane–methanol giving primary alcohol 27 as a mixture of anomers. Reaction with triflic anhydride then gave the unstable triflate derivative 28 which was used immediately in the subsequent coupling reaction.
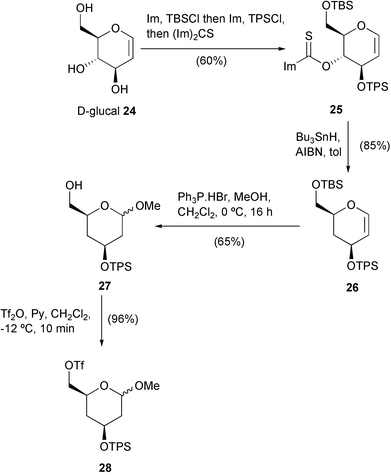 | ||
| Scheme 4 | ||
Meanwhile, the aryl iodide coupling partner 30 was successfully prepared from o-anisic acid 29viao-lithiation and iodination,16 followed by Mitsunobu esterification (Scheme 5). Disappointingly, none of the desired product 32 could be isolated following treatment of iodide 30 with triflate 28 under the previously-devised conditions for Grignard formation and copper-catalysed coupling. Presumably the intermediate Grignard reagent 31 was less stable than the model version 21 and failed to survive the elevated reaction temperatures required for reaction with the relatively unreactive electrophile 28.
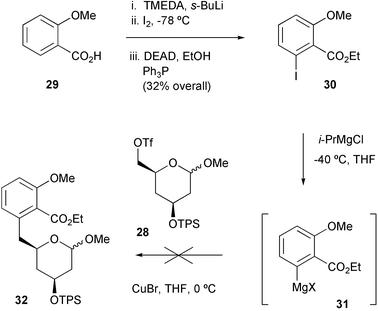 | ||
| Scheme 5 | ||
We therefore looked for a more stable alternative to Grignard reagent 31 and examined Kotsuki's benzofuryl reagent 34, which has been designed as a masked nucleophilic o-hydroxybenzoic acid synthon: the latent functionality can be revealed by oxidative furan cleavage (see later).17 The starting material required for this procedure, 4-bromo-2-methylbenzofuran 33, was prepared using the method published by Kotsuki et al.,17 and after some experimentation with reaction conditions, we discovered that it was possible to couple Grignard reagent 34 to triflate 28 using Cu(I)-catalysis producing adduct 35 in 59% yield (Scheme 6).
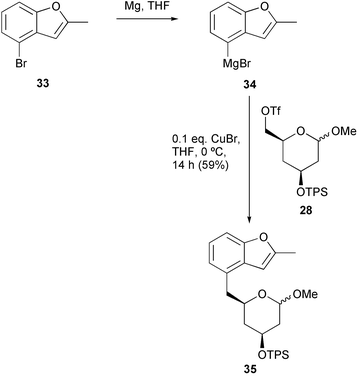 | ||
| Scheme 6 | ||
Having established a procedure for the introduction of the C-6 substituent, we turned our attention towards the introduction of a suitably functionalised side-chain at the C-1 position. Initial studies utilised the model compounds 36 (X = Cl, F) and focused on the incorporation of a 4-carbon fragment by use of silyl enol ether methodology (Scheme 2, Route A). Reactions of simple silyl enol ethers with sugar-derived electrophiles are well-documented,9 but extension of this technique to functionalised nucleophiles has seen little attention to date. We required a 4-carbon oxygenated silyl enol ether for later elaboration. To this end we examined a series of functionalised silyl enol ethers 37a–e prepared by LDA deprotonation of the corresponding ketones followed by enolate trapping using chlorotrimethylsilane.18 Unfortunately, this methodology gave a mixture of regioisomeric enol ethers, despite the use of hindered alcohol protecting groups, and all of the silyl enol ethers proved to be unstable to the coupling conditions. However, the known bis-silyl enol ether 3819 possesses higher stability, and reaction of this with a model deoxyglycosyl chloride 36 (X = Cl), mediated by silver triflate in dichloromethane, successfully generated the C-glycoside 39, in good yield but with the undesired β-stereochemistry predominating (Scheme 7). Efforts to bias the stereoselectivity of the reaction towards the α-anomer by use of boron trifluoride–diethyl ether in acetonitrile20 gave a poor yield of a mixture of anomers (<30%, α ∶ β >5:1) which were difficult to separate and purify.
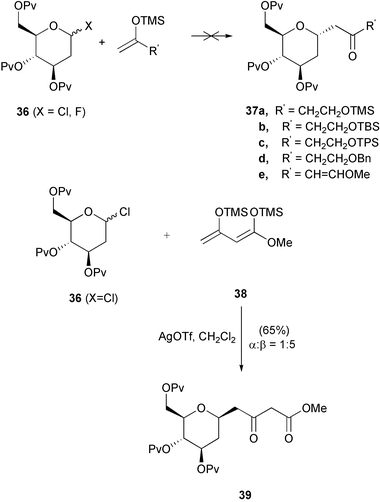 | ||
| Scheme 7 | ||
These disappointing results led us to examine the more stereochemically reliable reactions of allyltrimethylsilane as outlined in Scheme 2, Route B. We decided to retain the benzofuryl group during the anomeric coupling procedure and therefore investigated the allylation of methyl acetal 35 (Scheme 8). Treatment of acetal 35 with allyltrimethylsilane–TMSOTf–MeCN, conditions known10 to promote α-allylation, produced exclusively the required C-allylated adduct 40, after resilylation to restore the protection lost by partial cleavage during the Lewis acid-mediated coupling conditions. Adduct 40 was isolated in 84% yield over these two steps with complete α-stereocontrol (as deduced from anomeric coupling constants).
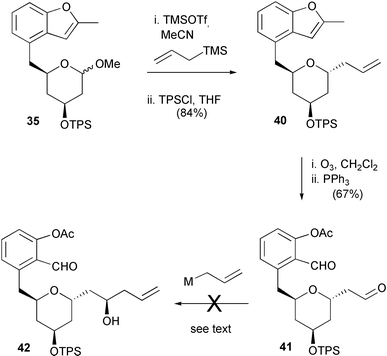 | ||
| Scheme 8 | ||
Subsequent ozonolysis of 40 resulted in the cleavage of both the terminal alkene and the benzofuran giving dialdehyde 41 in 67% yield. Given the expected lower reactivity of the aromatic aldehyde of 41, we had hoped to perform a regio- and stereo-selective allylation reaction at the aliphatic aldehydic centre. However, subjection of compound 41 to several allylation conditions [allylmagnesium bromide, allyltrimethylsilane–dimethylaluminium chloride,21 allyl(diisopinocampheyl)borane22] failed to give any of the desired allyl compound 42.
The inability to discriminate between the two aldehydes in compound 41 encouraged us to carry out these oxidative cleavage processes in a sequential manner (Scheme 9). Thus, intermediate 35, described earlier, was subjected to benzofuranyl ozonolytic cleavage to give mono-aldehyde 43. Subsequent aldehyde oxidation and acetyl saponification followed by concomitant methylation of both of the resultant benzoic acid and phenol groups led us to the required salicylate core 44 (Scheme 9).
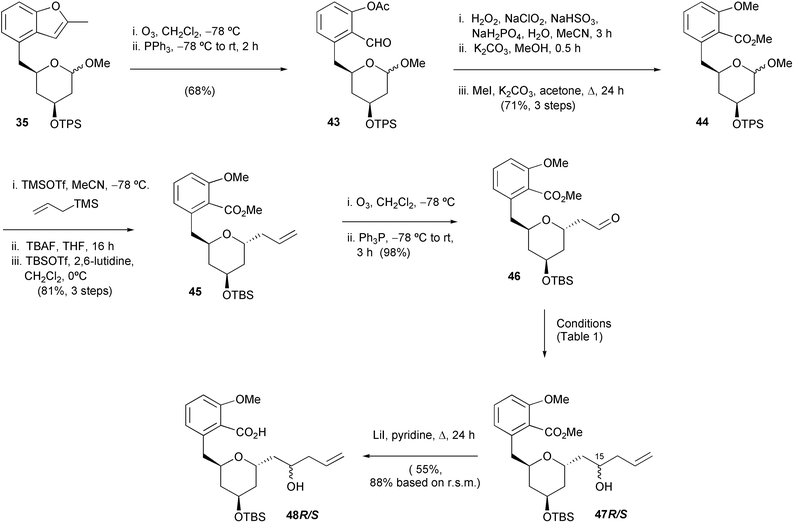 | ||
| Scheme 9 | ||
Reaction of allyltrimethylsilane with acetal 44 using the allyltrimethylsilane–TMSOTf–MeCN conditions described above again gave the desired α-C-allylated product, this time with a significant amount of TPS group cleavage. Unfortunately, the resultant secondary alcohol could not be reprotected using TPSCl, presumably for steric reasons. Therefore, complete removal of the TPS group was carried out using fluoride and then the product was treated with the more reactive silylating agent TBSOTf to give the TBS ether 45 in 81% yield over the three steps (Scheme 9). The next step in this revised synthetic strategy was the oxidative cleavage of the terminal double bond of alkene 45. This was again achieved using ozone, with a reductive work up, giving a quantitative yield of aldehyde 46 (Scheme 9).
We were then ready for a second attempt at stereoselective allylation (cf.Scheme 8), this time without the problem of regioselectivity. Initial attempts (Scheme 9, and Table 1) with aldehyde 46, utilised allyltrimethylsilane in the presence of chelating Lewis acids, with a view to obtaining the 15S-diastereomer of product 47 for subsequent Mitsunobu lactonisation.23 The best result (entry i) was obtained using dimethylaluminium chloride giving the 1,3-anti and 1,3-syn addition products 47S and 47R in a ratio of 85 ∶ 15.21 The structure of the major isomer was assigned with the help of Mosher ester studies.24 Interestingly (entry ii), the use of titanium(IV) chloride in this reaction lead to no diastereoselective bias at the C-15 centre, which is unusual, although a similar result was reported by De Brabander during his total synthesis of apicularen A.5d
| Conditions | Yield 47(%) | Ratio of 15S ∶ 15R |
|---|---|---|
| (i) Allyltrimethylsilane, −78 °C, Me2AlCl, 30 min | 85 | 85 ∶ 15 |
| (ii) TiCl4, −78 °C, allyltrimethylsilane, 1 h | 35 | 50 ∶ 50 |
| (iii) (−)-B-Allyl-β-(diisopinocampheyl)borane, Et2O, −78 °C, 3 h | 77 | 10 ∶ 90 |
The use of reagent control was also successful (entry iii), Brown's allyl(diisopinocampheyl)borane22 reacting with aldehyde 46 to give a 90 ∶ 10 predominance of the required 1,3-syn addition product 47R in good yield.
Cleavage of the methyl ester in 47 was then attempted using hydrolytic conditions [e.g. LiOH, Ba(OH)2] but without success. However, we were pleased to find that lithium iodide-induced O-alkyl ester cleavage allowed access to the macrocyclisation precursors 48R and 48S (90 ∶ 10) in good yield.
Several different lactonisation methods were assessed in order to close the macrocyclic ring-system. Whilst the procedures of Mitsunobu23 and Yamaguchi and co-workers25 gave none of the desired lactone, the DCC–DMAP procedure developed by Boden and Keck26 successfully afforded the lactone 49, together with a small amount of the C-15 epimer 50, which was easily separated by chromatography (Scheme 10).
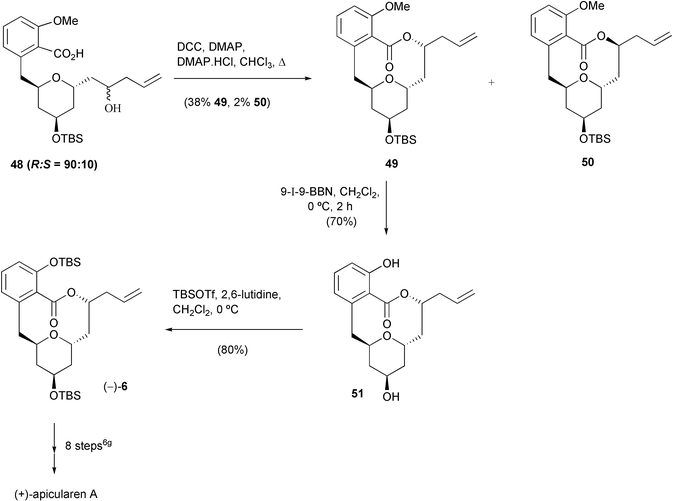 | ||
| Scheme 10 | ||
Finally, silyl and methyl group deprotection of 49 with iodo-9-BBN27 gave the diol 51, which was fully characterised and found to have comparable spectroscopic/analytical data to those reported by De Brabander et al. for the enantiomeric compound.5d,6g For example, the 13C NMR data for 51 are compared to published data5d in Table 2.
| a Corrected for calibration at acetone, 29.84 (rather than 30.385d). b The peak at δ 125.5 was not reported in the original paper5d where an erroneous peak at δ 160.7 was included. In a personal communication Professor De Brabander confirmed the revised data shown above. |
|---|
| δ C (125 MHz, acetone-d6): 169.28 (169.3), 154.31 (154.3), 140.23 (139.6), 135.32 (135.4), 130.38 (130.3), 125.47 (125.5),b 122.34 (122.4), 117.48 (117.5), 114.43 (114.4), 73.65 (73.7), 73.61 (73.6), 68.08 (68.1), 64.91 (64.9), 40.36 (40.4), 40.10 (40.1), 39.96 (40.0), 39.72 (39.8), 39.13 (39.1). |
Silylation of both the phenolic and secondary hydroxy groups then gave the corresponding bis-TBS ether (−)-6. The enantiomeric compound, (+)-6, has been converted into (−)-apicularen A by De Brabander et al.5d,6g The chemistry described herein, by preparation of the advanced apicularen intermediate (−)-6, thus constitutes a formal total synthesis of (+)-apicularen A.
(ii) Tetrahydropyran-ring cleavage: conversion of the apicularen nucleus into salicylihalamide-type products
As a brief extension to our apicularen synthetic studies, we decided to explore the ring-opening of the tetrahydropyran ring, possibly leading to salicylihalamide-type products, and then, if successful, the re-closure of the tetrahydroyran ring system returning to the apicularen nucleus. We felt that such studies would be valuable in terms of evaluating the possibility of devising a unified approach to both types of natural products, and could provide information concerning possible biosynthetic pathways.We decided to attempt this interconversion via tetrahydropyranone 52, which we aimed to subject to ring-opening conditions (Scheme 11). The regioselectivity in this proposed enolate β-elimination process would obviously be of interest, but we were hoping to observe at least partial conversion into enone 53 which possesses a salicylihalamide-like nucleus. In turn, we hoped to investigate the ring closure of hydroxy enone 53 to return to the apicularen nucleus 52 in what appears to be a likely biosynthetic sequence.5p,r
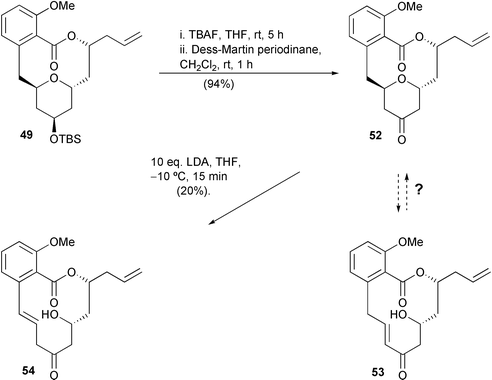 | ||
| Scheme 11 | ||
The key pyranone 52 was readily prepared from silyl ether 49 in 94% overall yield by fluoride-mediated deprotection followed by Dess–Martin oxidation. Initial attempts to ring-open the tetrahydropyran moiety of 52 involved the use of weak acids and bases but were not successful. However, when pyranone 52 was treated with an excess of LDA a hydroxy-alkene was formed in 20% yield. To our initial surprise, the product was not enone 53, nor the regioisomeric enone, but the styrene 54. The identity of compound 54 was confirmed unambiguously by X-ray crystallography.28 It is possible that pyranone 52 is first converted into enone 53 which subsequently undergoes isomerisation to give styrene 54: however, it seems more likely that styrene 54 is generated directly from 52 by benzylic deprotonation. We next attempted to recyclise 54 by treatment with base, but unfortunately without success. Lack of material precluded further studies such as the base-mediated ring-opening of 49 and the acid-catalysed ring-closure of 54. Although further research is obviously required, these preliminary experiments demonstrate the first conversion of the apicularen nucleus into a salicylihalamide-type product thus suggesting an alternative synthetic approach to the salicylihalamides, and may be instructive in terms of biosynthetic studies.
Conclusions
Compound (+)-6 has been converted into (−)-apicularen A in 8 steps (4% overall yield) by a published procedure.6a,5a The synthetic studies described herein therefore constitute a formal total synthesis of (+)-apicularen A. Tetrahydropyran ring opening has been achieved using pyranone 52, providing interesting mechanistic and biosynthetic insights.Experimental
General
All reactions were carried out under a nitrogen atmosphere. THF and diethyl ether (Et2O) were dried over Na–benzophenone ketyl; CH2Cl2 was dried over CaH2 and distilled immediately before use. Where necessary, acetonitrile and toluene were purchased in anhydrous form. Column chromatography was carried out using 70–200 or 33–63 micron (60 Å) silica gel. NMR spectra were recorded using JEOL GX-270 or Bruker AMX-500 instruments in the solvents indicated; chemical shifts (δ) in ppm are given relative to Me4Si, coupling constants (J) in Hz. Infra-red spectra were recorded on an API Mattson Genesis FT-IR. Melting points were obtained using an Electrothermal IA9100 digital apparatus (uncorrected). Optical rotations: JASCO DIP-370 digital polarimeter at 20 °C (Na-D line, 589 nm). Mass spectra were obtained on a Fisons analytical (VG) autospec instrument. CuBr was purified by precipitation from conc. HBr.29 Tf2O was re-distilled from P2O5 before use. All other commercially available compounds were used as received.Ethyl 2-(tetrahydro-2H-pyran-2-ylmethyl)benzoate (23)
To a stirred solution of ester 20 (750 mg, 2.66 mmol) in THF (5 mL) at −40 °C was added isopropylmagnesium chloride (1.5 mL, 3.0 mmol, 2 M in diethyl ether) dropwise. The mixture was stirred at this temperature for 40 min then copper(I) bromide (85 mg, 0.59 mmol) was added followed by a solution of triflate 2213 (400 mg, 1.61 mmol) in THF (3 mL). The mixture was warmed to 0 °C over 30 min, stirred at this temperature for 5 h then quenched with sat. NH4Cl(aq) and extracted with diethyl ether. The organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure to give an oil which was purified by flash chromatography (petroleum ether ∶ EtOAc, 8 ∶ 1) to give the title compound23 (202 mg, 51%) as a colourless oil; Rf 0.50 (petroleum ether ∶ EtOAc, 7 ∶ 1); νmax/cm−1 (thin film) 2980, 2636, 2848, 1717, 1601, 1447, 1365, 1256, 1088, 1047; δH (270 MHz, CDCl3): 7.89 (dd, 1 H, J = 8.0, 1.5 Hz), 7.42 (ddd, 1 H, J = 8.0, 8.0, 1.5 Hz), 7.32–7.24 (m, 2 H), 4.37 (q, 2 H, J = 7.0 Hz), 3.98–3.91 (m, 1 H), 3.58–3.48 (m, 1 H), 3.35 (ddd, 1 H, J 11.5, 11.5, 2.5 Hz), 3.17 (dd, 1 H, J = 13.0, 5.0 Hz), 3.09 (dd, 1 H, J = 13.0, 7.5 Hz), 1.85–1.79 (m, 1 H), 1.65–1.27 (m, 5 H), 1.41 (t, 3 H, J = 7.0 Hz); δC (67.9 MHz, CDCl3): 168.3, 140.8, 132.8, 131.9, 130.9, 130.8, 126.6, 79.0, 68.9, 61.2, 41.7, 32.2, 26.5, 24.0, 14.7. Found: C, 72.54; H, 8.37%. C15H20O3 requires C, 72.55; H, 8.12%. Found (CI): 249.1489 [MH]+, C15H20O3 requires 249.1491 (0.7 ppm error); m/z (CI) 249 (100%, [MH]+), 203 (15).1,5-Anhydro-6-O-[tert-butyl(dimethyl)silyl]-3-O-[tert-butyl(diphenyl)silyl]-2-deoxy-4-O-(1H-imidazol-1-ylthiocarbonyloxy)-D-arabino-hex-1-enitol (25)
To a stirred solution of D-glucal 24 (8.98 g, 61.4 mmol) in THF (180 mL) at rt was added imidazole (4.60 g, 68.0 mmol) followed by the slow addition of a solution of tert-butyldimethylsilyl choride (9.53 g, 61.4 mmol) in THF (10 mL). After 4.5 h another portion of imidazole (4.60 g, 68.0 mmol) and a solution of tert-butyldiphenylsilyl chloride (15.4 mL, 16.5 g, 58.9 mmol) in THF (10 mL) was added. The reaction mixture was stirred for 24 h then filtered and thiocarbonyldiimidazole (14.1 g, 71.8 mmol) added to the filtrate. After 20 h at rt the solvent was evaporated under reduced pressure and the residue purified by column chromatography (petroleum ether ∶ EtOAc, 9 ∶ 1) to give the title compound25 (22.5 g, 60%) as a pale yellow gum; Rf 0.56 (petroleum ether ∶ EtOAc, 4 ∶ 1); [α]D +23.7 (c 2.50, CHCl3); νmax/cm−1 (thin film) 2953, 2932, 2858, 1646, 1391, 1247, 1112; δH (270 MHz, CDCl3): 8.20 (1 H, s), 7.72–7.66 (4 H, m), 7.51–7.32 (7 H, m), 7.00–6.98 (1 H, m), 6.40 (1 H, d, J = 6.5 Hz), 5.94 (1 H, td, J = 4.0, 1.0 Hz), 4.63 (1 H, ddd, J = 6.5, 4.5, 1.0 Hz), 4.41–4.34 (1 H, m), 4.24–4.21 (1 H, m), 4.14 (1 H, dd, J = 7.0, 11.5 Hz), 4.03 (1 H, dd, J = 4.5, 11.5 Hz), 1.08 (9 H, s), 0.93 (9 H, s), 0.11 (3 H, s), 0.09 (3 H, s); δC (67.9 MHz, CDCl3): 182.4, 143.5, 136.8, 135.8, 133.3, 132.5, 130.7, 130.0, 129.9, 127.8, 127.7, 117.9, 101.1, 78.5, 76.3, 63.4, 61.5, 26.8, 25.9, 19.1, 18.3, −5.3, −5.4. Found (CI): 609.2631 [MH]+, C32H45N2O4SSi2 requires 609.2639 (1.3 ppm error); m/z (CI) 609 (25%, [MH]+), 69 (100).2,6-Anhydro-1-O-[tert-butyl(dimethyl)silyl]-4-O-[tert-butyl(diphenyl)silyl]-3,5-dideoxy-D-threo-hex-5-enitol (26)
A stirred solution of D-glucal derivative 25 (1.40 g, 2.23 mmol) in toluene (40 mL) was degassed by the successive evacuation of the reaction flask (water pump), then flushing with Ar (3 times). The mixture was heated to reflux then a solution of tributyltin hydride (1.26 mL, 1.36 g, 4.68 mmol) and AIBN (95 mg) in toluene (10 mL) was added dropwise over 30 min. Heating was continued for a further 30 min then the reaction was cooled to rt and the solvent evaporated under reduced pressure to give an orange oil. This was purified by column chromatography (petroleum ether to petroleum ether ∶ EtOAc, 9 ∶ 1) to give the title compound26 (920 mg, 85%, containing a small amount of tributyltin-derived impurity) as a pale yellow gum; Rf 0.83 (petroleum ether ∶ EtOAc, 4 ∶ 1); νmax/cm−1 (thin film) 2957, 2931, 2856, 1644, 1471, 1427, 1251, 1110, 840, 701; δH (270 MHz, CDCl3): 7.70–7.62 (4 H, m), 7.47–7.34 (6 H, m), 6.28 (1 H, dd, J = 6.5, 1.0 Hz), 4.67 (1 H, ddd, J = 6.5, 2.5, 1.5 Hz), 4.44–4.38 (1 H, m), 3.92–3.80 (2 H, m), 3.64–3.59 (1 H, m), 1.98–1.90 (1 H, m), 1.81–1.75 (1 H, m), 1.07 (9 H, s), 0.89 (9 H, s), 0.05 (6 H, s); δC (67.9 MHz, CDCl3): 144.3, 136.1, 136.1, 134.5, 134.4, 130.0, 127.9, 105.9, 75.5, 65.6, 63.9, 34.4, 27.3, 26.3, 19.4, 18.7, −4.3, −5.0. Found (CI): 500.3016 [MNH4]+, C28H42O3Si2 requires 500.3016 (0.1 ppm error); m/z (CI) 500 (<1%, [MNH4]+), 483 (<1), 425 (1), 244 (6), 227 (100), 196 (6).Methyl 3-O-[tert-butyl(diphenyl)silyl]-2,4-dideoxy-D-threo-hexopyranoside (27)
To an ice-cooled, stirred solution of compound 26 (3.04 g, 6.30 mmol) and methanol (300 mg, 9.4 mmol) in dichloromethane (25 mL) was added triphenylphosphine hydrobromide (110 mg, 0.32 mmol). After 3 h at 0 °C, methanol (0.9 mL) and triphenylphosphine hydrobromide (290 mg) were added and the mixture was allowed to warm to rt then stirred for 2 h. Sat. NaHCO3(aq) (15 mL) was then added and the mixture extracted with dichloromethane. The organic layers were dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The crude product was purified by column chromatography (petroleum ether ∶ EtOAc, 4 ∶ 1) to give the title compound27 (1.44 mg, 65%, mixture of anomers) as a colourless gum; Rf 0.17 (petroleum ether ∶ EtOAc, 4 ∶ 1); νmax/cm−1 (thin film) 3466, 2931, 1428, 1112, 1047, 703; δH (270 MHz, CDCl3): 7.69–7.63 (4 H, m), 7.45–7.33 (6 H, m), 4.78 (1 H, d, J = 3.5 Hz), 4.23–4.08 (1 H, m), 3.64–3.42 (3 H, m) 3.20 (3 H, s), 2.02–1.95 (1 H, m), 1.89 (1 H, dd, J = 7.5, 5.5 Hz), 1.67–1.57 (2 H, m), 1.49–1.36 (1 H, dt, J = 11.5. 11.5 Hz), 1.04 (9 H, s); δC (67.9 MHz, CDCl3): 136.0, 134.6, 134.5, 129.9, 127.9, 127.9, 99.5, 68.6, 65.9, 65.4, 54.2, 39.7, 37.0, 27.2, 19.4. Found (CI): 401.2157 [MH]+, C23H32O4Si requires 401.2148 (2.1 ppm error); m/z (CI) 401 (1%, [MH]+), 386 (3), 291 (59), 207 (67), 187 (100), 133 (67).Ethyl 2-iodo-6-methoxybenzoate (30)
(a) To a stirred solution of TMEDA (3.40 mL, 22.5 mmol) in THF (50 mL) was added sec-butyllithium (1.4 M in cyclohexane, 16.0 mL, 22.4 mmol) at ca. −60 °C under N2. The resultant yellow solution was cooled to ca. −100 °C (EtOH/liq. N2 cooling bath) and a solution of 2-methoxybenzoic acid 29 (1.45 g, 9.53 mmol) in THF (10 mL) was added dropwise over 30 min. The orange–yellow mixture was stirred for a further 30 min at this temperature then warmed to −78 °C over 30 min. A solution of iodine (9.00 g, 35.4 mmol) in THF (10 mL) was then added until the brown colour persisted and the mixture was warmed to −60 °C then stirred at this temperature for a further 1 h. Sat. NH4Cl(aq) (5 mL) was then added slowly followed by sat. Na2SO3(aq) until the brown colour had dissipated. The solution was then extracted with diethyl ether (100 mL) and the organic extract washed with 1 M NaOH (10 mL). The aqueous layers were then combined and acidified (10% HCl) and extracted with dichloromethane (4 × 100 mL), these organic extracts were dried (MgSO4), filtered and the solvent evaporated under reduced pressure to give the crude product as a yellow oil containing unreacted 2-methoxybenzoic acid. This mixture was re-dissolved in methanol (15 mL) and conc. H2SO4 (0.75 mL), then stirred at reflux for 4 h. After cooling to rt, water (50 mL), then diethyl ether (50 mL) were added and the mixture was made alkaline with 1 M NaOH. The organic layer was separated and the aqueous phase further extracted with diethyl ether (50 mL). The aqueous layer was then acidified (10% HCl) and extracted with dichloromethane (3 × 50 mL). These organic extracts were dried (MgSO4), filtered and the solvent evaporated under reduced pressure to give 2-iodo-6-methoxybenzoic acid (1.01 g, 39%) as colourless crystals; Rf 0.22 (EtOAc); mp 128–130 °C (lit.30 mp 131.5–132 °C).(b) To a stirred solution of the acid from (a) (526 mg, 1.91 mmol), triphenylphosphine (650 mg, 2.48 mmol) and ethanol (0.17 mL, 2.90 mmol) in THF (15 mL) at 0 °C was added dropwise diethyl azodicarboxylate (0.39 mL, 2.48 mmol). After 4 h, the solvent was evaporated and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 30 ∶ 1) to give the title compound30 (485 mg, 83%) as a colourless oil; Rf 0.84 (petroleum ether ∶ EtOAc, 1 ∶ 1); νmax/cm−1 (thin film) 2979, 2940, 1731, 1584, 1567, 1461, 1430, 1264, 1104; δH (270 MHz, CDCl3): 7.37 (dd, 1 H, J = 8.0, 0.5 Hz), 7.03 (t, 1 H, J = 8.0 Hz), 6.87 (d, 1 H, J = 8.0 Hz), 4.41 (q, 2 H, J = 7.0 Hz), 3.79 (s, 3 H), 1.40 (t, 3 H, J = 7.0 Hz); δC (67.9 MHz, CDCl3): 167.7, 157.0, 131.7, 131.1, 130.6, 111.0, 92.7, 62.2, 56.7, 14.4. Found: C, 39.63; H, 3.55%. C10H11IO3 requires C, 39.24; H, 3.62%). Found (EI): 305.9756 (M+), C10H11IO3 requires 305.9753 (1.0 ppm error); m/z (EI) 306 (40%, M+), 261 (100), 218 (13), 203 (10), 151 (5).
{(4S,6R)-2-Methoxy-6-[(2-methyl-1-benzofuran-4-yl)methyl]tetrahydro-2H-pyran-4-yloxy}(tert-butyl)diphenylsilane (35)
(a) Formation of Grignard reagent 34: to a suspension of magnesium turnings (180 mg, 7.40 mmol) in THF (2 mL) was added catalytic iodine and the mixture stirred at rt, under N2 until the iodine colour had been consumed. A solution of 4-bromo-2-methylbenzofuran 3317 (1.04 g, 4.91 mmol) in THF (5 mL) was then added over 6 h via syringe pump and the mixture stirred for a further 1 h giving the Grignard reagent 34 as a grey–green cloudy solution which was used immediately.(b) Synthesis of triflate 28: to a stirred solution of alcohol 27 (1.12 g, 2.80 mmol) and pyridine (0.68 mL, 660 mg, 8.4 mmol) in dichloromethane (10 mL) at −12 °C (ice–salt) under N2 was added triflic anhydride (0.60 mL, 1.0 g, 3.6 mmol) in dichloromethane (2 mL) over 2 min. After a further 15 min at this temperature the mixture was diluted with dichloromethane (5 mL) and washed with sat. NaHCO3(aq) (5 mL). The aqueous layer was then extracted with dichloromethane (2 × 5 mL). The organic layers were washed with brine (10 mL), dried (MgSO4), and filtered through a short plug of silica, eluting with dichloromethane (15 mL). The solvent was evaporated under reduced pressure (below 20 °C), azeotroping the remaining pyridine with toluene (1 mL), at 0.1 mmHg to give triflate 28 (1.07 g, 96%, mixture of anomers) as a colourless oil. (This material darkened in colour upon standing and was dried for no more than 30 min at rt, then used immediately.) Rf 0.37 (dichloromethane ∶ petroleum ether, 1 ∶ 1); δH (270 MHz, C6D6): 7.75–7.69 (4 H, m), 7.26–7.19 (6 H, m), 4.39 (1 H, d, J = 3.0 Hz), 4.28–4.15 (1 H, m), 3.81 (1 H, dd, J = 7.5, 10.5 Hz), 3.55 (1 H, dd, J = 2.5, 10.5 Hz), 3.23 (1 H, ddt, J = 12.0, 7.5, 2.5 Hz), 1.91 (1 H, ddt, J = 13.0, 5.0, 1.5 Hz), 1.44 (1 H, ddd, J = 13.0, 10.5, 3.0 Hz), 1.34–1.27 (1 H, m), 1.16 (10 H, m).
(c) The Grignard reagent 34, freshly prepared, was cooled to 0 °C, then copper(I) bromide (140 mg, 1.00 mmol) was added and the mixture stirred for 2 min under N2. Triflate 28 was then dissolved in THF (10 mL) and added to the Grignard reagent dropwise over 5 min. The resultant mixture was stirred overnight at 0 °C then quenched with sat. NH4Cl(aq) ∶ NH4OH(aq) (10 ∶ 1, 2 mL) and the mixture filtered through Celite and the filtrate extracted with EtOAc. The organic extract was then dried (MgSO4), filtered and the solvent evaporated under reduced pressure. Flash chromatography (petroleum ether ∶ dichloromethane, 2 ∶ 1) gave the title compound35 (852 mg, 59% from alcohol 27, mixture of anomers) as a pale brown oil; Rf 0.26 (dichloromethane ∶ petroleum ether, 1 ∶ 1); νmax/cm−1 (thin film) 2932, 1428, 1113, 1070, 1048, 702; δH (270 MHz, CDCl3): 7.63–7.57 (4 H, m), 7.41–7.23 (7 H, m), 7.08 (1 H, t, J = 8.0 Hz), 6.91 (1 H, dd, J = 8.0, 0.5 Hz), 6.30 (1 H, t, J = 1.0 Hz), 4.72 (1 H, d, J = 2.5 Hz), 4.12–4.01 (1 H, m), 3.69–3.79 (1 H, m), 2.99 (3 H, s), 2.97 (1 H, dd, J = 13.5, 6.5 Hz), 2.76 (1 H, dd, J = 13.5, 6.5 Hz), 2.44 (3 H, d, J = 1.0 Hz), 1.95 (1 H, ddt, J = 13.0, 4.5, 1.5 Hz), 1.63–1.71 (2 H, m), 1.35 (1 H, dt, J = 11.5, 11.5 Hz), 1.02 (9 H, s); δC (67.9 MHz, CDCl3): 155.0, 154.8, 136.1, 134.5, 129.9, 129.8, 128.0, 127.9, 127.7, 155.0, 154.8, 130.7, 129.2, 123.4, 123.1, 108.9, 101.7, 99.5, 68.3, 66.0, 54.5, 41.3, 39.8, 39.7, 27.2, 19.3, 14.4. Found (CI): 532.2878 [MNH4]+, C32H38O4Si requires 532.2883 (1.0 ppm error); m/z (CI) 532 (3%, [MNH4]+), 500 (2), 483 (9), 457 (3), 405 (4), 274 (17), 227 (100), 216 (30), 196 (42), 183 (17).
{(2S,4R,6R)-2-Allyl-6-[(2-methyl-1-benzofuran-4-yl)methyl]tetrahydro-2H-pyran-4-yloxy}(tert-butyl)diphenylsilane (40)
To a stirred solution of acetal 35 (420 mg, 0.760 mmol) and allyltrimethylsilane (0.43 mL, 2.7 mmol) in acetonitrile (4 mL) at −35 °C was added trimethylsilyl triflate (60 µL, 0.34 mmol) dropwise. The reaction mixture was warmed to ca. −25 °C, stirred for 40 min then sat. NaHCO3 (6 mL) was added and the mixture extracted with EtOAc. The organic extracts were washed with brine, dried (MgSO4), filtered and the solvent removed under reduced pressure to give an oil which was re-dissolved in DMF (3 mL).The resultant solution was cooled to 0 °C then imidazole (50 mg, 0.73 mmol) and tert-butyldiphenylsilyl chloride (200 mg, 0.73 mmol) were added. The mixture was allowed to warm to rt and stirred for 15 h. The solvent was then evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 30 ∶ 1) to give the title compound40 (337 mg, 84%) as a colourless gum; Rf 0.52 (petroleum ether ∶ EtOAc, 9 ∶ 1); [α]D +59.3 (c 0.750, CHCl3); νmax/cm−1 (thin film) 3071, 2932, 2857, 1589, 1428, 1251, 1111, 1073; δH (270 MHz, CDCl3): 7.67–7.60 (m, 4 H), 7.44–7.26 (m, 7 H), 7.11 (t, 1 H, J = 8.0 Hz), 6.96 (d, 1 H, J = 8.0 Hz), 6.35 (s, 1 H), 5.60–5.47 (m, 1 H), 4.92 (dd, 1 H, J = 10.0, 2.0 Hz), 4.85 (dd, 1 H, J = 17.0, 1.5 Hz), 4.10–3.90 (m, 2 H), 3.81–3.70 (m, 1 H), 3.18 (dd, 1 H, J = 13.5, 6.5 Hz), 2.86 (dd, 1 H, J = 13.5, 7.0 Hz), 2.46 (s, 3 H), 2.25–2.14 (m, 1 H), 2.20 (ddd, 1 H, J = 14.0, 7.0, 7.0 Hz), 1.99–1.88 (ddd, 1 H, J = 14.0, 7.0, 7.0 Hz), 1.79–1.72 (m, 1 H), 1.70–1.58 (m, 2 H), 1.51–1.40 (m, 1 H), 1.07 (s, 9 H); δC (67.9 MHz, CDCl3): 154.9, 154.7, 136.1, 135.3, 134.5, 131.1, 130.0, 129.2, 127.9, 123.4, 123.2, 116.9, 109.0, 101.7, 71.7, 70.3, 66.4, 40.4, 40.0, 37.6, 37.1, 27.3, 19.4, 14.5. Found: C, 77.92; H, 7.37%. C34H40O3Si requires C, 77.82; H, 7.68%. Found (CI): 542.3097 [MNH4]+, C34H40O3Si requires 542.3090 (1.2 ppm error); m/z (CI) 542 (12%, [MNH4]+), 525 (2, [MH]+), 467 (15), 447 (18), 379 (15), 269 (50), 251 (30), 227 (20), 199 (100), 185 (24), 145 (30).
3-{(2R,4S,6R)-4-[tert-Butyl(diphenyl)silyloxy]-6-(2-oxoethyl)tetrahydro-2H-pyran-2-ylmethyl}-2-formylphenyl acetate (41)
Ozone was bubbled through a stirred solution of benzofuran 40 (195 mg, 0.372 mmol) in dichloromethane (6.5 mL) at −78 °C for ca. 20 min, until a pale blue colour appeared. Oxygen was then bubbled through the solution for a further 5 min and triphenylphosphine (420 mg, 1.60 mmol) was added. The reaction mixture was allowed to warm to 4 °C then stirred for a further 15 h. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 3 ∶ 1) to give the title compound41 (140 mg, 67%) as a colourless gum; Rf 0.12 (petroleum ether ∶ EtOAc, 4 ∶ 1); [α]D +54.6 (c 1.00, CHCl3); νmax/cm−1 (thin film) 2930, 2857, 1768, 1723, 1695, 1603, 1468, 1427, 1368, 1188, 1110, 1059; δH (270 MHz, CDCl3): 10.31 (s, 1 H), 9.38 (t, 1 H, J = 3.0 Hz), 7.69–7.60 (m, 4 H), 7.51–7.35 (m, 7 H), 7.13 (d, 1 H, J = 7.5 Hz), 7.03 (d, 1 H, J = 7.5 Hz), 4.63–4.53 (m, 1 H), 4.02–3.92 (m, 1 H), 3.73–3.65 (m, 1 H), 3.33 (dd, 1 H, J = 14.0, 8.5 Hz), 3.17 (dd, 1 H, J = 14.0, 4.0 Hz), 2.41 (ddd, 1 H, J = 16.0, 9.0, 3.0 Hz), 2.37 (s, 3 H), 2.06 (ddd, 1 H, J = 16.0, 5.5, 3.0 Hz), 1.95–1.87 (m, 1 H), 1.74–1.50 (m, 2 H), 1.50–1.41 (m, 1 H), 1.09 (s, 9 H); δC (67.9 MHz, CDCl3): 200.9, 190.3, 169.7, 152.9, 143.2, 136.0, 136.0, 134.2, 134.1, 130.2, 130.1, 128.0, 126.6, 122.0, 71.2, 66.3, 66.0, 46.5, 39.6, 38.7, 38.2, 27.2, 19.3. Found (CI): 576.2772 [MNH4]+, C33H38O6Si requires 576.2784 (1.6 ppm error); m/z (CI) 576 (80%, [MNH4]+), 559 (25, [MH]+), 517 (65), 303 (37), 274 (35), 261 (30), 216 (37), 196 (100).3-{(2R,4S)-4-[tert-Butyl(diphenyl)silyloxy]-6-methoxytetrahydro-2H-pyran-2-ylmethyl}-2-formylphenyl acetate (43)
Oxygen was bubbled through a stirred solution of benzofuran 35 (403 mg, 0.783 mmol) in dichloromethane (15 mL) for 10 min, with cooling to −78 °C. Ozone in oxygen was then bubbled through the solution for ca. 30 min until a pale blue colour appeared. Oxygen was then bubbled through the solution for a further 10 min after which time triphenylphosphine (637 mg, 2.43 mmol) was added. The cooling bath was then removed and the reaction mixture allowed to warm to rt over 3 h. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 9 ∶ 1) to give the title compound43 (280 mg, 68%, mixture of anomers) as a yellowish gum; Rf 0.18 (petroleum ether ∶ EtOAc, 9 ∶ 1); νmax/cm−1 (thin film) 2933, 1773, 1697, 1197, 1112; δH (270 MHz, CDCl3): 10.34 (1 H, s), 7.75–7.65 (4 H, m), 7.51–7.34 (7 H, m), 7.14 (1 H, d, J = 7.0 Hz), 7.03 (1 H, dd, J = 8.0, 1.0 Hz), 4.68 (1 H, d, J = 3.0 Hz), 4.20–4.08 (1 H, m), 3.68–3.55 (1 H, m), 3.18 (1 H, dd, J = 14.0, 8.0 Hz), 3.01 (1 H, dd, J = 14.0, 4.5 Hz), 2.87 (3 H, s), 2.38 (3 H, s), 2.00–1.94 (1 H, m), 1.89–1.82 (1 H, m), 1.64 (1 H, ddd, J = 13.0, 11.0, 3.5 Hz), 1.45 (1 H, dd, J =11.5, 11.5 Hz), 1.09 (9 H, s); δC (67.9 MHz, CDCl3): 190.0, 169.5, 151.9, 143.0, 135.8, 134.5, 134.4, 133.8, 129.7, 127.7, 126.8, 121.9, 99.0, 68.3, 65.6, 54.2, 41.1, 39.4, 38.6, 27.0, 21.0, 19.2. Found (CI): 564.2790 [MNH4]+, C32H38O6Si requires 564.2781 (1.5 ppm error); m/z (CI) 564 (12%, [MNH4]+), 529 (6), 515 (100), 473 (10), 259 (25), 196 (8).Methyl 2-{(2R,4S)-4-[tert-butyl(diphenyl)silyloxy]-6-methoxytetrahydro-2H-pyran-2-ylmethyl}-6-methoxybenzoate (44)
(a) To a solution of sodium chlorite (83 mg, 0.91 mmol), sodium dihydrogen orthophosphate (176 mg, 1.47 mmol) and hydrogen peroxide (30% v/v in water, 75 mg) in water (2 mL) at 0 °C was added sodium hydrogen sulfite (48 mg, 0.40 mmol) portion-wise. This mixture was slowly added to a stirred solution of aldehyde 43 (230 mg, 0.409 mmol) in acetonitrile (5 mL) at rt. After 1.5 h a mixture of sodium chlorite (40 mg), sodium dihydrogen orthophosphate (85 mg), hydrogen peroxide (30% v/v in water, 35 mg) and sodium hydrogen sulfite (25 mg) was again prepared and added to the aldehyde mixture. After a further 2 h, sat. sodium sulfite (2 mL) was added cautiously and the reaction mixture extracted with EtOAc (4 × 20 mL). The organic extracts were dried (MgSO4), filtered and the solvent evaporated under reduced pressure to give 2-(acetyloxy)-6-{(2R,4S)-4-[tert-butyl(diphenyl)silyloxy]-6-methoxytetrahydro-2H-pyran-2-ylmethyl}benzoic acid (222 mg, 94%, mixture of anomers) as a colourless oil; Rf 0.19 (petroleum ether ∶ EtOAc, 2 ∶ 1); νmax/cm−1 (thin film) 3070, 2930, 2858, 1771, 1730, 1702, 1472, 1427, 1371, 1198, 1112; δH (270 MHz, CDCl3): 8.50 (1 H, br s), 7.70–7.62 (4 H, m), 7.48–7.33 (7 H, m), 7.05 (1 H, d, J = 8.0 Hz), 7.00 (1 H, dd, J = 8.0, 1.0 Hz), 4.71 (1 H, d, J = 3.5 Hz), 4.20–4.07 (1 H, m), 3.80–3.68 (1 H, m), 2.93 (1 H, dd, J = 14.0, 9.0 Hz), 2.81 (1 H, dd, J = 14.0, 4.0 Hz), 2.77 (3 H, s), 2.27 (3 H, s), 1.99–1.84 (2 H, m), 1.65 (1 H, ddd, J = 12.0, 11.0, 4.0 Hz), 1.53 (1 H, dt, J = 12.0, 12.0 Hz), 1.05 (9 H, s); δC (67.9 MHz, CDCl3): 169.6, 167.0, 148.7, 135.8, 134.3, 134.1, 131.2, 129.8, 127.9, 127.7, 121.8, 99.4, 69.8, 65.1, 54.5, 40.9, 40.6, 39.2, 27.0, 21.0, 19.2. Found (CI): 580.2726 [MNH4]+, C32H38O7Si requires 580.2731 (0.8 ppm error); m/z (CI) 580 (22%, [MNH4]+), 453 (75), 247 (75), 196 (100).(b) To a stirred solution of the acid from (a) (222 mg, 0.395 mmol) in methanol (6 mL) was added potassium carbonate (290 mg, 2.10 mmol) at rt. After 1 h, the basic reaction mixture was neutralised with 1 M HCl and extracted with EtOAc. The aqueous layer was then acidified to pH 2.0 and again extracted with EtOAc. The organic extracts were combined, dried (MgSO4), filtered and the solvent evaporated under reduced pressure to give crude hydroxy-acid as a pale brown gum (203 mg). This was re-dissolved in acetone (5 mL) and potassium carbonate (216 mg, 1.56 mmol) added, followed by methyl iodide (0.49 mL, 1.1 g, 7.8 mmol). The mixture was stirred under N2 at reflux for 2 d. After cooling to rt the reaction mixture was filtered, the solvent evaporated under reduced pressure and the crude product purified by flash chromatography (petroleum ether ∶ EtOAc, 6 ∶ 1) to give the title compound44 (161 mg, 75%, mixture of anomers) as a yellowish gum; Rf 0.26 (petroleum ether ∶ EtOAc, 6 ∶ 1); νmax/cm−1 (thin film) 2932, 1726, 1472, 1279, 1113; δH (270 MHz, CDCl3): 7.77–7.60 (4 H, m), 7.43–7.30 (6 H, m), 7.22 (1 H, t, J = 8.0 Hz), 6.77 (1 H, d, J = 8.0 Hz), 6.76 (1 H, d, J = 8.0 Hz), 4.68 (1 H, d, J = 3.0 Hz), 4.05–4.14 (1 H, m), 3.81 (3 H, s), 3.80 (3 H, s), 3.67–3.57 (1 H, m), 2.99 (3 H, s), 2.77 (1 H, dd, J = 14.0, 7.5 Hz), 2.57 (1 H, dd, J = 14.0, 6.0 Hz), 1.96 (1 H, dd, J = 12.5, 5.0 Hz), 1.73–1.56 (2 H, m), 1.32 (1 H, dt, J = 11.5, 11.5 Hz), 1.04 (9 H, s); δC (67.9 MHz, CDCl3): 168.7, 156.5, 137.3, 135.8, 134.6, 134.5, 130.1, 129.8, 129.6, 127.6, 127.0, 122.8, 109.1, 99.2, 67.9, 65.7, 56.0, 56.3, 52.1, 41.1, 39.5, 39.5, 27.0, 19.2. Found (CI): 566.2963 [MNH4]+, C32H40O6Si requires 566.2938 (4.4 ppm error); m/z (CI) 566 (3%, [MNH4]+), 517 (95), 491 (100), 261 (95).
Methyl 2-{(2R,4R,6S)-6-allyl-4-[tert-butyl(dimethyl)silyloxy]tetrahydro-2H-pyran-2-ylmethyl}-6-methoxybenzoate (45)
To a stirred solution of acetal 44 (139 mg, 0.253 mmol) in acetonitrile (1.5 mL) at −20 °C, under N2, was added allyltrimethylsilane (0.20 mL, 150 mg, 1.3 mmol) followed by trimethylsilyl triflate (46 µL 57 mg, 0.26 mmol) dropwise over 10 min. The reaction mixture was warmed to 0 °C over 45 min then quenched with sat. NaHCO3(aq) and extracted with EtOAc (2 × 10 mL). The organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure to give a colourless gum. This was re-dissolved in THF (1.0 mL) and TBAF (1 M in THF + 5 wt% water, 0.75 mL, 0.75 mmol) added. This was stirred overnight at rt. The solvent was then evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 1 ∶ 1) to give the corresponding alcohol (83 mg) as a colourless gum. This was re-dissolved in dichloromethane (2.0 mL), cooled to 0 °C and 2,6-lutidine (56 µL, 51 mg, 0.48 mmol) followed by tert-butyl(dimethyl)silyl triflate (52 µL, 61 mg, 0.23 mmol) were added with stirring. After 45 min, a further portion of tert-butyl(dimethyl)silyl triflate (5 µL, 6 mg, 0.02 mmol) was added and stirring continued for a further 45 min. The solvent was then evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 6 ∶ 1) to give the title compound45 (89 mg, 81%) as a colourless gum; Rf 0.69 (petroleum ether ∶ EtOAc, 2 ∶ 1); [α]D +35.2 (c 1.34, CHCl3); νmax/cm−1 (thin film) 2950, 2855, 1733, 1585, 1472, 1268, 1114, 1073; δH (270 MHz, CDCl3): 7.27 (1 H, t, J = 8.0 Hz), 6.90 (1 H, d, J = 8.0 Hz), 6.80 (1 H, d, J = 8.0 Hz), 5.70 (1 H, dddd, J = 17.0, 10.0, 7.5, 6.5 Hz), 5.08–4.98 (2 H, m), 4.05–3.98 (1 H, m), 3.97–3.90 (1 H), 3.91 (3 H, s), 3.91–3.84 (1 H, m), 3.82 (3 H, s), 3.00 (1 H, dd, J = 14.0, 7.5 Hz), 2.67 (1 H, dd, J = 14.0, 6.0 Hz), 2.45–2.33 (1 H, m), 2.23–2.13 (1 H, m), 1.87–1.78 (1 H, m), 1.76–1.54 (2 H, m), 1.34 (1 H, dt, J = 13.0, 9.0 Hz), 0.89 (9 H, s), 0.05 (6 H, s); δC (67.9 MHz, CDCl3): 168.9, 156.5, 137.7, 135.2, 130.2, 124.1, 123.0, 116.8, 109.0, 70.8, 70.4, 65.1, 56.0, 52.3, 40.1, 39.5, 37.8, 37.3, 25.9, 18.2, −4.5, −4.6. Found (CI): 435.2568 [MH]+, C24H38O5Si requires 435.2567 (0.3 ppm error); m/z (CI) 435 (100%, [MH]+), 403 (50), 377 (18), 255 (20).Methyl 2-{(2R,4S,6S)-4-[tert-butyl(dimethyl)silyloxy]-6-(2-oxoethyl)tetrahydro-2H-pyran-2-ylmethyl}-6-methoxybenzoate (46)
Oxygen was bubbled through a stirred solution of ester 45 (136 mg, 0.313 mmol) in dichloromethane (10 mL) at −78 °C for 10 min. Ozone in oxygen was then bubbled through the solution for ca. 25 min until a pale blue colour appeared. Oxygen was bubbled through the solution for a further 10 min then triphenylphosphine (246 mg, 0.939 mmol) was added. The cooling bath was then removed and the reaction mixture allowed to warm to rt over 3 h. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 4 ∶ 1) to give the title compound46 (133 mg, 98%) as a colourless gum; Rf 0.52 (petroleum ether ∶ EtOAc, 2 ∶ 1); [α]D +36.3 (c 2.20, CHCl3); νmax/cm−1 (thin film) 2951, 2935, 2856, 1738, 1585, 1471, 1267, 1247, 1074; δH (270 MHz, C6D6): 9.36 (1 H, dd, J = 3.0, 2.0 Hz), 7.05 (1 H, t, J = 8.0 Hz), 6.87 (1 H, dd, J = 8.0, 1.0 Hz), 6.64 (1 H, dd, J = 8.0, 1.0 Hz), 4.40 (1 H, m), 4.01–3.85 (1 H, m), 3.73 (1 H, m), 3.65 (3 H, s), 3.25–3.15 (4 H, m), 2.76 (1 H, dd, J = 14.0, 5.0 Hz), 2.37 (1 H, ddd, J = 16.0, 9.0, 3.0 Hz), 1.81 (1 H, ddd, J = 16.0, 5.5, 2.0 Hz), 1.85–1.72 (1 H, m), 1.53 (1 H, ddd, J = 13.0, 8.5, 5.0 Hz), 1.40–1.30 (2 H, m), 0.94 (9 H, s), 0.00 (3 H, s), −0.01 (3 H, s); δC (67.9 MHz, C6D6): 199.7, 168.6, 157.0, 138.1, 130.1, 125.2, 122.9, 109.3, 71.0, 65.9, 65.3, 55.3, 51.8, 46.7, 39.9, 39.5, 38.7, 26.1, 18.2, −4.5, −4.6. Found (CI): 437.2374 [MH]+, C23H36O6Si requires 437.2359 (3.2 ppm error); m/z (CI) 437 (1, [MH]+), 401 (6), 379 (100), 257 (46).Methyl 2-{(2R,4S,6R)-4-[tert-butyl(dimethyl)silyloxy]-6-[(2S)-2-hydroxypent-4-enyl]tetrahydro-2H-pyran-2-ylmethyl}-6-methoxybenzoate (47)
(a) To a solution of aldehyde 46 (334 mg, 0.765 mmol) in dichloromethane (15 mL) at −78 °C was added allyltrimethylsilane (0.36 mL, 2.3 mmol) followed by dimethylaluminium chloride (1 M in hexane, 4.59 mL, 4.59 mmol) with stirring under N2. After 30 min, sat. NH4Cl(aq) (0.5 mL) was added and the reaction allowed to warm to rt. Brine (5 mL) was added and the mixture extracted with dichloromethane (4 × 10 mL). The organic extracts were dried (MgSO4), filtered and the solvent evaporated under reduced pressure to give the crude product as a cloudy gum. Flash chromatography (petroleum ether ∶ EtOAc, 2 ∶ 1) gave the title compound47 (310 mg, 85%, 15S ∶ 15R = 85 ∶ 15) as a colourless oil; Rf 0.34 (petroleum ether ∶ EtOAc, 2 ∶ 1); [α]D +18.0 (c 1.50, CHCl3); νmax/cm−1 (thin film) 3460, 2927, 2853, 1721, 1583, 1468, 1433, 1264, 1104, 1070; δH (270 MHz, CDCl3): 7.28 (1 H, t, J = 8.0 Hz), 6.87 (1 H, d, J = 8.0 Hz), 6.80 (1 H, d, J = 8.0 Hz), 5.76–5.61 (1 H, m), 5.05–4.96 (2 H, m), 4.34–4.25 (1 H, m), 4.02–3.93 (1 H, m), 3.89 (3 H, s), 3.82 (3 H, s), 3.82–3.73 (1 H, m), 3.43–3.34 (1 H, m), 3.06 (1 H, dd, J = 14.0, 8.5), 2.66 (1 H, dd, J = 14.0, 4.5), 2.09 (2 H, t, J = 7.0 Hz), 1.84 (1 H, dt, J = 13.0, 3.5 Hz), 1.75 (1 H, ddd, J = 14.0, 11.0, 3.0 Hz), 1.62 (2 H, t, J = 5.0 Hz), 1.45–1.26 (2 H, m), 0.89 (9 H, s), 0.05 (6 H, 2 × s); δC (67.9 MHz, CDCl3): 168.9, 156.6, 138.2, 135.3, 130.2, 124.0, 122.7, 117.0, 109.2, 70.6, 67.0, 66.8, 65.2, 55.9, 52.2, 41.6, 39.8, 39.2, 38.8, 38.7, 25.8, 18.1, −4.7, −4.7. Found (CI): 479.2825 [MH]+, C26H42O6Si requires 479.2829 (0.9 ppm error); m/z (CI) 479 (100%, [MH]+), 447 (42), 429 (47), 167 (33).(b) To a stirred solution of (+)-B-methoxydiisopinocampheyl borane (475 mg, 1.50 mmol) in Et2O (10 mL) was added allylmagnesium bromide (1 M in Et2O, 1.5 mL, 1.50 mmol) at −78 °C under N2. After 15 min, the mixture was allowed to warm to rt and stirred for 2 h. The mixture was again cooled to −78 °C and aldehyde 46 (437 mg, 1.5 mmol) in Et2O (5 mL) was added dropwise. After 3 h at −78 °C, the mixture was allowed to warm to rt and 1 M NaOH (5 mL) then 30% H2O2 (5 mL) was added; stirring was continued for a further 45 min. Sat. Na2SO3(aq) (2 mL) was then added and the reaction mixture extracted with EtOAc (3 × 100 mL). The organic extracts were combined and dried (MgSO4), filtered and evaporated under reduced pressure. Flash chromatography (petroleum ether ∶ EtOAc, 4 ∶ 1) gave the title compound47 (416 mg, 77%, 15R ∶ 15S = 90 ∶ 10) as a colourless oil; Rf 0.35 (petroleum ether ∶ EtOAc, 2 ∶ 1); [α]D +28.0 (c 0.95, CHCl3); νmax/cm−1 (thin film) 3496, 2949, 2929, 2856, 1732, 1585, 1471, 1267, 1074; δH (270 MHz, CDCl3): 7.28 (1 H, t, J = 8.0 Hz), 6.87 (1 H, d, J = 8.0 Hz), 6.80 (1 H, d, J = 8.0 Hz), 5.86–5.70 (1 H, m), 5.10–5.00 (2 H, m), 4.30–4.19 (1 H, m), 4.04–3.94 (2 H, m), 3.89 (3 H, s), 3.81 (3 H, s), 3.80–3.68 (1 H, m), 3.10 (1 H, dd, J = 14.0, 8.0 Hz), 2.75 (1 H, dd, J = 14.0, 5.5 Hz), 2.20–2.13 (2 H, m), 1.82 (1 H, dt, J = 13.0, 4.0 Hz), 1.75–1.66 (1 H, m), 1.59 (2 H, t, J = 6.0 Hz), 1.43–1.36 (2 H, m), 0.88 (9 H, s), 0.05 (3 H, s), 0.04 (3 H, s); δC (67.9 MHz, CDCl3): 168.9, 156.7, 137.6, 135.2, 130.4, 124.1, 122.8, 117.3, 109.4, 71.1, 71.0, 69.8, 64.9, 56.1, 52.4, 41.7, 39.7, 39.4, 38.9, 38.6, 26.0, 18.2, −4.5, −4.6. Found (CI): 479.2818 [MH]+, C26H42O6Si requires 479.2829 (2.2 ppm error); m/z (CI) 479 (100%, [MH]+), 447 (15), 421 (10), 180 (15), 167 (18).
2-{(2R,4S,6R)-4-[tert-Butyl(dimethyl)silyloxy]-6-[(2R)-2-hydroxypent-4-enyl]tetrahydro-2H-pyran-2-ylmethyl}-6-methoxybenzoic acid (48)
To a stirred solution of ester 47 (15R ∶ 15S = 90 ∶ 10, 32 mg, 0.067 mmol) in pyridine (2 mL) was added lithium iodide (105 mg, 0.784 mmol), and the solution was heated at 100 °C under N2 for 24 h. The mixture was cooled to rt and diluted with EtOAc (5 mL) and 0.5 M HCl (1 mL). This was extracted with EtOAc (6 × 5 mL) and the organic extracts dried (MgSO4), filtered and the solvent evaporated under reduced pressure. Flash chromatography (petroleum ether ∶ EtOAc, 2 ∶ 1 to EtOAc + 1% AcOH) gave first, recovered starting material 47 (12 mg) and then the title compound48 (17 mg, 55%, 88% with respect to the recovered starting material, 15R ∶ 15S = 90 ∶ 10) as a yellowish gum; Rf 0.45 (EtOAc + 1% AcOH); [α]D +64.4 (c 1.00, CHCl3); νmax/cm−1 (thin film) 3440, 2951, 2929, 2856, 1726, 1585, 1471, 1263, 1074; δH (270 MHz, CDCl3): 7.29 (1 H, dd, J = 7.5, 8.5 Hz), 6.84 (1 H, d, J = 8.5 Hz), 6.82 (1 H, d, J = 7.5 Hz), 5.76–5.59 (1 H, m), 5.08–5.00 (2 H, m), 4.28–4.18 (1 H, m), 4.11–4.01 (1 H, m), 4.01–3.88 (1 H, m), 3.85 (3 H, s), 3.67–3.58 (1 H, m), 3.07 (1 H, dd, J = 14.0, 4.0 Hz), 2.86 (1 H, dd, J = 14.0, 10.0 Hz), 2.20–2.10 (2 H, m), 2.00–1.62 (2 H, m), 1.69–1.63 (2 H, m), 1.47–1.26 (2 H, m), 0.88 (9 H, s), 0.06 (3 H, s), 0.05 (3 H, s); δC (67.9 MHz, CDCl3): 156.7, 137.0, 134.3, 130.7, 123.6, 117.8, 109.8, 72.3, 71.8, 70.1, 64.6, 56.1, 41.2, 40.5, 39.9, 39.6, 37.3, 25.9, 18.1, −4.8, −4.9. Found (CI): 465.2664 [MH+], C25H40O6Si requires 465.2672 (1.8 ppm); m/z (CI) 465 (100%, [MH]+), 447 (30), 421 (25), 184 (30), 167 (35).(1R,11R,13R,15S)-11-Allyl-15-[tert-butyl(dimethyl)silyloxy]-7-methoxy-10,17-dioxatricyclo[11.3.1.03,8]heptadeca-3,5,7-trien-9-one (49)
To a solution of DCC (365 mg, 1.77 mmol), DMAP (252 mg, 2.07 mmol) and DMAP·HCl (280 mg, 1.77 mmol) in CHCl3 (spectrophotometric grade 99.8%, 105 mL) at reflux with stirring under N2 was added carboxylic acid 48 (15R ∶ 15S = 90 ∶ 10, 137 mg, 0.295 mmol) via syringe pump over 5 h. This was stirred for a further 16 h at reflux then cooled to rt and methanol (2 mL) and acetic acid (0.2 mL) were added. After stirring for a further 30 min at rt the solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 6 ∶ 1) giving first the cyclised compound 49 (50 mg, 38%) as a white solid; Rf 0.40 (petroleum ether ∶ EtOAc, 4 ∶ 1); mp 149–150 °C (EtOAc); [α]D −6.3 (c 0.50, CHCl3); νmax/cm−1 (thin film) 2928, 2913, 1719, 1583, 1471, 1276, 1252, 1117; δH (500 MHz, CHCl3): 7.22 (1 H, t, J = 8.0 Hz), 6.76 (1 H, d, J = 8.0 Hz), 6.73 (1 H, d, J = 8.0 Hz), 5.87 (1 H, dddd, J = 16.0, 10.0, 6.0, 6.0 Hz), 5.61–5.55 (1 H, m), 5.13 (1 H, dd, J = 16.0, 2.0), 5.09 (1 H, d, J = 10.0), 4.56–4.49 (1 H, m), 4.08–4.01 (1 H, m), 4.00–3.93 (1 H, m), 3.78 (3 H, s), 3.54 (1 H, dd, J = 15.0, 10.5 Hz), 2.37 (1 H, dd, J = 15.0, 1.5 Hz), 2.47–2.43 (1 H, m), 2.36–2.28 (1 H, m), 1.87 (1 H, ddd, J = 13.5, 5.5, 4.0 Hz), 1.82 (1 H, ddd, J = 11.0, 11.0, 14.5), 1.66–1.48 (4 H, m), 0.91 (9 H, s), 0.06 (6 H, s); δC (125 MHz, CHCl3): 169.59, 156.01, 139.31, 133.92, 130.02, 125.92, 122.90, 117.66, 109.50, 74.06, 73.12, 65.87, 65.69, 55.98, 39.77, 39.43, 39.32, 39.11, 39.44, 25.97, 18.18, −4.74. Found (CI): 447.2566 [MH]+, C25H38O5Si requires 447.2567 (0.1 ppm error); m/z (FAB) 469 (100%, [MNa]+), 176 (45), 149 (17).Further elution gave a trace of diastereomer 50 (2 mg, 2%) as a white solid; Rf 0.31 (petroleum ether ∶ EtOAc, 4 ∶ 1); mp 93–95 °C; [α]D +7.9 (c 0.20, CHCl3); νmax/cm−1 (thin film) 2951, 2924, 2856, 1718, 1585, 1471, 1458, 1263, 1110, 1070; δH (500 MHz, CDCl3): 7.29 (1 H, t, J = 8.0 Hz), 6.82 (1 H, d, J = 8.0 Hz), 6.74 (1 H, d, J = 8.0 Hz), 5.83–5.74 (1 H, m), 5.06 (1 H, dd, J = 17.0, 1.5 Hz), 5.05 (1 H, d, J = 11.0 Hz), 4.66 (1 H, m), 4.24 (1 H, t, J = 10.5 Hz), 4.14 (1 H, m), 3.99–3.92 (1 H, m), 3.81 (3 H, s), 3.61 (1 H, t, J = 13.0 Hz), 2.56–2.50 (2 H, m), 2.44 (1 H, dt, J = 7.5, 7.5 Hz), 1.95 (1 H, ddd, J = 13.5, 6.5, 4.0 Hz), 1.67–1.60 (3 H, m), 1.55–1.50 (1 H, m), 1.47 (1 H, dt, J = 9.5, 3.5 Hz), 0.92 (9 H, s), 0.06 (6 H, s); δC (125 MHz, CDCl3): 170.22, 155.64, 140.23, 133.80, 130.64, 124.52, 122.66, 117.75, 108.28, 74.20, 74.06, 65.72, 60.67, 55.71, 39.77, 38.98, 37.37, 36.73, 25.95, 18.11, −4.64, −4.78. Found (CI): 447.2565 [MH]+, C25H38O5Si requires 447.2567 (0.3 ppm error); m/z (CI) 447 (100%, [MH]+), 429 (32), 389 (13), 297 (10), 148 (10).
(1R,11R,13R,15S)-11-Allyl-7,15-dihydroxy-10,17-dioxatricyclo[11.3.1.03,8]heptadeca-3,5,7-trien-9-one (51)
To a stirred solution of methyl ether 49 (8 mg, 0.018 mmol) in dichloromethane (1 mL) was added 9-iodo-9-BBN (1 M in hexanes, 40 µL, 0.040 mmol) at 0 °C under N2. After 1 h a further portion of 9-iodo-9-BBN (1 M in hexanes, 10 µL, 0.010 mmol) was added and the reaction was stirred for a further 1 h. Water (2 drops) was added, stirring continued for 15 min, then the mixture extracted with EtOAc. The organic extracts were dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by flash chromatography (dichloromethane ∶ EtOH, 20 ∶ 1) to give the title compound51 (4 mg, 70%) as a pale brown gum; Rf 0.30 (dichloromethane ∶ EtOH, 20 ∶ 1); [α]D −4.5 (c 0.15, MeOH); lit. (enantiomer)5d [α]D +6.8 (c 0.16, MeOH); νmax/cm−1 (thin film) 3262, 2924, 1716, 1583, 1464, 1294; δH (500 MHz, acetone-d6) 8.39 (1 H, s), 7.11 (1 H, dd, J = 8.0, 7.7 Hz), 6.77 (1 H, d, J = 8.0 Hz), 6.69 (1 H, d, J = 7.7 Hz), 5.92 (1 H, dddd, J = 6.4, 7.6, 10.2, 17.2 Hz), 5.48 (1 H, dddd, J = 2.4, 5.6, 5.6, 10.0 Hz), 5.13 (1 H, dddd, J = 1.6, 1.6, 2.4, 17.2 Hz), 5.03 (1 H, dddd, J = 1.6, 1.6, 2.4, 10.2 Hz), 4.24–4.30 (1 H, m), 3.96–4.03 (1 H, m), 3.85–3.91 (1 H, m), 3.77–3.81 (1 H, m), 3.34 (1 H, dd, J = 9.8, 15.0 Hz), 2.44 (1 H, dd, J = 1.0, 15.0 Hz), 2.30–2.41 (2 H, m), 1.93 (1 H, ddd, J = 4.5, 4.5, 12.7 Hz), 1.77–1.86 (1 H, m), 1.40–1.71 (4 H, m); δC (125 MHz, acetone-d6) 169.28, 154.31, 140.23, 135.32, 130.38, 125.47, 122.34, 117.48, 114.43, 73.65, 73.61, 68.08, 64.91, 40.36, 40.10, 39.96, 39.72, 39.13. Found (CI): 319.1545 [MH]+, C18H22O5 requires 319.1546 (0.1 ppm error); m/z (CI) 336 (20%, [MNH4]+), 319 (100, [MH]+), 301 (70), 283 (20), 162 (20), 134 (25).1H NMR, 13C NMR and IR spectroscopic data were in agreement with published values (see Table 2).5d
(1R,11R,13R,15S)-11-Allyl-7,15-bis[tert-butyl(dimethyl)silyloxy]-10,17-dioxatricyclo[11.3.1.03,8]heptadeca-3,5,7-trien-9-one [(−)-6]
To a solution of diol 51 (3 mg, 0.009 mmol) in dichloromethane (0.5 mL) was added 2,6-lutidine (0.2 mL) and tert-butyl(dimethyl)silyl triflate (10 µL 12 mg, 0.044 mmol) with stirring at 0 °C under N2. After 2 h a further portion of tert-butyl(dimethyl)silyl triflate (10 µL, 12 mg, 0.044 mmol) was added and stirring was continued for a further 2 h. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 9 ∶ 1) to give the title compound (−)-6 (3 mg, 80%) as a colourless oil; Rf 0.71 (petroleum ether ∶ EtOAc, 2 ∶ 1); [α]D −20 (c 0.10, CHCl3); νmax/cm−1 (thin film) 2927, 2856, 1722, 1579, 1463, 1285, 1255, 1106; δH (500 MHz, acetone-d6): 7.18 (1 H, t, J = 8.0 Hz), 6.83 (1 H, d, J = 8.0 Hz), 6.79 (1 H, d, J = 8.0 Hz), 5.90 (1 H, dddd, J = 17.0, 10.0, 7.0, 7.0 Hz), 5.58–5.52 (1 H, m), 5.14 (1 H, dd, J = 17.0, 1.0 Hz), 5.07 (1 H, dt, J = 10.0, 1.0 Hz), 4.33–5.28 (1 H, m), 4.18–4.13 (1 H, m), 3.93–3.88 (1 H, m), 3.46 (1 H, dd, J = 15.0, 10.5 Hz), 2.44 (1 H, J = 15.0, 1.5 Hz), 2.48–2.35 (2 H, m), 1.92 (1 H, ddd, J = 13.5, 5.0, 5.0 Hz), 1.81 (1 H, dt, J = 14.5, 10.5 Hz), 1.65–1.58 (1 H, m), 1.60 (1 H, dt, J = 14.5, 2.0 Hz), 1.56–1.47 (2 H, m), 0.99 (9 H, s), 0.92 (9 H, s), 0.26 (3 H, s), 0.20 (3 H, s), 0.10 (3 H, s), 0.09 (3 H, s); δC (125 MHz, acetone-d6): 169.54, 152.45, 140.94, 134.98, 130.17, 129.85, 124.08, 117.98, 117.67, 74.20, 73.75, 66.75, 66.55, 40.22, 40.10, 39.97, 39.47, 38.87, 26.25, 26.16, 18.88, 18.55, −3.79, −4.22, −4.61, −4.64. Found (CI): 547.3275 [MH]+, C30H50O5Si2 requires 547.3275 (0.0 ppm error); m/z (CI) 564 (12%, [MNH4]+), 547 (85, [MH]+), 529 (100), 489 (14), 428 (30), 261 (25), 137 (30).(1R,11R,13R)-11-Allyl-7-methoxy-10,17-dioxatricyclo[11.3.1.03,8]heptadeca-3,5,7-triene-9,15-dione (52)
(a) To a solution of lactone 49 (50 mg, 0.11 mmol) in THF (2 mL) was added TBAF (1 M in THF, 0.22 mL, 0.22 mmol) with stirring at rt. After 5 h the solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 4 ∶ 1 to EtOAc) to give (1R,11R,13R,15S)-11-allyl-15-hydroxy-7-methoxy-10,17-dioxatricyclo[11.3.1.03,8]heptadeca-3,5,7-trien-9-one (36 mg, 97%) as a colourless oil; Rf 0.50 (EtOAc); [α]D −10.6 (c 1.50, CHCl3); νmax/cm−1 (thin film) 3460, 2946, 2923, 2841, 1732, 1598, 1581, 1470, 1272, 1119, 1089, 1071; δH (270 MHz, CDCl3): 7.21 (1 H, t, J = 8.0 Hz), 6.77 (1 H, d, J = 8.0 Hz), 6.72 (1 H, d, J = 8.0 Hz), 5.86 (1 H, dddd, J = 17.0, 10.0, 7.0, 7.0 Hz), 5.62–5.52 (1 H, m), 5.18–5.08 (2 H, m), 4.34–4.25 (1 H, m), 4.05–3.93 (2 H, m, 9-H), 3.77 (3 H, s), 3.37 (1 H, dd, J = 15.0, 10.0 Hz), 2.50–2.27 (2 H, m), 2.44 (1 H, d, J = 14.5 Hz), 2.08 (1 H, broad s), 2.00–182 (2 H, m), 1.77–1.70 (1 H, dt, J = 13.0, 6.0 Hz), 1.58–1.46 (3 H, m); δC (67.9 MHz, CDCl3): 169.2, 156.1, 138.5, 133.7, 130.0, 125.5, 122.9, 117.8, 109.5, 73.0, 72.7, 67.6, 65.1, 55.9, 39.7, 39.5, 38.9, 38.7, 38.2. Found (CI): 333.1701 [MH]+, C19H24O5 requires 333.1702 (0.4 ppm error); m/z (CI) 350 (10%, [MNH4]+), 333 (100, [MH]+), 315 (63), 245 (10), 148 (25).(b) To a solution of the alcohol from (a) (30 mg, 0.090 mmol) in dichloromethane (2 mL) was added Dess–Martin periodinane (42 mg, 0.099 mmol) with stirring under N2 at rt. After 1 h the solvent was evaporated under reduced pressure and the residue purified by flash chromatography (petroleum ether ∶ EtOAc, 1 ∶ 1) to give the title compound52 (29 mg, 97%) as a white crystalline solid; Rf 0.40 (EtOAc); mp 184–185 °C; [α]D −140 (c 0.60, CHCl3); νmax/cm−1 (thin deposit) 2934, 2908, 1709, 1598, 1470, 1355, 1278, 1254, 1116, 1071; δH (270 MHz, CDCl3): 7.28 (1 H, t, J = 8.0 Hz), 6.84 (1 H, d, J = 8.0 Hz), 6.79 (1 H, d, J = 8.0 Hz), 5.88 (1 H, dddd, J = 17.0, 10.0, 7.0, 7.0 Hz), 5.68–5.58 (1 H, m), 5.17 (1 H, ddd, J = 17.0, 3.0, 1.5 Hz), 5.17–5.12 (1 H, m), 4.50 (1 H, tddd, J = 10.0, 3.5, 1.5, 0.5), 4.25 (1 H, dddd, J = 11.0, 8.5, 6.5, 2.0), 3.81 (3 H, s), 3.46 (1 H, dd, J = 14.0, 11.0 Hz), 2.67 (1 H, ddd, J = 15.5, 6.5, 0.5 Hz), 2.52 (1 H, dd, J = 15.5, 8.5 Hz), 2.51–2.31 (4 H, m), 2.22 (1 H, dd, J = 17.0, 10.0 Hz), 1.85 (1 H, ddd, J = 15.0, 11.0, 10.0 Hz), 1.71 (1 H, ddd, J = 15.0, 3.5, 1.5 Hz); δC (67.9 MHz, CDCl3): 207.2, 169.9, 156.1, 137.1, 133.5, 130.6, 125.8, 122.7, 118.1, 110.2, 74.2, 72.2, 68.3, 55.9, 47.2, 45.2, 40.2, 39.1, 39.0. Found (CI): 331.1546 [MH]+, C19H22O5 requires 331.1545 (0.2 ppm error); m/z (CI) 348 (11%, [MNH4]+), 331 (100, [MH]+), 313 (63), 177 (12), 148 (24).
(3R,5R)-3-Allyl-5-hydroxy-14-methoxy-3,4,5,6-tetrahydro-1H-2-oxabenzocyclododecene-1,7(8H)-dione (54)
To a freshly prepared stirred solution of LDA (0.1 M in THF, 3.0 mL, 0.30 mmol) was added a solution of pyranone 52 (10 mg, 0.030 mmol) in THF (0.6 mL) under N2 at −10 °C. After 15 min the reaction mixture had darkened considerably. Sat. NH4Cl(aq) (0.1 mL) was then added, the mixture stirred at −10 °C for a further 10 min, then the solvent evaporated under reduced pressure. Flash chromatography (petroleum ether: EtOAc, 3 ∶ 1 to 1 ∶ 1) gave first recovered starting material 52 (2 mg, 20%), then the title compound54 (2 mg, 20%) as a yellowish oil, which was recrystallised from ether to give a yellowish solid; Rf 0.27 (petroleum ether ∶ EtOAc, 1 ∶ 1); mp 123–124 °C; [α]D −110 (c 0.20, MeOH); νmax/cm−1 (thin deposit): 3448, 2941, 1707, 1644, 1575, 1467, 1268, 1110; δH (500 MHz, acetone-d6): 7.37 (1 H, t, J = 8.0 Hz), 7.01 (1 H, d, J = 8.0 Hz), 6.86 (1 H, d, J = 8.0 Hz), 6.71 (1 H, d, J = 16.0 Hz), 6.00 (1 H, ddd, J = 16.0, 8.0, 6.5 Hz), 5.87 (1 H, dddd, J = 16.0, 10.0, 6.5, 6.5 Hz), 5.31 (1 H, m), 5.11 (1 H, dq, J = 17.0, 1.5 Hz), 5.08 (1 H, ddt, J = 10.0, 2.5, 1.0 Hz), 4.46 (1 H, m), 3.81 (1 H, d, J = 6.0 Hz), 3.23 (2 H, m), 2.99 (1 H, dd, J = 13.5, 3.5 Hz), 2.43 (2 H, tq, J = 6.0, 1.5 Hz), 2.26 (1 H, dd, J = 13.5, 9.5 Hz), 1.99 (1 H, ddd, J = 14.0, 11.0, 5.0 Hz), 1.76 (1 H, ddd, J = 14.0, 9.5, 3.5 Hz); δC (125 MHz, acetone-d6): 206.75, 167.87, 157.29, 136.96, 134.60, 134.04, 131.06, 128.85, 124.16, 121.23, 118.05, 111.13, 71.78, 65.21, 56.33, 50.15, 47.93, 41.77, 40.36. Found (CI): 348.1812 [MNH4]+, C19H22O5 requires 348.1811 (0.2 ppm error); m/z (CI) 348 (10%, [MNH4]+), 331 (100, [MH]+), 313 (85), 295 (15), 234 (15), 217 (35), 174 (20), 148 (15).Acknowledgements
We thank the BBSRC and GlaxoSmithKline for a CASE studentship (I. S.) and Elsevier Science for additional financial support (A. L. and S. A. S.).References
- (a) R. Jansen, B. Kunze, F. Sasse, H. Reichenbach and G. Höfle, Drugs from Natural Products Conference, Dublin, 1998 Search PubMed; (b) B. Kunze, R. Jansen, F. Sasse, G. Höfle and H. Reichenbach, J. Antibiotics, 1998, 51, 1075–1080 Search PubMed; (c) R. Jansen, B. Kunze, H. Reichenbach and G. Höfle, Eur. J. Org. Chem., 2000, 913–919 CrossRef CAS . The absolute stereochemistry of (−)-apicularen A in the original publication (ref. 1a) was subsequently revised to the enantiomeric form (ref. 1c).
- F. Sasse, personal communication, referred to in ref. 1c.
- (a) K. L. Erickson, J. A. Beutler, J. H. Cardellina II and M. R. Boyd, J. Org. Chem., 1997, 62, 8188–8192 CrossRef CAS; K. L. Erickson, J. A. Beutler, J. H. Cardellina II and M. R. Boyd, J. Org. Chem., 2001, 66, 1532 CrossRef CAS; (b) T. C. McKee, D. L. Galinis, L. K. Pannell, J. H. Cardellina II, J. Lakkso, C. M. Ireland, L. Murray, R. J. Capon and M. R. Boyd, J. Org. Chem., 1998, 63, 7805–7810 CrossRef CAS; (c) K. A. Dekker, R. J. Aiello, H. Hirai, T. Inagaki, T. Sakakibara, Y. Suzuki, J. F. Thompson, Y. Yamaguchi and N. Kojima, J. Antibiot., 1998, 51, 14–20 Search PubMed; (d) J. W. Kim, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, J. Org. Chem., 1999, 64, 153–155 CrossRef CAS.
- M. R. Boyd, C. Farina, P. Belfiore, S. Gagliardi, J. W. Kim, Y. Hayakawa, J. A. Beutler, T. C. McKee, B. J. Bowman and E. J. Bowman, J. Pharmacol. Exp. Ther., 2001, 297, 114–120 Search PubMed.
- (a) A. Fürstner, G. Seidel and N. Kindler, Tetrahedron, 1999, 55, 8215–8230 CrossRef CAS; (b) A. Fürstner, O. R. Thiel and G. Blanda, Org. Lett., 2000, 2, 3731–3734 CrossRef CAS; (c) Y. Wu, O. R. Seguil and J. K. De Brabander, Org. Lett., 2000, 2, 4241–4244 CrossRef CAS; (d) A. Bhattacharjee and J. K. De Brabander, Tetrahedron Lett., 2000, 41, 8069–8073 CrossRef CAS; (e) J. T. Feutrill, G. A. Holloway, F. Hilli, H. M. Hügel and M. A. Rizzacasa, Tetrahedron Lett., 2000, 41, 8569–8572 CrossRef CAS; (f) B. B. Snider and F. Song, Org. Lett., 2000, 2, 407–408 CrossRef CAS; (g) I. Stefanuti, S. A. Smith and R. J. K. Taylor, Tetrahedron Lett., 2000, 41, 3735–3738 CrossRef CAS; (h) S. A. Raw and R. J. K. Taylor, Tetrahedron Lett., 2000, 41, 10357–10361 CrossRef CAS; (i) K. Kuramochi, H. Watanabe and T. Kitahara, Synlett, 2000, 397–399 CAS; (j) R. Shen and J. A. Porco Jr, Org. Lett., 2000, 2, 1333–1336 CrossRef CAS; (k) Y. Wu, L. Esser and J. K. De Brabander, Angew. Chem., Int. Ed., 2000, 39, 4308–4310 CrossRef CAS; (l) G. L. Georg, Y. M. Ahn, B. Blackman, F. Farokhi, P. T. Flaherty, C. J. Mossman, S. Roy and K. Yang, Chem. Commun., 2001, 255–256 RSC; (m) F. Scheufler and M. E. Maier, Synlett, 2001, 1221–1224 CrossRef CAS; (n) R. S. Coleman and R. Garg, Org. Lett., 2001, 3, 3487–3490 CrossRef CAS; (o) A. Fürstner, C. Brehm and Y. Cancho-Grande, Org. Lett., 2001, 3, 3955–3957 CrossRef CAS; (p) S. M. Kühnert and M. E. Maier, Org. Lett., 2002, 4, 643–646 CrossRef; (q) M. Bauer and M. E. Maier, Org. Lett., 2002, 4, 2205–2208 CrossRef CAS; (r) F. Hilli, J. M. White and M. A. Rizzacasa, Tetrahedron Lett., 2002, 43, 8507 CrossRef CAS.
- (a) Y. Wu, X. Liao, R. Wang, X.-S. Xie and J. K. De Brabander, J. Am. Chem. Soc., 2002, 124, 3245–3253 CrossRef CAS; (b) A. Fürstner, T. Dierkes, O. R. Thiel and G. Blanda, Chem. Eur. J., 2001, 7, 5286–5298 CrossRef CAS; (c) D. Labrecque, S. Charron, R. Rej, C. Blais and S. Lamothe, Tetrahedron Lett., 2001, 42, 2645–2648 CrossRef CAS; (d) A. B. Smith III and J. Zheng, Tetrahedron, 2002, 58, 6455–6571 CrossRef; (e) B. B. Snider and F. Song, Org. Lett., 2001, 3, 1817–1820 CrossRef CAS; (f) R. Shen, C. T. Lin and J. A. Porco, J. Am. Chem. Soc., 2002, 124, 5650–5651 CrossRef CAS; (g) A. Bhattacharjee, O. R. Seguil and J. K. De Brabander, Tetrahedron Lett., 2001, 42, 1217–1220 CrossRef CAS; (h) For a second Total Synthesis of Apicularen A, see: K. C. Nicolaou, D. W. Kim and R. Baati, Angew. Chem., Int. Ed., 2002, 41, 3701 Search PubMed.
- Preliminary communication: A. Lewis, I. Stefanuti, S. A. Swain, S. A. Smith and R. J. K. Taylor, Tetrahedron Lett., 2001, 42, 5549–5552 Search PubMed.
- M. Furber, R. J. K. Taylor and S. C. Burford, J. Chem. Soc., Perkin Trans. 1, 1986, 1809–1815 RSC.
- P. Allevi, M. Anastasia, P. Ciuffreda, A. Fiecchi and A. Scala, J. Chem. Soc., Chem. Commun., 1987, 101–102 RSC.
- O. Gaertzen, A. M. Misske, P. Wolbers and H. M. R. Hoffmann, Tetrahedron Lett., 1999, 40, 6359–6363 CrossRef CAS.
- L. Boymond, M. Rottländer, G. Cahiez and P. Knochel, Angew. Chem., Int. Ed., 1998, 37, 1701 CrossRef CAS.
- K. Kitagawa, A. Inoue, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2000, 39, 2481–2483 CrossRef CAS.
- H. Kotsuki, I. Kadota and M. Ochi, Tetrahedron Lett., 1989, 30, 1281–1284 CrossRef CAS.
- J. R. Rasmussen, C. J. Slinger, R. J. Kordish and D. D. Newman-Evans, J. Org. Chem., 1981, 46, 4843–4846 CrossRef CAS.
- V. Bolitt and C. Mioskowski, J. Org. Chem., 1990, 55, 5812–5813 CrossRef.
- B. Bennetau, J. Mortier, J. Moyroud and J.-L. Guesnet, J. Chem. Soc., Perkin Trans. 1, 1995, 1265–1271 RSC.
- H. Kotsuki, T. Araki, A. Miyazaki, M. Iwasaki and P. K. Datta, Org. Lett., 1999, 1, 499–502 CrossRef CAS.
- V. A. Martin, D. H. Murray, N. E. Pratt, Y. Zhao and K. F. Albizati, J. Am. Chem. Soc., 1990, 112, 6965–6978 CrossRef CAS.
- P. Brownbridge, T. H. Chan, M. A. Brook and G. J. Kang, Can. J. Chem., 1983, 61, 688–693 CAS.
- A. Hosomi, Y. Sakata and H. Sakurai, Tetrahedron Lett., 1984, 22, 2383–2386 CrossRef CAS.
- D. A. Evans, B. D. Allison and M. G. Yang, Tetrahedron Lett., 1999, 40, 4457–4460 CrossRef CAS.
- P. K. Jadhav, K. S. Bhat, P. T. Perumal and H. C. Brown, J. Org. Chem., 1986, 51, 432–439 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CAS.
- E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394–2395 CrossRef CAS.
- A. Fürstner and G. Seidel, J. Org. Chem., 1997, 62, 2332–2336 CrossRef.
- The EPSRC crystallography service (Southampton) and Dr P. Timmins (University of York) obtained and refined the crystal structure of compound 54.
- Organocopper Reagents: A Practical Approach, Ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994 Search PubMed; R. K. Dieter, L. A. Silks III, J. R. Fishpaugh and M. E. Kastner, J. Am. Chem. Soc., 1985, 107, 4679 Search PubMed.
- S. Kobayashi, S. Tagawa and S. Nakajima, Chem. Pharm. Bull., 1963, 11, 123–126 CAS.
| This journal is © The Royal Society of Chemistry 2003 |