Peptides to peptidomimetics: towards the design and synthesis of bioavailable inhibitors of oligosaccharyl transferase
Eranthie Weerapana and Barbara Imperiali*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: imper@mit.edu
First published on 26th November 2002
Abstract
Oligosaccharyl transferase (OT) is the enzyme responsible for asparagine-linked glycosylation in the lumen of the endoplasmic reticulum, which is a subcellular compartment within eukaryotic cells. Inhibition of this enzyme within a cellular environment would provide a valuable investigative tool for glycobiology. Due to the limitations of peptides, none of the existing peptide-based inhibitors of OT demonstrate activity in cell-based enzyme assays. We report herein the design, synthesis and preliminary biological characterization of a family of peptidomimetics that inhibit OT with Ki values in the nanomolar range. The hexapeptide Bz-Dab-Ala-Thr-Val-Thr-Nph-NH2 (Ki = 69 nM) was used as the prototype for the design of bioavailable inhibitors. Several aminobenzoic acid spacer groups were evaluated as potential isosteres of the Val-Thr dipeptide unit and the peptidomimetic incorporating 3-aminobenzoic acid proved to inhibit OT with similar potency to the parent compound (Ki = 84 nM). Further modifications explored the effects of size, hydrophobicity and conformational rigidity on enzyme affinity. This study yielded a family of potent non-peptidic inhibitors that are viable candidates for the in vivo inhibition of OT.
Introduction
Protein glycosylation plays a key role in numerous cellular processes including immune response, intracellular targeting, intercellular recognition and protein folding and stability.1 Asparagine-linked glycosylation is a major class of glycosylations mediated by a single enzyme, oligosaccharyl transferase (OT) that is localized in the lumen of the endoplasmic reticulum (ER). This multimeric membrane-bound enzyme catalyzes the co-translational transfer of a dolichol-linked tetradecasaccharide (GlcNAc2Man9Glc3) to an asparagine side chain in the Asn-Xaa-Thr/Ser consensus sequence within a nascent polypeptide chain2 as shown in Fig. 1. This step constitutes the first committed step in N-linked glycosylation; the resulting glycoprotein is further processed by various glycosyltransferases in the ER and Golgi apparatus to create a diverse array of oligosaccharide units.3 The availability of OT-specific inhibitors that function in vivo may ultimately provide an insight into this vital protein modification reaction.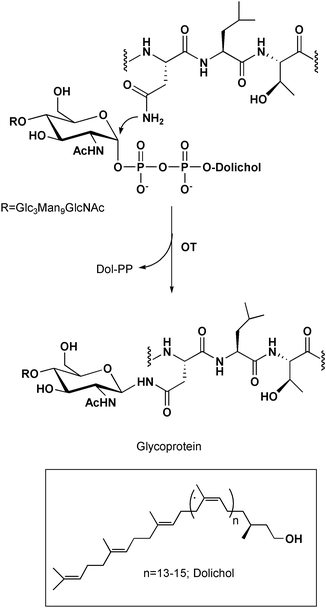 | ||
| Fig. 1 The co-translational transfer of a dolichol-linked tetradecasaccharide to an asparagine side chain catalyzed by oligosaccharyl transferase. | ||
Previous studies in the Imperiali group have yielded inhibitors with low nanomolar affinity for OT. These inhibitors are based on the Asn-Xaa-Thr/Ser consensus sequence of the natural substrate. The capped tripeptide, Bz-Asn-Leu-Thr-NHMe is a substrate for the enzyme (Km = 20 µM),4 and the first generation of OT inhibitors was based on this truncated substrate. Replacing the amide in the asparagine side chain with an amine yielded a substrate mimic Bz-Dab-Leu-Thr-NHMe (1,3-diaminobutanoic acid, Dab) that is a weak competitive inhibitor of OT.5 Studies extending from this initial discovery exploited binding interactions between OT and residues beyond the consensus sequence6 to yield a hexapeptide Bz-Dab-Ala-Thr-Val-Thr-Nph-NH2 (Nph = p-nitrophenylalanine) that is a potent inhibitor of yeast OT in vitro (Ki = 69 nM).7 Additionally, peptides that are structurally constrained in an Asx-turn conformation show enhanced affinity for the enzyme. The above inhibitor demonstrates a slightly enhanced potency when constrained via a side-chain to backbone macrocyclization. Inhibitors derived from these initial lead compounds were potent, yet none displayed any activity in vivo. In general, peptidic compounds exhibit poor absorption properties and limited proteolytic stability and are not optimal for studies under cell-based conditions. This created the need to obtain non-peptidic, bioavailable inhibitors of OT that would enable the study of N-linked glycosylation and its role in cellular processes in vivo.
Currently, the only inhibitor of N-linked protein glycosylation to function within a cellular environment is the microbial product tunicamycin. Tunicamycin is a bisubstrate analog that inhibits the first step in the assembly of the dolichol-linked oligosaccharide donor.8 Since this transformation occurs numerous steps prior to the reaction catalyzed by OT, the effect of the inhibitor on the actual glycosylation step is not immediate, nor does it have the potential to reveal the specific consequences of blocking N-glycosylation. The focus of this study is the generation of rationally designed, non-peptidic inhibitors that would have the physical characteristics to be able to function in vivo.
The linear hexapeptide Bz-Dab-Ala-Thr-Val-Thr-Nph-NH2 was used as the prototype for designing an inhibitor of OT that would function in vivo. The readily modifiable and modular peptide platform provided a foundation for the rational design of a peptidomimetic inhibitor. Toward this goal, we have modified this structure via isosteric replacement of the peptide backbone to yield a non-peptidic entity that demonstrates higher proteolytic stability and increased lipophilicity to enable passive permeation of the cellular and ER membranes. There has been no previous record of peptidomimetics or pseudopeptides that act as potent inhibitors of OT. In this paper we report the systematic introduction of non-peptidic character into our previous inhibitors to yield several compounds with nanomolar inhibition potency for OT.
Results
Design and synthesis of inhibitors
The class of dipeptide isosteres that is the focus of this study is based on an aminobenzoic acid framework. Similar scaffolds have been successfully incorporated within the structures of Src SH2 domain antagonists,10 growth hormone-releasing peptide receptor agonists11 and Ras farnesyl protein transferase inhibitors.12 These isosteres introduce rigidity and hydrophobicity to the system as well as reducing the total amide bond content. We replaced the Val-Thr unit with a variety of spacers that incorporated 3- or 4-aminobenzoic acid and 3- or 4-aminomethylbenzoic acid. All the unprotected aminobenzoic acids were commercially available except for 3-aminomethylbenzoic acid, which was prepared from 3-cyanobenzoic acid by catalytic hydrogenation (Pd/C). The amine or aniline groups of the building blocks were then protected as the Alloc (allyloxycarbonyl) derivative using allyl chloroformate. The synthesis of the peptidomimetic analogs includes the solid phase coupling of the corresponding Alloc protected aminobenzoic acid to the p-nitrophenylalanine residue using O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) as a coupling agent. The coupling step was followed by Alloc deprotection using tetrakis(triphenylphosphine)palladium(0) and phenylsilane and subsequent coupling of the threonine, alanine and diaminobutyric acid residues using standard peptide synthesis protocols.
The four inhibitors synthesized via this method are illustrated in Fig. 2. These compounds were then assayed for their in vitro potency as OT inhibitors using solubilized yeast (S. cerevisiae) microsomes. It was found that the 3-aminobenzoic acid dipeptide isostere (4) was the most effective mimic of the Val-Thr unit with inhibitory activity similar to the parent peptidic inhibitor. The reduced amide bond character and increased rigidity of the 3-aminobenzoic acid isostere has minimal effects on affinity to OT, confirming that the amide bond between the Val-Thr unit can be replaced without deleterious consequences for inhibitor binding. Due to the high affinity of compound 4 for OT, it is an excellent candidate for further modification toward the goal of generating a more hydrophobic non-peptide inhibitor.
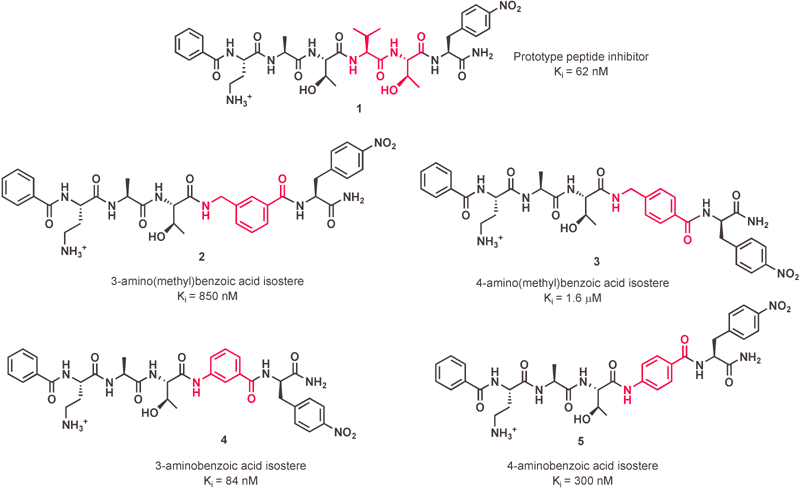 | ||
| Fig. 2 Inhibitors including dipeptide isosteres in place of the Val-Thr dipeptide. | ||
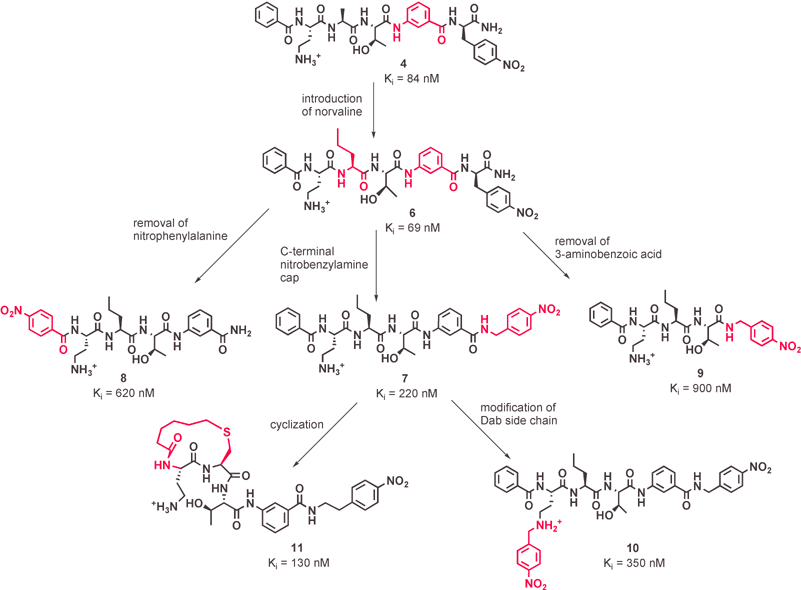 | ||
| Fig. 3 Further modifications of the peptidomimetic compound. | ||
In general, fewer amide bonds are desirable so as to impart proteolytic stability as well as lipophilicity.14a In accordance with this, further modifications were investigated to systematically reduce the number of amide bonds within the inhibitor structure and to determine which of these bonds are important for interaction with the enzyme. The dipeptide isostere studies removed the amide between the Val-Thr unit with minimal effect on binding. The next amide bond that was targeted was the amide C-terminal to the p-nitrophenylalanine residue. Replacement of this amide was achieved by using a 4-nitrobenzylamine capping group instead of the intact p-nitrophenylalanine. The resulting inhibitor 7 shows a 3-fold loss in affinity, suggesting that there is a weak interaction between this terminal amide and residues at the OT active site.
A second factor that promotes cell permeability is the overall size of the molecule and studies show that higher molecular weight compounds are in general less likely to be membrane permeable relative to smaller entities.14a In accordance with this hypothesis, two inhibitors, 8 and 9 were synthesized with molecular weights in the 500 Da range. Inhibitor 8 omits the p-nitrophenylalanine residue that is used for quantification purposes, but installs an N-terminal nitrobenzyl cap instead (to enable accurate quantitation as described previously). Previous studies14b in the group have shown that substitution of nitrobenzyl for benzyl at the N-terminal cap causes minimal effects in enzyme affinity. These two changes to the inhibitor structure resulted in a compound with a Ki of 620 nM, a seven-fold decrease in enzyme affinity. This result indicates that the C-terminal p-nitrophenylalanine provides valuable interactions with residues at the enzyme active site to increase potency in addition to supplying a quantification tool. Inhibitor 9 installs the nitrophenyl group adjacent to the consensus sequence and excludes the Val-Thr dipeptide isostere. The resulting molecule is small and contains only 4 amide bonds, factors that enhance its drug-like character. The lower Ki value (900 nM) of this compound confirms that the Val-Thr dipeptide and hence the aminobenzoic acid unit is important for enzyme binding as it orients the nitrophenyl group at the optimal distance for favorable interactions as well as providing additional hydrophobic contacts. The synthesis of inhibitors 7 and 9 involved the use of an aldehyde-functionalized resin, to which the nitrobenzyl functionality was introduced via a reductive amination using 4-nitrobenzylamine. The peptide was then extended from the resulting secondary amine using standard peptide synthesis protocols.
Of the changes that were made to increase the bioavailability of the initial lead compound (4), the installation of the nitrobenzyl cap (7) had the least effect on enzyme affinity. This compound displays decreased amide bond character relative to the first generation inhibitors. Further modifications were then performed on this inhibitor skeleton, to add more ‘drug-like’ character and increase enzyme affinity.
Hydrophobicity is an important concept in bioavailability.14a Throughout this study, theoretical water-octanol partition coefficients were calculated for each of the inhibitors using the ACD/LogP software that calculates log P values based on an algorithm that uses a database of over 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 compounds.15 These computed values of log P were used to obtain a very rough estimate of the hydrophobicity of the synthesized compounds. The values of log P for the inhibitors discussed in this paper ranged from 1.19 to 5.04 and even the most hydrophobic inhibitors were well behaved under the aqueous assay conditions at concentrations up to 10 µM. The most hydrophobic of the inhibitors synthesized was 10, which installed a nitrobenzyl group at the amine terminus of the Dab side chain. This modification was chosen due to information suggesting the presence of aromatic amino acids at the site of interaction of carbohydrate binding proteins.16,17 It was hypothesized that the sugar-binding site of OT consists of similar aromatic side chains and previous studies in the group demonstrated that incorporating a naphthyl group at this position enhances inhibition.18 The synthesis of this compound used Fmoc-Dab(Alloc)-OH instead of the Boc-protected unit used in the other syntheses. This orthogonal protection scheme enabled the selective deprotection of the amino side chain using tetrakis(triphenylphosphine)palladium(0) and phenylsilane. The nitrobenzyl moiety was installed via reductive amination with nitrobenzaldehyde and sodium triacetoxyborohydride. The resulting inhibitor 10 shows a Ki value of 350 nM with no solubility problems under the aqueous assay conditions. The added aromatic group does not increase affinity as expected, suggesting that the nitrobenzyl group is not oriented optimally for π-stacking interactions at the active site.
600 compounds.15 These computed values of log P were used to obtain a very rough estimate of the hydrophobicity of the synthesized compounds. The values of log P for the inhibitors discussed in this paper ranged from 1.19 to 5.04 and even the most hydrophobic inhibitors were well behaved under the aqueous assay conditions at concentrations up to 10 µM. The most hydrophobic of the inhibitors synthesized was 10, which installed a nitrobenzyl group at the amine terminus of the Dab side chain. This modification was chosen due to information suggesting the presence of aromatic amino acids at the site of interaction of carbohydrate binding proteins.16,17 It was hypothesized that the sugar-binding site of OT consists of similar aromatic side chains and previous studies in the group demonstrated that incorporating a naphthyl group at this position enhances inhibition.18 The synthesis of this compound used Fmoc-Dab(Alloc)-OH instead of the Boc-protected unit used in the other syntheses. This orthogonal protection scheme enabled the selective deprotection of the amino side chain using tetrakis(triphenylphosphine)palladium(0) and phenylsilane. The nitrobenzyl moiety was installed via reductive amination with nitrobenzaldehyde and sodium triacetoxyborohydride. The resulting inhibitor 10 shows a Ki value of 350 nM with no solubility problems under the aqueous assay conditions. The added aromatic group does not increase affinity as expected, suggesting that the nitrobenzyl group is not oriented optimally for π-stacking interactions at the active site.
Molecular rigidity is proposed to play a role in cell permeation by locking out access to clearance enzymes while retaining inhibitory potency against the target.19 Conformational studies on different OT substrates have shown that peptides constrained to an Asx-turn motif through side chain to main chain macrocyclization are more competent substrates for OT compared to the corresponding linear analogs.5 When applied to inhibitor design, the cyclized hexapeptide cyclo(Hex-Dab-Cys)-Thr-Val-Thr-Nph-NH2 was found to be twice as potent as its linear counterpart.4 Due to this observation, it was decided that the macrocycle motif be incorporated into the non-peptidic inhibitor that is the focus of this study. The cyclization, together with a modified C-terminal cap, 4-nitrophenethylamine instead of nitrobenzylamine, resulted in a 2-fold increase in enzyme affinity. This resulted in inhibitor 11 that displays very little peptidic character, yet high affinity for OT (Ki = 130 nM), one of the most hydrophobic and conformationally constrained inhibitors to demonstrate activity against this enzyme. This inhibitor fulfils some of the key prerequisites for bioavailability such as reduced amide bond character, increased hydrophobicity and rigidity as well as being smaller in size relative to the parent peptidic compound.
The synthesis of inhibitor 11 utilized an aldehyde-functionalized resin (2-(4-formyl-3-methoxyphenoxy)ethylpolystyrene) that installed the C-terminal 4-nitrophenethylamine cap via a reductive amination as illustrated in Scheme 1. The Alloc-protected 3-aminobenzoic acid moiety was coupled to the resulting secondary amine using HATU as a coupling reagent. Subsequent Alloc deprotection was then followed by coupling of threonine, 4-methoxytrityl (Mmt) protected cysteine and diaminobutyric acid residues using standard peptide synthesis procedures. The peptide was capped with 6-bromohexanoic acid, the Mmt protection of the cysteine side chain was removed with 1% TFA and the cyclization between the resultant thiolate and the 6-bromohexanoyl group was achieved in degassed DMF, using an excess of 1,1,3,3-tetramethylguanidine as base.20
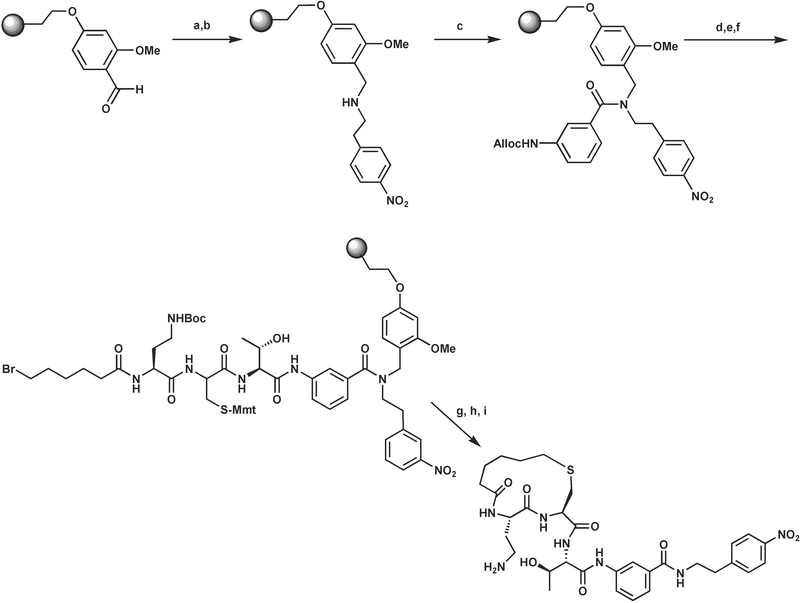 | ||
| Scheme 1 Synthesis of the cyclized inhibitor (11). Reagents and conditions: a). nitrophenethylamine, trimethyl orthoformate, dichloroethane; b). sodium triacetoxyborohydride; c). Alloc-3-aminobenzoic acid, HATU, N,N-diisopropylethylamine (DIEA) ; d). Pd(PPh3)4, phenylsilane; e). standard solid phase peptide coupling of Fmoc-Thr(t-Bu)-OH, Fmoc-Cys(Mmt)-OH, Fmoc-Dab(Boc)-OH; f). 6-bromohexanoic acid, benzotriazol-1-yloxytrispyrrolidinophosphonium hexafluorophosphate (PyBOP), DIEA; g) 1% TFA; h) 1,1,3,3-tetramethylguanidine; i) cleavage from resin (95% TFA). | ||
Since these compounds will eventually be tested as potential inhibitors of OT within mammalian cells, it was necessary to confirm that they inhibit the mammalian as well as the yeast (S. cerevisiae) OT. Hence assays were carried out as before but using pig liver microsomes instead of those derived from yeast, to determine the Ki for a representative mammalian OT. These studies show that there is no significant change in Ki values between the yeast and mammalian enzymes; hence none of the inhibitors are selective for either species although inhibition in yeast is generally more efficient. The results of these enzyme assays are summarized in Table 1.
The OT inhibitors developed in these studies will now be implemented into a cellular assay for OT function. The current focus of the studies is therefore the development of a convenient and high throughput assay system for analyzing OT inhibition in a stable mammalian cell line.
Discussion
Protein N-glycosylation is a crucial post-translational modification that is mediated by the enzyme oligosaccharyl transferase (OT). Due to its multimeric, membrane-bound nature, there is no structural data available on the enzyme, and any studies into its inhibition could provide valuable information regarding the structure and function of this elusive enzyme. In particular, inhibition of this enzyme within a cellular environment could be the means of obtaining knowledge on the immediate biological implications of N-linked glycosylation. Although potent inhibitors of OT exist, they all display peptidic character with poor proteolytic stability and membrane permeability, which render them unsuitable for in vivo studies. For this reason, this study describes efforts made towards the design and synthesis of a non-peptidic, hydrophobic OT inhibitor that still demonstrates affinity for the enzyme in the nanomolar range.In order to follow a rational approach to peptidomimetic inhibitor design, the hexapeptide Bz-Dab-Ala-Thr-Val-Thr-Nph-NH2 was used as the prototype. Studies on various Val-Thr dipeptide mimetics proved 3-aminobenzoic acid to be the most suitable isostere for this unit with a Ki value that is almost identical to the parent peptide. Further modifications conferred structural features on the inhibitor to improve its bioavailability. The amide bond character of the inhibitor was significantly reduced thus increasing the proteolytic stability of the molecules. Since smaller size is favored for cell permeation, modular deletion of units from the prototype inhibitor yielded compounds with low molecular weight. Hydrophobicity as measured by log P values is also an important factor in cell permeation. Removal of hydrogen-bond donor groups and the introduction of aromatic groups assist in increasing the log P values whilst maintaining solubility under the assay conditions. Finally, rigidity was introduced to the inhibitor by an aminobenzoic acid scaffold as well as cyclization into an Asx turn conformation. This study yielded a family of non-peptidic inhibitors that display varying degrees of hydrophobicity, size and rigidity as viable candidates for the in vivo inhibition of OT. In addition to providing potential bioavailable inhibitors of OT, the structure–activity relationships that result from this study provide valuable clues to the nature of the interactions occurring at the OT active site. The prime candidate for cellular studies is inhibitor 11, which displays several properties that are desired of in vivo inhibitors whilst maintaining a very high affinity for the enzyme (130 nM). The success of such a bioavailable inhibitor on a cellular level would enable studies into the effects of inhibiting the biosynthesis of N-linked glycoproteins in specific cellular situations.
Experimental
General procedures for solid-phase peptide synthesis
All peptides were synthesized by manual solid phase methods using Fmoc-PAL-PEG resin and using Fmoc (fluoren-9-ylmethoxycarbonyl) as the protecting group for the α-amino functionalities. Amino acids were coupled using PyBOP as the coupling reagent and were used with the following side-chain protection forms: Dab(Boc), Cys(Mmt), Thr(t-Bu). (All amino acid derivatives were obtained from commercial sources.) At the conclusion of the peptide synthesis, the peptides were capped with a large excess (10 eq.) of benzoic anhydride and pyridine. Cleavage from the resin was performed with trifluoroacetic acid (TFA) ∶ CH2Cl2 ∶ triisopropylsilane ∶ H2O (90 ∶ 5 ∶ 2.5 ∶ 2.5). All peptides were purified by preparative HPLC with a gradient of increasing acetonitrile–0.1% TFA (solvent A) in water–0.1% TFA (solvent B).Bz-Dab-Ala-Thr-(3-aminomethylbenzoyl)-Nph-NH2 (2)
Fmoc-PAL†–PEG–PS resin was used and the p-nitrophenylalanine residue was coupled using standard Fmoc-based peptide synthesis methods. After Fmoc-deprotection, Alloc protected 3-aminomethylbenzoic acid was added in excess (4 eq.) with HATU (4 eq.) and N,N-diisopropylethylamine (DIEA) (8 eq.). The coupling was allowed to proceed overnight, after which Alloc deprotection was performed with tetrakis(triphenylphosphine)palladium(0) (0.2 eq.) and phenylsilane (25 eq.) in CHCl2. Three 20 minute deprotections were performed under a N2 atmosphere. Fmoc-Thr(t-Bu)-OH coupling was performed using HATU as above, and the subsequent alanine and diaminobutyric acid residues were coupled using standard procedures. The peptide was capped with benzoic anhydride, cleaved from resin and purified by HPLC.HPLC tR = 21.60 min (C18, 7–100% B in 28 min).
ES MS for 2 (C35H43N8O9+): calcd 719.76; obsd [M]+ 719.5.
Bz-Dab-Ala-Thr-(4-aminomethylbenzoyl)-Nph-NH2 (3)
The standard procedure outlined above was used, where Alloc protected 4-aminomethylbenzoic acid was coupled to the p-nitrophenylalanine on PAL–PEG–PS resin.HPLC tR = 22.60 min (C18, 7–100% B in 28 min).
ES MS for 3 (C35H43N8O9+): calcd 719.76; obsd [M]+ 719.5.
Bz-Dab-Ala-Thr-(3-aminobenzoyl)-Nph-NH2 (4)
The standard procedure outlined above was used, where Alloc protected 3-aminobenzoic acid was coupled to the p-nitrophenylalanine on the PAL–PEG–PS resin.HPLC tR = 21.43 min (C18, 7–100% B in 28 min).
ES MS for 4 (C34H41N8O9+): calcd 705.74; obsd [M]+ 705.5.
Bz-Dab-Ala-Thr-(4-aminobenzoyl)-Nph-NH2 (5)
The standard procedure outlined above was used, where Alloc protected 4-aminobenzoic acid was coupled to the p-nitrophenylalanine on PAL–PEG–PS resin.HPLC tR = 19.22 min (C18, 7–100% B in 28 min).
ES MS for 5 (C34H41N8O9+): calcd 705.74; obsd [M]+ 705.1.
Synthesis of Bz-Dab-Nva-Thr-(3-aminobenzoyl)-Nph-NH2 (6)
The standard procedure for the synthesis of inhibitor 4 was applied here, except that Fmoc-Ala-OH was replaced with the commercially available unnatural amino acid Fmoc-Nva-OH (norvaline).HPLC tR = 20.26 min (C18, 7–100% B in 28 min).
ES MS for 6 (C36H45N8O9+): calcd 733.79; obsd [M]+ 733.3.
Synthesis of Bz-Dab-Nva-Thr-(3-aminobenzoyl)-NHCH2PhNO2 (7)
This peptide was synthesized on 2-(4-formyl-3-methoxyphenoxy)ethylpolystyrene (FMPE) resin available from Novabiochem, which is an aldehyde-functionalized resin. The resin was swollen in trimethyl orthoformate (TMOF) and dichloroethane (DCE) (3 ∶ 2) for 20 min. p-Nitrobenzylamine (10 eq.) was added to the resin and stirred under N2 for 3 hours, then 10 eq. of sodium triacetoxyborohydride was added and the mixture shaken overnight. Alloc-protected 3-aminobenzoic acid (4 eq.) was coupled to the resulting secondary amine using HATU and DIEA. Fmoc-Thr(t-Bu)-OH, Fmoc-Nva-OH and Fmoc-Dab(Boc)-OH were coupled using standard procedures. The peptide was capped with benzoic anhydride and purified by HPLC as before.HPLC tR = 23.85 min (C18, 7–100% B in 28 min).
ES MS for 7 (C34H41N7O8+): calcd 676.73; obsd [M]+ 676.4.
Synthesis of Bz(NO2)-Dab-Nva-Thr-(3-aminobenzoyl)-NH2 (8)
Alloc-protected 3-aminobenzoic acid was coupled to PAL–PEG–PS resin, followed by the coupling of Fmoc-Thr(t-Bu)-OH, Fmoc-Nva-OH and Fmoc-Dab(Boc)-OH using standard peptide synthesis procedure. The peptide was capped with 4-nitrobenzoic (2 eq.) acid, using PyBOP (2 eq.) as the coupling reagent and DIEA (4 eq.).HPLC tR = 23.02 min (C18, 7–100% B in 28 min).
ES MS for 8 (C27H36N7O8+): calcd 586.62; obsd [M]+ 586.4.
Synthesis of Bz-Dab-Nva-Thr-NH-CH2PhNO2 (9)
The procedure used was the same as for inhibitor 7 above, except that Fmoc-Thr(t-Bu)-OH was coupled to the resin after the reductive amination, instead of 3-aminobenzoic acid. The peptide was cleaved and purified as before.HPLC tR = 23.20 min (C18, 7–100% B in 28 min).
ES MS for 9 (C27H36N6O7+): calcd 557.62; obsd [M]+ 557.3.
Synthesis of Bz-Dab(4-nitrobenzyl)-Nva-Thr-(3-aminobenzoyl)-NHCH2CH2PhNO2 (10)
FMPE resin was used to couple the p-nitrobenzylamine as before. The residues, Alloc-3-aminobenzoic acid, Fmoc-Thr(t-Bu)-OH, Fmoc-Nva-OH and Fmoc-Dab(Alloc)-OH were coupled using the standard procedures outlined previously. The peptide was capped with benzoic anhydride and pyridine as before. The orthogonal Alloc protection on the Dab side chain was removed with Pd(PPh3)4 and phenylsilane as before and a mixture of DCE–TMOF (3 ∶ 2) with 10% acetic acid was added to the resin. p-Nitrobenzaldehyde (10 eq.) was added to the resin and shaken for 5 hours. The reagent mixture was drained and the resin washed with TMOF. NaBH(OAc3) (10 eq.) was added and the resin stirred under N2 overnight. The resin was washed with TMOF, DCE and dichloromethane and cleaved and purified as before.HPLC tR = 27.70 min (C18, 7–100% B in 28 min).
ES MS for 10 (C41H46N8O10): calcd 810.85; obsd [M]+ 811.3.
Synthesis of cyc[Hex-Dab-Cys]-Thr-(3-aminobenzoyl)-NHCH2CH2PhNO2 (11)
This peptide was synthesized on FMPE resin swollen with TMOF and DCE. p-Nitrophenethylamine (10 eq.) was added to the resin and stirred under N2 for 3 hours, then 10 eq. of sodium triacetoxyborohydride was added and the mixture shaken overnight. Alloc-protected 3-aminobenzoic acid (4 eq.) was coupled to the resulting secondary amine using HATU and DIEA. After Alloc deprotection, as before, Fmoc-Cys(Mmt)-OH and Fmoc-Dab(Boc)-OH were coupled using the standard peptide coupling procedures described above. The peptide was capped with 6-bromohexanoic acid and the Mmt protecting group was removed using 1% TFA in CH2Cl2. Cyclization was effected between the thiolate of the cysteine and the 6-bromohexanoyl group in degassed DMF using a large excess of 1,1,3,3-tetramethylguanidine as a base (24 hours). The peptide was cleaved and purified as before.HPLC tR = 23.90 min (C18, 7–100% B in 28 min).
ES MS for 11 (C41H46N8O10+): calcd 686.80; obsd [M]+ 686.3.
Determination of Ki values
In a typical yeast OT assay, [3H]DPPC (dolichol pyrophosphate chitobiose) (50000 dpm, 60 Ci mmol−1) was aliquoted from a chloroform–methanol stock solution into a microcentrifuge tube, and residual solvent was removed under a gentle stream of nitrogen. Mixtures of increasing amounts of inhibitor (in 10 µl of DMSO) and a constant amount of enzyme in buffer were incubated on ice for 30 min. This incubation time was required to ensure pre-equilibration of the inhibitor with the enzyme. The reaction was initiated by adding 10 μL of a solution of Bz-Asn-Leu-Thr-NHMe (4Km)
(2 mM for yeast and 10 mM for pig liver microsomes) substrate in DMSO. Aliquots (40 μL) of the reaction mixture were quenched after 2, 4, 6 and 8 min, and the aqueous phase containing the radioactive N-glycosylated peptide was extracted and quantified using a scintillation counter. Preliminary experiments employed a broad range of inhibitor concentrations for each peptide to afford a rough estimate of the IC50. Three concentrations were then selected to give between 30–70% inhibition. All experiments were run in duplicate and in each case the Ki was determined using the following equation: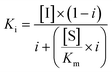 | (1) |
Acknowledgements
This research was supported by the NIH (GM 39334) and Merck Research Laboratories.References
- A. Varki, Glycobiology, 1993, 3, 97–130 Search PubMed.
- B. Imperiali and T. L. Hendrickson, Biorg. Med. Chem., 1995, 3, 1565–1578 Search PubMed.
- K. A. Presper and E. C. Heath, in The Enzymology of Post-translational Modification of Proteins;Academic Press, London, 1985, pp. 54–93 Search PubMed.
- T. L. Hendrickson, J. R. Spencer, M. Kato and B. Imperiali, J. Am. Chem. Soc., 1996, 118, 7636–7637 CrossRef CAS.
- B. Imperiali, K. L. Shannon and K. W. Rickert, J. Am. Chem. Soc., 1992, 114, 7942–7943 CrossRef CAS.
- C. Kellenberger, T. L. Hendrickson and B. Imperiali, Biochemistry, 1997, 36, 12554–12559 CrossRef CAS.
- S. Peluso, M. de L. Ufret and B. Imperiali, unpublished results.
- A. P. Elbein, Trends Biochem. Sci., 1981, 6, 291–293 CrossRef.
- Y. Gavel and G. von Heijne, Protein Eng., 1990, 3, 433–442 CAS.
- E. A. Lunney, K. S. Para, J. R. Rubin, C. Humblet, J. Hl Fergus, J. S. Marks and T. K. Sawyer, J. Am. Chem. Soc, 1997, 119, 12471–12476 CrossRef CAS.
- B. Peschke, K. Madsen, B. S. Hansen and N. L. Johansen, Biorg. Med. Chem. Lett., 1997, 7, 1969–1972 Search PubMed.
- Y. Qian, M. A. Blaskovich, M. Saleem, C. M. Seong, S. P. Wathen, A. D. Hamilton and S. M. Sebti, J. Biol. Chem., 1994, 269, 12410–12413 CAS.
- B. Imperiali and K. L. Shannon, Biochemistry, 1991, 30, 4374–4380 CrossRef CAS.
- (a) C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef; (b) B. Imperiali and S. Peluso, unpublished results.
- ACD/LogP software, Advanced Chemistry Development Inc, Toronto, Ontario, Canada, 2001 (http://www.acdlabs.com).
- P. E. Johnson, P. Tomme, M. D. Joshi and L. P. McIntosh, Biochemistry, 1996, 35, 13895–13906 CrossRef CAS.
- C. Wiesmann, W. Hengstenberg and G. E. Schulz, J. Mol. Biol., 1997, 269, 851–860 CrossRef CAS.
- M. de L. Ufret and B. Imperiali, Biorg. Med. Chem. Lett., 2000, 10, 281–284 Search PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- A. A. Virgilio and J. A. Ellman, J. Am. Chem. Soc., 1994, 116, 11580–11581 CrossRef CAS.
Footnote |
| † PAL = 5-[4-(9-fluorenylmethoxycarbonylamino)-3,5-dimethoxyphenoxy]pentanoic acid. |
| This journal is © The Royal Society of Chemistry 2003 |