Antisense oligonuclotides with oxetane-constrained cytidine enhance heteroduplex stability, and elicit satisfactory RNase H response as well as showing improved resistance to both exo and endonucleases†
Pushpangadan. I.
Pradeepkumar
,
Nariman V.
Amirkhanov
and
Jyoti
Chattopadhyaya
*
Department of Bioorganic Chemistry, Biomedical Center, University of Uppsala, Box 581, S-751 23 Uppsala, Sweden. E-mail: jyoti@boc.uu.se; Fax: +4618554495
First published on 26th November 2002
Abstract
Antisense oligonucleotides (AONs) with single and double oxetane ![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) modifications [1′,2′-oxetane constrained cytidine, 1-(1′,3′-O-anhydro-β-D-psicofuranosyl)cytosine] have been evaluated, in comparison with the corresponding
modifications [1′,2′-oxetane constrained cytidine, 1-(1′,3′-O-anhydro-β-D-psicofuranosyl)cytosine] have been evaluated, in comparison with the corresponding ![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) -modified AONs, for their antisense potentials by targeting to a 15mer complementary RNA. Although the modified mixmer AONs show ∼3 °C drop per modification in melting temperature (Tm) of their hybrid AON–RNA duplexes, they are found to be good substrates for RNase H, in comparison with the native AON–RNA duplex. An AON with double modifications along with 3′-DPPZ (dipyridophenazine) conjugation shows the Tm of the hybrid duplexes as high as that of the native, and the RNase H activity as good as its unconjugated counterpart. A detailed Michaelis–Menten kinetic analysis of RNase H cleavage showed that the single and double modified AON–RNA duplexes as well as double modifications along with 3′-DPPZ have catalytic activities (kcat) close to the native. However, the RNase H binding affinity (1/Km) showed a slight decrease with increase in the number of modifications, which results in less effective enzyme activity (kcat/Km) for modified AON–RNA duplexes. All oxetane modified AON–RNA hybrids showed a correlation of Tm with the 1/Km, Vmax, or Vmax/Km. The modified AONs (with 3′-DPPZ), as in the counterpart, showed an enhanced tolerance towards the endonuclease and exonuclease degradation compared to the native (the oxetane-sugar and the DPPZ based AONs are non-toxic to K562 cell growth, ref. 18). Thus a balance has been found between exo and endonuclease stability vis-a-vis thermostability of the heteroduplex and the RNase H recruitment capability and cleavage with the oxetane-constrained cytidine incorporated AONs as potential antisense candidates with a fully phosphate backbone for further biological assessment.
-modified AONs, for their antisense potentials by targeting to a 15mer complementary RNA. Although the modified mixmer AONs show ∼3 °C drop per modification in melting temperature (Tm) of their hybrid AON–RNA duplexes, they are found to be good substrates for RNase H, in comparison with the native AON–RNA duplex. An AON with double modifications along with 3′-DPPZ (dipyridophenazine) conjugation shows the Tm of the hybrid duplexes as high as that of the native, and the RNase H activity as good as its unconjugated counterpart. A detailed Michaelis–Menten kinetic analysis of RNase H cleavage showed that the single and double modified AON–RNA duplexes as well as double modifications along with 3′-DPPZ have catalytic activities (kcat) close to the native. However, the RNase H binding affinity (1/Km) showed a slight decrease with increase in the number of modifications, which results in less effective enzyme activity (kcat/Km) for modified AON–RNA duplexes. All oxetane modified AON–RNA hybrids showed a correlation of Tm with the 1/Km, Vmax, or Vmax/Km. The modified AONs (with 3′-DPPZ), as in the counterpart, showed an enhanced tolerance towards the endonuclease and exonuclease degradation compared to the native (the oxetane-sugar and the DPPZ based AONs are non-toxic to K562 cell growth, ref. 18). Thus a balance has been found between exo and endonuclease stability vis-a-vis thermostability of the heteroduplex and the RNase H recruitment capability and cleavage with the oxetane-constrained cytidine incorporated AONs as potential antisense candidates with a fully phosphate backbone for further biological assessment.
Introduction
Antisense Oligonucleotides (AONs) down-regulate gene expression (Fig. 1) by complexing with the target mRNA in two different ways:1 (1) Translational arrest of the message by tight binding to the target mRNA (steric blocking of ribosal machinery), which requires at least stoichiometric amounts of AON to the target. (2) The catalytic cleavage of the target mRNA in the AON–RNA hybrid by the activation of RNase H, which is a ubiquitous enzyme in all cells.2 The latter strategy based on RNase H activation has intrinsic advantages over the steric blocking because it requires only a relatively small amount of AON, compared to the former, causing permanent destruction of the translated message with catalytic turnover, provided the AON has enough life time (stability) in cells.1 The RNase H insensitive AONs employed so far for the translational arrest have relatively high melting temperature (Tm), which may bind to other targets with one or two mismatches owing to poor discrimination. Hence, considerable attention has been directed in the design, synthesis and evaluation of AONs which are capable to recruit RNase H keeping the target affinity close to the unmodified duplex.2b The first generation AONs, the nucleolytically stable phosphorothioates, although working by utilizing the RNase H mechanism, its low binding affinity to the target RNA, less sequence specificity and strong affinity to heparin-binding proteins3 (one to three order magnitude higher than the corresponding phosphodiester oligonucleotide) generate plenty of non-antisense effects, which in turn limit its therapeutic potential.3 Chimeric oligonucleotides (gapmers) based on a phosphorothioate backbone (second generation AONs) with various 2′-O-akyl modifications4,5 at the 3′- and 5′-ends and 4 to 6 unmodified nucleotides in the middle, although showing enhanced binding affinity, fail to keep the RNase H cleavage as good as the native phosphorothioate or phosphodiester counterparts.5 An increment of gapsize has been shown to increase the RNase H activity;6 however, it has the disadvantage that these gapmers are more exposed to endonuclease degradations.7 Chimeric AONs based on methylphosphonates8 and AONs based on boranophosphates9 show enhanced RNase H activity but poor affinity to the RNA target.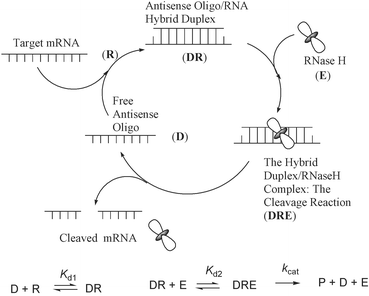 | ||
| Fig. 1 The catalytic RNase H promoted cleavage of the target mRNA through the formation of an antisense oligonucleotide–RNA hybrid duplex. The kinetic scheme of the RNase H hydrolysis is shown in the bottom part of the cartoon, where D is AON (antisense oligo); R is the target RNA; Kd1 is the equilibrium constant of dissociation of the heteroduplex DR; Kd2 is equilibrium constant of dissociation of the substrate–enzyme complex DRE. | ||
The search for an ideal chemistry (without changing the phosphodiester backbone) which is capable of giving bona fide antisense action has intensified in the past decade: oligos incorporating conformationally (3′-endo) constrained nucleosides10,11 show enhanced stability of the resulting AON–RNA hybrid duplex. The enhanced stability of conformationally constrained AONs is attributed to the formation of a rigid A type AON–RNA duplex (reminiscent of an RNA–RNA type duplex) which accounts for the loss of RNAse H recruiting capability.2b,4
Within the family of conformationally constrained nucleotides, we have recently12 reported the RNase H recruiting capabilities of north-east constrained oxetane thymidine,
[1-(1′,3′-O-anhydro-β-D-psicofuranosyl)thymine, Fig. 2] modified mixmer AONs where we have shown that, although Tm drops by ∼6 °C per modification, the RNase H cleavage of the target RNA remain intact or increased compared to the wild type hetroduplex, under enzyme saturation conditions12c
(enzyme is completely saturated by substrate, AON–RNA duplex). Although the modification did not alter the global helical structure of the modified AON–RNA duplex in comparison with the wild type duplex12
(as revealed by CD spectra), the target RNA strand in the AON–RNA duplex was resistant to RNase H promoted cleavage, up to 5 nucleotides towards the 3′-end from the site opposite to the introduction, because of subtle alteration of the microenvironment as a result of incorporation.12
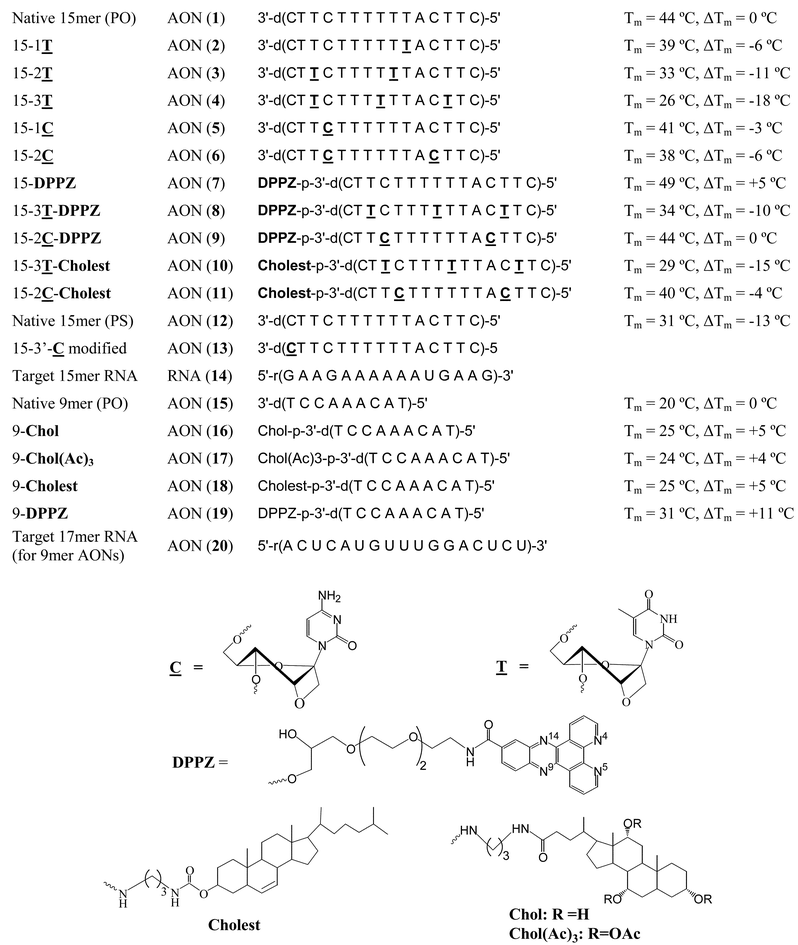 | ||
| Fig. 2 Sequences of various AONs and their target RNAs. The melting temperature (Tm) and the difference in Tm (ΔTm) with respect to the native phosphodiester AON 1 for 15mer AONs or AON 15 for 9mer AONs are also shown. | ||
A detailed Michaelis–Menten type kinetic analysis of triple modified AON–RNA hybrid has also been carried out13 to understand the mechanism of RNAse H recruitment, substrate recognition, specificity and cleavage vis-à-vis conformational preorganization of the hybrid duplex. It has emerged that the Vmax and the Km were respectively ∼2 and ∼10 times more for the triple oxetane modified AON–RNA duplex than those for the native counterpart, which shows the increase in the catalytic activity of the oxetane modified AON–RNA duplex almost 2 fold owing to the decreased affinity of the substrate toward the enzyme.13
The single, double and triple oxetane modified AONs have been shown to have 3 to 5 fold protection12c towards endonuclease which augments its antisense potential through the RNase H assisted pathway. We have also shown that the Tm loss, owing to oxetane modifications, can be regained (+8 °C) by the introduction of a DPPZ14
(dipyridophenazine) moiety at the 3′-end of the AON without sacrificing RNase H eliciting and cleavage power.12c
The 3′-DPPZ moiety also renders the AONs stable towards 3′-exonucleases.12c,14b Interestingly, the tethering of DPPZ in a native 9mer AON has been shown15 to increase the effective enzyme activity (Vmax/Km) of the RNase H by six fold compared to the native 9mer AON and two fold compared to the high hydrophobic 3′-tethers like cholesterol15,16a–e and cholic acid17 because of the high binding affinity (1/Km) of DPPZ modified AON–RNA duplex to the enzyme.15 It has also been found that the introduction of DPPZ moiety to oxetane modified mixmer AONs to be non-toxic in various cell culture assays.18
In view of the novel antisense properties of oxetane modified AONs containing modifications, we here report the synthesis of oxetane
[1-(1′, 3′-O-anhydro-β-D-psicofuranosyl)cytosine, (24) in Scheme 1] modified AONs (5, 6, 13, Fig. 2) and their 3′-DPPZ (9) and cholesterol 11 conjugated AONs. We also report the hybridization properties of these AONs to the target RNA 14, as well as their exo and endonuclease resistance properties along with a detailed kinetic analysis of RNase H cleavage of single and double modified AON–RNA hybrid duplexes in comparison with the corresponding modified AON (2–4)–RNA (14) and the native 15mer AON (1)–RNA (14) duplexes.
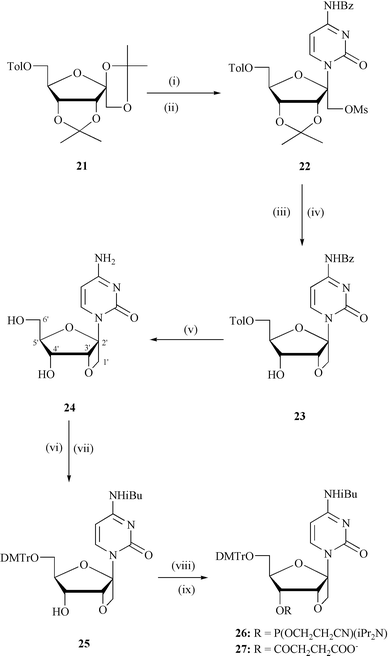 | ||
| Scheme 1 Reagents and conditions: (i) persilylated N4-Bz-cytosine, acetonitrile, TMsOTf, 0 °C to rt, 18 h; (ii) MsCl, pyridine, 4 °C, 12 h; (iii) 90% TFA–water, rt, 20 min, (iv) NaH, DMF, 4 °C, 9 h; (v) methanolic NH3, rt, 48 h ; (vi) TMsCl, pyridine, rt, 30 min followed by iBuCl, rt, 3 h; (vii) DMTrCl, pyridine, rt, 12 h; (viii) (2-cyanoethoxy)bis(N,N- diisopropylamino)phosphine, N,N-diisopropylammonium tetrazolide, DCM, 12 h for 26; (ix) succinic anhyride, DMAP, DCM, 4 h, rt, for 27. Abbreviations: Tol = 4-methylbenzoyl, Bz = benzoyl, TMSOTf = trimethylsilyl trifluoromethane sulfonate, rt = room temperature, Ms = methanesulfonyl, DMTr = dimethoxytrityl, Et = ethyl, TFA = trifluroacetic acid, iBu = isobutyryl, DCM = dichloromethane, TMS = trimethylsilyl, DMAP = dimethylaminopyridine. | ||
Results
(1) Endonuclease (DNase I) stability
When tested for endonuclase stability using DNase I, the single modified AON 5 showed more than 3 fold protection (60% left after 1 h), while the double modified AON 6 gave 4–5 fold protection (93% left), whereas two modifications with 3′-terminal DPPZ showed also 4–5 fold protection (98% left) compared with native AON 1
(24% left after 1 h)
(Fig. 3). The stability achieved by double modifications was comparable to, or better than, that of the phosphorothioate AON 12)(88% of AON left after 1 h incubation)
(Fig. 3). The similar behaviour of endonuclease tolerence has been reported for various modified AONs.12c
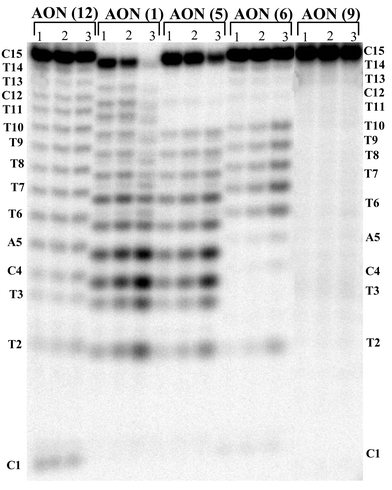 | ||
| Fig. 3 PAGE analysis of the DNase I degradation of AON 1, 5, 6, 9 and 12. Lanes 1 to 3 represent the time points after 30 min., 1 h and 3 h of reaction. The % of AON left after 1 h of incubation: 24% of AON 1, 88% of AON 12, 60% of AON 5, 93% of AON 6 and 98% of AON 9. Note a comparison of relative cleavage after 3 h that AON 9 shows even more stability to endonuclease (94%) than phosphorothioate AON 12 (82%). | ||
(2) 3′-Exonuclase stability
We have subsequently examined the effect of 3′-exonuclase (snake venom phosphodiesterase, SVPDE) on modification. For this, we have incorporated one modification at the 3′ end of AON 13. After 15 min, the native AON 1 with the phosphodiester backbone was found to be completely hydrolysed by the enzyme, whereas 60% of 3′
modified AON was intact after 1 h (Fig. 4). On the other hand, ca. 20% of interior double modified AON 6 was left undigested by SVPDE after 1 h. (Fig. 4). As reported earlier, complete exonuclase resistance was achieved with DPPZ moiety at the 3′-end (97% of AON 9 left after 1 h of incubation), which was comparable to that of PS AON (96% of AON left after 1 h of incubation)
(Fig. 4).
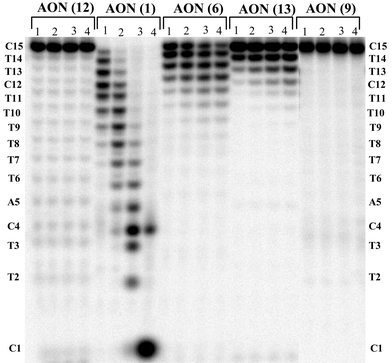 | ||
| Fig. 4 PAGE analysis of the snake venom phosphodiesterase (SVPDE) degradation of AONs 1, 6, 9, 12 and 13. Lanes 1 to 4 represent the time points after 15 min, 30 min, 1 h and 3 h of reaction. The % of AON left after 1 h of incubation with enzyme: 0% of AON 1, 95% of AON 12, 20% of AON 6, 60% of AON 13 and 97% of AON 9. | ||
(3) Human serum stability
The stability of AONs wa also checked in human serum, where the major degrading enzymes present are 3′-exonucleases.7 All DPPZ modified AONs (7–9) (62–68 % AON left after 1 h) showed comparable stabilities to that of PS AON (63% of AON left after 1 h) (Fig. 5), whereas the native AON was fully digested under identical conditions in 15 min.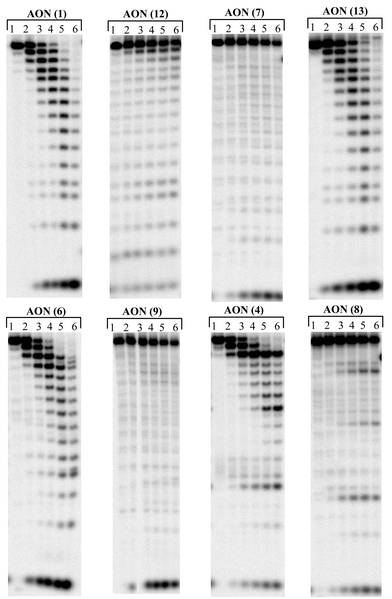 | ||
| Fig. 5 PAGE analysis of the degradation of AON 1, 12, 7, 13, 6, 9, 4 and 8 in human serum. Lanes 1 to 6 represents the time points after 0, 5, 15 and 30 min, 1 h and 2 h of reaction. The % of AON left after 1 h of incubation: 0% of AON 1, 63% of AON 12, 62% of AON 7, 0% of AON 13, 0% of AON 6, 64% of AON 9, 0% of AON 4 and 68% of AON 8. | ||
(4) Binding affinity of the oxetane-modified antisense oligos to the target RNA
The incorporation of single modification as in AON 5–RNA 14 causes 3 °C drop in Tm
(Tm
= 41 °C) with respect to the unmodified AON 1–RNA 14 hybrid duplex (Tm
= 44 °C, Fig. 1). The incorporation of two modifications caused 6 °C drop in Tm of the AON 6–RNA duplex. This should be compared with the corresponding modification12, which leads to a ∼6 °C drop in Tm per modification. However, the loss in Tm owing to two modifications, for example, is fully restored to the level of native hybrid duplex by the introduction of a dipyridophenazine (DPPZ) moiety at the 3′ end (Tm
= 44 °C), as in AON 9. It has also been found that the introduction of DPPZ moiety to oxetane and oxetane modified mixmer AONs to be non-toxic in various cell culture assays.18 Conjugation of the cholesterol15,16 moiety at the 3′-end of the and modified oxetane also increases Tm of the hybrid AON–RNA duplexes in comparison to their unconjugated counterparts (Fig. 2). However, increase in Tm in the case of the cholesterol conjugated AONs (10 and 11)
(ΔTm
=
∼2.5 °C) is less than in the case of DPPZ conjugated ones (8 and 9)
(ΔTm
= 5 to 8 °C). Thus incorporation of modifications has clear advantages over modifications in the construction of AONs as far as the target RNA affinity is concerned.
(5) RNase H cleavage pattern of complementary RNA in heteroduplexes containing ![[C with combining low line]](https://www.rsc.org/images/entities/h3_b_char_0043_0332.gif) oxetane modified AONs and their DPPZ and cholesterol conjugates
oxetane modified AONs and their DPPZ and cholesterol conjugates
The single or double modified AON (5 or 6)–RNA (14) hybrids were found to be as good a substrate for RNase H as found for the modified AONs12
(Fig. 6, see also Fig. S1-S3, S4-S14 in supplementary information†). In all the modified hybrid duplexes including DPPZ and cholesterol conjugates, 5 nucleotides towards the 3′-end from the site opposite to the introduction were found to be insensitive towards RNase H promoted cleavage (Fig. 6, see also Fig. S1-S3, S15-S23), presumably owing to adoption of a local pseudo RNA–RNA type conformational character.12 This 5-nucleotide footprint generated owing to RNase H resistance to cleavage reaction is identical to what has been earlier found for modified AONs12
(see also Fig. S4-S14). Conjugation of the DPPZ to triple oxetane modified AON 8 results in an additional cleavage site compared to the unconjugated counterpart AON12c4
(see also Figs S15-S23). No such additional cleavage site was found in the case of the DPPZ conjugated double modified AON 9 compared to the unconjugated counterpart AON 6
(Fig. 6, see also Fig. S1-3). Conjugation of cholesterol moiety at the 3′-end of the or modified oxetane does not change RNA cleavage sites (Fig. 6, see also Fig. S15–23).
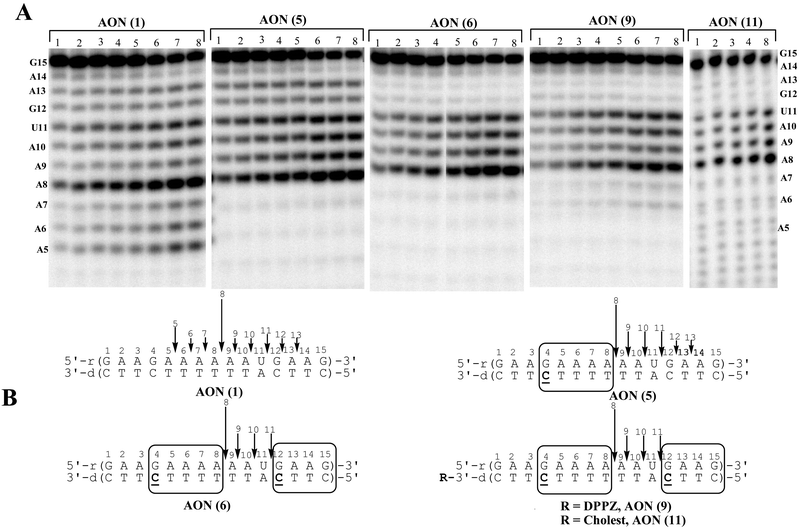 | ||
| Fig. 6
(A): Autoradiograms of 20% denaturing PAGE, showing the cleavage kinetics of 5′-32P-labelled target RNA 14 by RNase H1 in the native AON 1–RNA 14 and oxetane modified AON (5,6,9,11)–RNA 14 hybrid duplexes. Lanes 1 to 8 represents the aliquots of the digest taken after 2, 3, 4, 5, 6, 20, 40 and 60 min respectively. Conditions of cleavage reaction: RNA (0.04 µM) and AONs (5 µM) in buffer, containing 20 mM Tris-HCl (pH 8.0), 20 mM KCl, 10 mM MgCl2 and 0.1 mM DTT at 21 °C, 0.06 U of RNase H for AON 1, 5, 6, 9 and 0.12 U of RNase H for AON 11. Total reaction volume is 30 µl. The % of RNA left after 1 h: 38% of AON 1, 36% of AON 5, 54% of AON 6 56% of AON 9 and 32% of AON 11. (B): RNase H cleavage pattern of hybrid duplexes. The vertical arrows show the RNA cleavage sites and relative length of an arrow shows the relative extent of cleavage at that site. | ||
(6) Michaelis–Menten kinetics of modified AONs and their DPPZ and cholesterol conjugates and its comparison with the ![[T with combining low line]](https://www.rsc.org/images/entities/h3_b_char_0054_0332.gif) modified and native AONs
modified and native AONs
To understand the substrate specificity (Km, Vmax and kcat) of RNA cleavage by RNase H in modified AON–RNA hybrids, a detailed kinetic analysis of RNase H promoted cleavage of the RNA target in modified hybrid duplexes has been carried out, and subsequently compared with modified AONs 2–4 and the native AON 1
(Fig. 6 and see also Figs S1–23†). The initial velocities (v0) were calculated at different RNA concentration (at least in five different RNA concentrations, ([S0])), ranging from 0.008 to 3 µM at 5 µM AON under RNA 14 saturation conditions by AONs.13,15
The plot of v0 as a function of [S0] (Fig. 7) (materials and methods section) yielded the Michaelis–Menten parameters,19Km, Vmax and kcat (Table 1).
| AONs | T m/°C | V max/10−2 µM min−1 | K m/10−2 µM | k cat/min−1 | (Vmax/Km)/min−1 | (kcat/Km)/µM−1 min−1 | Relative (kcat/Km) |
|---|---|---|---|---|---|---|---|
a
Vmax
=
kcat
×
E0
(E0
= 0.06 U per 30 µl = 2 × 10−3 U µl−1
= 2.26757 × 10−4
µM ), specific activity = 420![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U/mg or 2.38 × 10−6 mg U−1
= 1.13386 × 10−13 moles per unit, MW = 21000 g mol−1. 000 U/mg or 2.38 × 10−6 mg U−1
= 1.13386 × 10−13 moles per unit, MW = 21000 g mol−1.
|
|||||||
| Native 15mer (1) | 44 | 1.9 ± 0.3 | 5.7 ± 1.1 | 84.4 ± 11.5 | 0.34 | 1476.9 | 1 |
|
-Oxetane modified AONs |
|||||||
| 15-1T (2) | 39 | 2.0 ± 0.1 | 8.3 ± 2.2 | 90.0 ± 5.3 | 0.25 | 1083.8 | 0.73 |
| 15-2T (3) | 33 | 2.4 ± 0.1 | 11.2 ± 1.7 | 104.9 ± 4.0 | 0.21 | 934.5 | 0.63 |
| 15-3T (4) | 26 | 3.4 ± 0.3 | 40 ± 2.9 | 165.8 ± 11.2 | 0.10 | 446.3 | 0.30 |
|
-Oxetane modified AONs |
|||||||
| 15-1C (5) | 41 | 2.0 ± 0.1 | 6.4 ± 0.9 | 88.6 ± 3.5 | 0.31 | 1376.3 | 0.93 |
| 15-2C (6) | 38 | 1.8 ± 0.1 | 9.6 ± 1.9 | 79.8 ± 4.9 | 0.19 | 828.8 | 0.56 |
| 3′-DPPZ conjugated AONs | |||||||
| 15-DPPZ (7) | 49 | 0.73± 0.06 | 1.5± 0.6 | 32.2 ± 2.6 | 0.50 | 2220 | 1.50 |
| 15-3T-DPPZ (8) | 34 | 1.4 ± 0.2 | 10.4 ± 3.7 | 62.2 ± 7.1 | 0.14 | 597.8 | 0.40 |
| 15-2C-DPPZ (9) | 44 | 1.9 ± 0.1 | 12.6 ± 2.1 | 81.6 ± 4.4 | 0.15 | 650.0 | 0.44 |
| 3′-Cholesterol conjugated AONs | |||||||
| 15-3T-Cholest (10) | 29 | 5.1 ± 0.4 | 66.3 ± 9.0 | 225.8 ± 15.9 | 0.08 | 340.7 | 0.23 |
| 15-2C-Cholest (11) | 40 | 3.5 ± 0.3 | 30.9 ± 6.6 | 153.5 ± 13.2 | 0.11 | 496.8 | 0.34 |
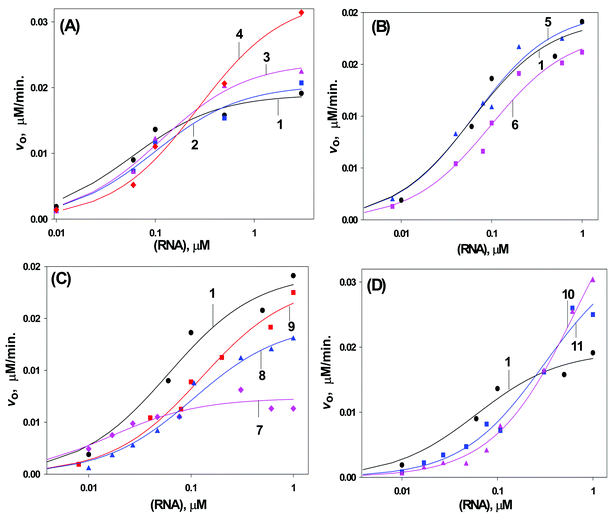 | ||
| Fig. 7 Initial velocity of the hydrolysis of the 15mer target RNA 14)in the AON (1–11)–RNA hybrids by RNase H as a function of the RNA concentration (logarithamic scale). Curves 1–11 correspond to the hybrid duplex formed by AONs 1–11 respectively. A: for unconjugated oxetane modified AONs 2–4. B: for unconjugated oxetane modified AONs 5 & 6. C: for DPPZ conjugated AONs 7–9. D: for cholesterol conjugated AONs 10, 11. Conditions of cleavage reaction: AONs (5 µM) and RNA 14 in buffer, containing 60 mMTris-HCl (pH 7.5), 60 mM KCl, 10 mM MgCl2 and 1 mM DTT at 21 °C, 0.06 U of RNase H. Total reaction volume is 30 µl. | ||
These data showed that the single and double modified AON (5 and 6)–RNA 14 duplexes have enzyme catalytic activity (Vmax or kcat) close to the native AON 1–RNA 14 duplexes (Table 1, Fig. 8A). However, the enzyme binding affinity (1/Km) showed a slight decrease with increase in number of modifications, which results in less effective enzyme activity (kcat/Km) for modified AON–RNA duplexes (Table 1, Figs. 8B and 8C). On the other hand, the kinetic data for modified AON (2–4)–RNA 14 duplexes showed that the enzyme catalytic activity (Vmax) increases, whereas the binding affinity (1/Km) decreases with the increase in the number of modifications, which gives less effective enzyme activity (kcat/Km) compared to the native counterpart (Table 1, Fig. 8).
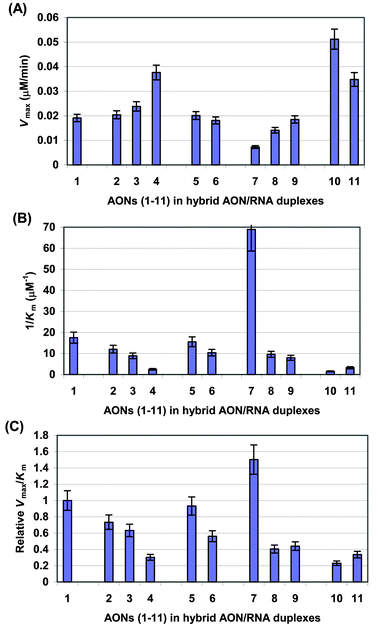 | ||
| Fig. 8 Bar graphs showing the Vmax
(A), 1/Km
(B) and relative Vmax/Km
(C) values for the RNase H promoted RNA hydrolysis in AON–RNA hybrid duplexes for native 15mer AON 1, and oxetane modified AONs 2–6, and their DPPZ and cholesterol conjugates 7–11. | ||
The introduction of a DPPZ moiety to the 3′-end of the double modified AON (9) does not change the catalytic activity (Vmax) of the enzyme in comparison with the native AON 1
(Table 1, Fig. 8A). It decreases the 1/Km and kcat/Km in comparison with the native AON (1)
(Table 1 and Fig. 8B, C). The introduction of a DPPZ moiety to the 3′-end of the triple modified AONs 8 slightly decreases Vmax value and the 1/Km of the enzyme, yielding a two fold lower kcat/Km value in comparison with the native counter part (Table 1 and Figs. 8A–C). In contrast to DPPZ conjugated AONs, conjugation of a cholesterol residue to the AONs (10 and 11) increases the Vmax values; however, the large drop in 1/Km results in the reduction of kcat/Km in comparison with the native AON 1–RNA duplex (Table 1, Fig. 8).
(7) Correlation of Michaelis–Menten parameters with the thermostability of the heteroduplexes.
With the exception of the 3′-conjugated counterparts, all and modified AON–RNA hybrids have shown a linear correlation of the Tm of the hybrid duplexes with [log(1/Km)], [log(Vmax)], or [log(Vmax/Km)]
(Fig. 9). The enzyme binding affinity (Fig. 9B) and effective enzyme activity (Fig. 9C) increase with increase of Tm, however, the catalytic activity (Fig. 9A) was found to decrease with rise in thermostability of the hybrid duplexes.
![The plot of catalytic activity [log (Vmax)]
(A), binding affinity [log (1/Km)]
(B) and effective activity [log(Vmax/Km)]
(C) of RNase H versus thermostability (Tm) of the C̲ and T̲ modified AON–RNA duplexes and their DPPZ and cholesterol conjugates. Linear correlation was found for all unconjugated AONs (1–6)–RNA duplexes, except for the DPPZ (■) and cholesterol (▲) conjugates 7–11. Note, those AONs (7–11) marked with (■) and (▲) are not included in the correlation equation. Correlation coefficients (R2) for the plots are: 0.92 for (A), 0.82 for (B) and 0.88 for (C).](/image/article/2003/OB/b210163g/b210163g-f9.gif) | ||
| Fig. 9 The plot of catalytic activity [log (Vmax)]
(A), binding affinity [log (1/Km)]
(B) and effective activity [log(Vmax/Km)]
(C) of RNase H versus thermostability (Tm) of the and modified AON–RNA duplexes and their DPPZ and cholesterol conjugates. Linear correlation was found for all unconjugated AONs (1–6)–RNA duplexes, except for the DPPZ (■) and cholesterol (▲) conjugates 7–11. Note, those AONs (7–11) marked with (■) and (▲) are not included in the correlation equation. Correlation coefficients (R2) for the plots are: 0.92 for (A), 0.82 for (B) and 0.88 for (C). | ||
(8) Synthesis of oxetane building blocks
1-(1′,3′-O-Anhydro-β-D-psicofuranosyl)cytosine (24) in Scheme 1 was prepared from the sugar, 6′-O-toluoyl-1,2:3,4-di-O-isopropylidene-β-D-psicofuranose12b
(21)
[Scheme 1]. Compound 21 was coupled with N,O-bis(trimethylsilyl)-N4-benzoylcytosine in the presence of TMSOTf as Lewis acid and acetonitrile as solvent to furnish an inseparable anomeric mixture of the protected psiconucleosides, which on mesylation gave 22 in 66% yield . Removal of the isopropylidene group from 22 using 90% CF3COOH in water at room temperature and subsequent cyclization with NaH in DMF gave 23 in moderate yield (49%). Detoluoylation of 23 furnished the desired bicyclic nucleoside 24
(96%) and its β configuration at anomeric carbon was confirmed by NOE measurements (Experimental section). The nucleoside was converted into the corresponding phosphoramidite building block 26
(93%)
viaN4-isobutyryl-6′-O-(4,4′-dimethoxytrityl) protected derivative 25 for incorporation of residues in the AONs (5, 6 and 9) and the 4′-succinate 27 for the preparation of CPG solid support, which was used for the synthesis of AON 13.
Discussion
Several attempts have been made to engineer nuclease resistant antisense oligonucleotides, which both show high thermodynamic stability when bound to the complementary RNA, and are capable of recruiting RNase H as well as the native AON.2b,4,10,11 Many of the conformationally constrained nucleotides in fact have been shown to enhance the stability of the modified AON–RNA heteroduplex, but failed to show any RNase H recruiting capability.2b,4,11 It has emerged that (a) the optimal minor grove width (close to A type duplex,2b 8.9–10.8 Å), (b) the flexibility of the AON–RNA duplex, as well as (c) minimal stereochemical interference by the antisense strand in the minor grove (both structural, chemical as well as hydration points of view) of the heteroduplex–enzyme complex dictate the optimal RNase H activity.2b The intrinsic flexibility of the hybrid duplex enables it to form a complex with RNase H, resulting in a more stabilized structure, which allows the reactive amino acid residues at the active site of RNase H to stereochemically position themselves at the minor groove of the duplex for the cleavage reaction to take place. Most of the north-constrained nucleosides were not able to fulfill these requirements.2,4,11b This was clearly evident with locked nucleic acids (LNA),10e,b,11b which showed high affinity to target RNA, but the mixmer of LNA–RNA heteroduplexes failed to elicit any RNase H activity.20,21 On the other hand, if in a gapmer the deoxynucleotide gaps between flanking LNAs are increased (6–10 native deoxynucleotide gaps), the flexibility in the deoxynucleotide gap is enhanced, which elicits the RNase H activity.6 We have earlier observed12 such footprints in the RNA cleavage pattern by RNase H owing to the altered AON–RNA conformation in the conformationally constrained oxetane-modified heteroduplex. It should be noted that the fifth internucleoside phosphate residue from the 5′-end of the complementary RNA strand is cleaved by RNase H in the case with our oxetane based AONs, whereas it is at the seventh phosphate where the enzymatic cleavage takes place with LNA incorporated AONs.6The oxetane based AONs have more affinity to the target RNA than oxetane modified AONs (Fig. 2). AONs with two modifications in conjunction with a tethered 3′-DPPZ group have a phosphodiester (PO) backbone, as in AON 9, which has an almost identical hybrid AON 9–RNA 14 stability to that of the native duplex AON 1–RNA 14; both have Tms of 44 °C (Fig. 2), thereby showing that the DPPZ conjugated modified AON (9) and the native counterpart (1) should be able to bind to the RNA target with equal specificity. Interestingly, DPPZ conjugated modified AON also fulfils the criteria for exo/endonucleolytic stability and RNase H recruitment capability, they are also found to be non-toxic.18 The summary of comparative Michaelis–Menten kinetic studies of and modified AONs in comparison with that of the native AON has shed light on the RNase H recruiting capability of AONs in depth in terms of substrate affinity and cleavage activity (Table 1).
(A) Unconjugated and oxetane modified AONs
The single modified AON 5–RNA hybrid elicits an RNase H response in a similar manner (Table 1, Fig. 6, 7B and 8) to that of the native in both low ([RNA]
≤ 0.01 µM) and high ([RNA]
≥ 1.0 µM) RNA concentration range (Fig. 6, 7B, 8A–C, see also Figs S1-S3, S4-S14 in the supplementary information†). On the other hand, the double modified AON 6–RNA hybrid elicits an RNase H response that is lower at low RNA concentration, [RNA]
≤ 0.01 µM)
(Figs. 7B, 8C), than the native whereas it is very comparable at high RNA concentration (Fig. 7B, 8A). This is in contrast to the behavior of modified AONs (2–5) where, at high RNA concentration, modified AONs have high target RNA cleavage activity by RNase H compared to the native (1)
(Fig. 7A and 8A), but at low RNA concentration enzyme activity is lower than that of the native AON (Fig. 7A and 8C).
The reason for the different behavior of and modified AON–RNA hybrids towards RNase H tolerance at high substrate concentration could be related to the Tm of the AON–RNA duplexes. All the -modified AONs were destabilized in a more pronounced manner (ΔTm
≈ 6 °C per modification) than the modified duplexes (ΔTm
≈ 3 °C per modification) and the native. This gives a less stable enzyme–substrate complex, and obviously a less stable enzyme–product complex for the -modified AONs. This means a low enzyme binding affinity of AON–RNA duplexes, which should give less 1/Km, and it is slightly less for -modified AON–RNA duplexes than that for the modified AONs and the native (Table 1 and Fig. 8B). Also, this means more turnover for the enzyme because of the high rate of dissociation of the enzyme–product complex, which should give more Vmax
(kcat) for -modified AON–RNA duplexes than for the modified AONs and the native (Table 1 and Fig. 8A).
The above explanation can be evidenced by a direct correlation between Vmax, kcat, 1/Km or Vmax/Km of RNase H with the Tm values of the corresponding and modified AON–RNA duplexes (Figs. 9A–C). The Vmax for the substrates (i.e. duplex), containing or modifications, increases with the decrease in Tm of the AON–RNA duplex (Fig. 9A) which is revealed by a linear dependence of [log(Vmax)] on Tm of AON–RNA duplexes (Fig. 9A). Conversely, the 1/Km and Vmax/Km values for the substrates increase with the increase in Tm of AON–RNA duplexes (Figs. 9B and C).
(B) DPPZ and cholesterol conjugated and oxetane modified AONs
The introduction of the π electron rich DPPZ or highly hydrophobic cholesterol residues in or modified AONs has different influences on the 1/Km and Vmax or kcat of RNase H (Table 1, Fig. 8A, B). DPPZ conjugation gives less binding affinity, and less or comparable catalytic activity of the RNase H compared with the native counterpart (Table 1, Fig. 8A, B). In contrast, conjugation of the hydrophobic cholesterol residue to AONs gives less binding affinity and more catalytic activity of RNase H (Table 1, Fig. 8A,B). Both DPPZ and cholesterol conjugation increase the Tm of the corresponding AON–RNA duplexes (Fig. 2), but no Tm correlation is found with the 1/Km, Vmax or Vmax/Km
(Figs. 9A–C), when unconjugated AONs (1–6) are taken into consideration.
A linear dependence of [log(Vmax)], [log(1/Km,)] or [log(Vmax/Km)] with the Tm of the corresponding AON–RNA heteroduplex has been also found for a short 9mer AON13 and its 3′-thethered conjugates13 having DPPZ or hydrophobic moieties like cholic acid, cholic acid triacetate or cholesterol residues (Fig. 10). In this group of AONs, only the DPPZ moiety has an additional influence on the binding affinity, catalytic activity and effective cleavage activity of the enzyme. DPPZ conjugated 9mer AON–RNA duplex has 6 times more 1/Km and Vmax/Km (and less Vmax) because of its high Tm compared to the native, hence the plots of [log(1/Km)] or [log(Vmax/Km)] versusTm (Figs. 10B–C) are not linearly correlated.
![The plot of catalytic activity [log (Vmax)]
(A), binding affinity [log (1/Km)]
(B) and effective activity [log(Vmax/Km)]
(C) of RNase H versus thermostability (Tm) of the AON–RNA duplexes formed with 17mer RNA 20 and native 9mer-AON 15, Chol-AON 16, Chol(Ac)3-AON 17, Cholest-AON 18 or DPPZ-AON 19. The linear correlation was found for all AONs (15–18)–RNA 20 duplexes and exceptions for their DPPZ (■)conjugate 19. Note, the AON 19 marked with (■) is not included in the correlation equation. Correlation coefficients (R2) for the plots are: 0.99 for (A), 0.97 for (B) and 0.98 for (C).](/image/article/2003/OB/b210163g/b210163g-f10.gif) | ||
| Fig. 10 The plot of catalytic activity [log (Vmax)] (A), binding affinity [log (1/Km)] (B) and effective activity [log(Vmax/Km)] (C) of RNase H versus thermostability (Tm) of the AON–RNA duplexes formed with 17mer RNA 20 and native 9mer-AON 15, Chol-AON 16, Chol(Ac)3-AON 17, Cholest-AON 18 or DPPZ-AON 19. The linear correlation was found for all AONs (15–18)–RNA 20 duplexes and exceptions for their DPPZ (■)conjugate 19. Note, the AON 19 marked with (■) is not included in the correlation equation. Correlation coefficients (R2) for the plots are: 0.99 for (A), 0.97 for (B) and 0.98 for (C). | ||
The reason that 3′-tethered 9mer AONs behave so differently from the corresponding 15mer AON conjugates may be owing to the size/volume of their respective duplex in the substrate–enzyme complex, which affects recognition and interaction by the enzyme. The differences in the enzymatic behavior of the DPPZ and cholesterol tethered AONs are perhaps owing to their different chemical nature: It is likely that the DPPZ group possibly interacts with the aromatic residues of the enzyme through π–π interactions (stacking), or/and through the coordination of Mg2+ with the nitrogen atoms (N4 and N5) in the fused phenanthroline system of the DPPZ moiety, which can act as reservoir of this important cofactor for the cleavage reaction.15 On the other hand, the poorer binding affinity of the cholesterol AON–RNA substrate to RNase H could be due to the undesirable interaction of the hydrophobic moiety with the enzyme, which can inhibit formation of the substrate–enzyme complex,16a giving poor binding affinity to the enzyme, as observed in this work.
Conclusions
(1) Antisense oligonucleotides (AONs) with single and double oxetane modifications [1′,2′-oxetane cytidine [1-(1′,3′-anhydro-β-D-psicofuranosyl)cytosine] were evaluated for their antisense potentials by targeting to a 15mer complementary RNA. Although the modified mixmer AONs showed ∼3 °C drop per modification in melting temperature (Tm) of their hybrid duplexes, they are found to be good substrate for RNase H, in comparison with the native AON–RNA duplex. The loss in Tm of interior modified AON–RNA hybrids was regained by the introduction of non-toxic dipyridophenazine (DPPZ) at the 3′-end (+6 °C).
(2) A detailed Michaelis–Menten kinetic analysis of RNase H cleavage showed that the single and double modified AON–RNA duplexes has catalytic activity (kcat) close to that of the native. However, the enzyme binding affinity (1/Km) showed a slight decrease with increase in the number of modifications, which results in less effective enzyme activity (kcat/Km) for modified AON–RNA duplexes.
(3) A comparative study with the corresponding single and double 1′,2′-oxetane thymidine, , modified AON–RNA duplexes, where the Tm drop was ∼6 °C per modification, however, showed that the kcat slightly increase and the 1/Km decrease with the increase in the number of modifications, which give a lower kcat/Km also for modified AON–RNA duplexes. The comparison of kcat/Km for both and modified AON–RNA duplexes show that RNase H activity can be achieved by the introduction of without sacrificing much of the target affinity which is an inherent drawback of modified AONs.
(4) All the and modified AON–RNA hybrids showed a correlation of Tm with 1/Km, Vmax, or Vmax/Km. The enzyme binding affinity and effective enzyme activity increase with increase of Tm; the catalytic activity was however found to decrease with a rise in Tm of the hybrid duplexes.
(5) The modified AONs, as in the counterpart, showed an enhanced tolerance towards the endonuclease degradation: single modification gives ∼3 fold protection and double modification gives ∼4 fold protection in comparison with the native AON.
(6) Even though the enzyme catalytic activity (kcat) of DPPZ conjugated double oxetane modified AON is comparable to the native, its enzyme binding affinity was two fold lower which in turn results in a the loss of kcat/Km by half. DPPZ also give enhanced protection against nucleases in human serum.
(7) 3′-Conjugation of a cholesterol moiety to the oxetane modified AONs has been found to have a less pronounced effect on RNA target affinity (ΔTm ≈ +2 to +3 °C) although these conjugated AON–RNA hybrids have shown high kcat of RNase H and low 1/Km and low kcat/Km, compared to the native counterpart.
(8) Thus, in this report, we show the utility of an AON having a fully phosphate backbone, by minimal (two) introduction of oxetane modifications in conjunction with DPPZ, as a potential antisense candidate with no toxicity. The candidacy of oxetane modified AONs with natural phosphate backbone should be considered as an alternative, keeping in view the non-antisense activity of the phosphorothioate based AONs (see ref. 3 for a critical review).
Materials and methods
Materials
All solvents were dried according to the reported procedures.22 The chromatographic separations were performed on Merck G60 silica gel. Thin layer chromatography (TLC) was performed on Merck pre-coated silica gel 60 F254 glass backed plates and developed in following dichloromethane–methanol mixtures. System (A): 97 : 3 (v/v), (B): 95 : 5 (v/v), (C) 90 : 10 (v/v). 1H-NMR spectra were recorded with Jeol GX 270 (if nothing else is indicated) and Bruker, DRX 600 spectrometers at 270 and 600 MHz, respectively, using TMS (0.0 ppm), methanol (3.4 ppm), DMSO-d6 (2.6 ppm) or acetonitrile (2.0 ppm, for D2O solutions) peaks as internal standards. 13C-NMR spectra were recorded with a Jeol GX 270 spectrometer at 67.9 MHz using the central peak of CDCl3 (76.9 ppm), DMSO-d6 (39.6 ppm) or acetonitrile (1.3 ppm, for D2O solutions) as internal standard. 31P-NMR spectra were recorded with a Jeol GX 270 spectrometer at 109.4 MHz using 85% phosphoric acid as an external standard. Chemical shifts are reported in ppm (δ scale). Thermal denaturation experiments were performed on a PC-computer interfaced Perkin Elmer UV/VIS spectrophotometer Lamda 40 with PTP-6 peltier temperature contoller. Fast deprotecting phosphoramidites were purchased from Glen Research Inc. (USA). T4 polynucleotide kinase, E. coli RNase H (5units/µl) and [γ-32P] ATP were purchased from Amersham Biosciences (Sweden). Phosphodiesterase I (Crotalus adamanteus venom), DNase 1 (Bovine pancreas) and Human Serum (AB male) were from SIGMAPreparation of oxetane modified building blocks
1H-NMR (CDCl3): 8.04–7.07 (m, 11H, 4-toluoyl, benzoyl, H-5, H-6), 5.48 (d, JH-3′, H-4′ = 5.8 Hz, 1H, H-3′), 5.16 (d, Jgem=11.1, 1H, H-1′), 4.96–4.91 (m, 2H, H-4′ & H-5′), 4.69 (dd, Jgem = 12.8 Hz, JH-5′, H-6″ = 2.5 Hz, 1H, H-6′), 4.52 (d, 1H, H-1″); 4.3 (dd, JH-5′, H-6′ = 3 Hz, 1H, H-6′b), 2.92 (s, 3H, CH3, OMs), 2.2 (s, 3H, CH3, 4-toluoyl), 1.64, 1.66 (s, 2 × 3H, CH3, isopropyl).
13C-NMR (CDCl3): 168.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 4-toluoyl), 165.7 (CO, benzoyl), 162.9 (C-4), 150.2 (C-2), 144.1 (C-6), 133.7, 133.3 (benzoyl), 129.5, 129.4, 129.1, 129, 128.8, 127.6, 125.9, (benzoyl, 4-toluoyl), 114.2 (C-5), 99.3 (C-2′), 86.5 (C-3′), 84.42 (C-5′), 81.9 (C-4′), 69.9 (C-1′), 64.7 (C-6′), 37.2 (CH3, mesyl), 25.8, 24 (CH3, isopropyl), 21.3 (CH3, 4-toluoyl).
O, 4-toluoyl), 165.7 (CO, benzoyl), 162.9 (C-4), 150.2 (C-2), 144.1 (C-6), 133.7, 133.3 (benzoyl), 129.5, 129.4, 129.1, 129, 128.8, 127.6, 125.9, (benzoyl, 4-toluoyl), 114.2 (C-5), 99.3 (C-2′), 86.5 (C-3′), 84.42 (C-5′), 81.9 (C-4′), 69.9 (C-1′), 64.7 (C-6′), 37.2 (CH3, mesyl), 25.8, 24 (CH3, isopropyl), 21.3 (CH3, 4-toluoyl).
1H-NMR (DMSO-d6): 8.08–7.39 (m, 11H, 4-toluoyl, benzoyl, H-5, H-6), 5.51 (d, JH-3′, H-4′ = 3.5 Hz, 1H, H-3′), 5.21 (d, Jgem = 8.3 Hz, 1H, H-1′), 4.8–4.76 (m, JH-5′, H-6′ = 4.9 Hz , 2H, H-1″, H-6′), 4.6–4.44 (m, Jgem = 12.4 Hz , 3H, H-4, H-5, H-6″), 2.3 (s, 3H, CH3, 4-toluoyl).
13C-NMR (DMSO-d6): 168.3 (CO, 4-toluoyl), 166.3 (CO, benzoyl), 164.5 (C-4), 154 (C-2), 147.2 (4-toluoyl), 144.5 (C-6), 133.7, 133.3 (benzoyl), 129.8, 129.7, 129, 128.8, 126.9 (benzoyl, 4-toluoyl), 97.7 (C-5), 92.7 (C-2′), 87.3 (C-3′), 81.1(C-5′), 78.1 (C-1′), 70.3 (C-4′), 64 (C-6′), 21.7 (CH3, 4-toluoyl).
1H-NMR (600 MHz, D2O): 7.28 (d, JH-5, H-6 = 7 .3 Hz, 1H, H-6), 5.94 (d, 1H, H-5), 5.44 (d, JH-3′, H-4′ = 3.1 Hz, 1H, H-3′), 5.14 (d, Jgem = 8.3 Hz, 1H, H-1′), 4.76 (d, 1-H, H-1″), 4.29–4.23 (m, JH-4′, H-5′ = 8.2 Hz, JH-5′, H-6′ = 4.9 Hz, 2H, H-4′ and H-5′), 3.9 (d, Jgem = 12.3 Hz, 1H, H-6′), 3.74 (dd, 1H, H-6″).
13C-NMR (D2O): 167.2 (C-4), 157.2 (C-2), 142.8 (C-6), 97.2 (C-5), 92.4 (C-2′), 88.3 (C-3′), 83.5 (C-5′), 79.5 (C-1′), 70.4 (C-4′), 61.3 (C-6′). 1D Diff. NOE shows 6% NOE enhancement for H6-H3′ and 4% NOE for H6-H1′. FAB-HRMS: [M + H]+ 256.0954; calcd 256.0933. For the corresponding thymine analog (ref 12b) FAB-HRMS: [M + H]+ 271.0922; calcd 271.0930.
1H-NMR (CDCl3): 7.51–7.15 (m, 11H, aromatic DMTr, H-6 and H-5), 6.81–6.76 (m, 4H, aromatic DMTr), 5.5 (d, JH-3′, H-4′ = 4.1 Hz, 1H, H-3′), 5.19 (d, Jgem = 7.8 Hz, 1H, H-1′), 4.81 (d, 1H, H-1″), 4.44 (dd, JH-4′, H-5 ′ = 7.9 Hz, 1H, H-4′), 4.37–4.29 (m, JH-5′, H-6′ = 2.97 Hz, JH-5′, H-6″ = 5.2 Hz, 1H, H-5′), 3.77 (s, 6H, OCH3, DMTr), 3.51 (dd, Jgem = 10.5 Hz, 1H, H-6′), 3.41 (dd, 1H, H-6″), 2.66–2.56 (m, 1H, CH, isobutyryl), 1.23, 1.21 (s, CH3, isobutyryl).
13C-NMR (CDCl3): 177.1(CO, isobutyryl), 163.1(C-4), 158.4 (DMTr), 153.7 (C-2), 144.7 (C-6), 144.4 ,135.8, 129.9, 128.9, 128, 127.5, 126.5, 125.1, 112.8 (DMTr), 97.5 (C-5), 92.3 (C-2′), 87.5 (C-3′), 86.5, 83.6 (C-5′), 78.8 (C-1′), 71.7 (C-4′), 62.7 (C-6′), 55.1 (DMTr), 36.5 (CH, isobutyryl), 18.9 (CH3, isobutyryl).
modified CPG solid support.
To a stirred solution of 25
(50 mg, 0.08 mmol) in 0.7 ml of CH2Cl2, succinic anhydride (16 mg, 0.16 mmol) and 4-(dimethylamino)pyridine (19.5 mg, 0.16 mmol) were added. The mixture was stirred for 4 h at rt. It was diluted with CH2Cl2 and then poured into saturated NaHCO3 solution. The organic phase was washed with 0.1 M citric acid (3 times), water and brine solution, dried over MgSO4, filtered and evaporated. The resulted succinate (27, 52 mg, 0.07 mmol) was dissolved in 6 ml acetonitrile and N,N-diisopropylethylamine (1.2 ml), benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (64 mg, 0.15 mmol), N-hydroxybenzotriazole (20 mg, 0.15 mmol) and 400 mg of aminopropyl CPG were added. The mixture was gently agitated for 2 h; the CPG was filtered, washed with acetonitrile, CH2Cl2, dry diethyl ether and assayed for loading (36 µmol per g of resin). The excess of amino groups on the support was then acetylated with 1 ml of acetic anhydride in 10 ml of pyridine containing 4,4-(dimethylamino)pyridine (180 mg) for 2 h. The support was then filtered and thoroughly washed with pyridine, CH2Cl2
(4 times) and diethyl ether, and then vacuum dried.
Synthesis, deprotection and purification of oligonucleotides
All oligonucleotides were synthesized using an Applied Biosystems 392 automated DNA/RNA synthesizer. Synthesis, deprotection and purification of all oxetane modified AONs,12b,c cholic acid,15 cholesterol15 conjugated AONs and their target RNAs were performed as previously described. For modified AONs containing oxetane , fast depropecting amidites were used, and they were deprotected by room temperature treatment of NH4OH for 16 h. All AONs were purified by reversed-phase HPLC eluting with the following systems: A (0.1 M triethylammonium acetate, 5% MeCN, pH 7) and B (0.1 M triethylammonium acetate , 50% MeCN, pH 7). The RNA target was purified by 20% 7 M urea polyacrylamide gel electrophoresis and its purity and of all AONs (greater than 95%) was confirmed by PAGE. MALDI-MS analysis: AON (5)
[MH]−
= 4477.1; calcd = 4474.74; AON (6)
[MH]−
= 4503.63; calcd = 4502.74; AON (9)
[MH]−
= 5096.55; calcd = 5094.74.
UV melting experiments
Determination of the Tms of the AON–RNA hybrids and RNA–RNA duplexes was carried out in the following buffer: 57 mM Tris-HCl (pH 7.5), 57 mM KCl, 1 mM MgCl2 and 2 mM DTT. Absorbance was monitored at 260 nm in the temperature range from 3 °C to 60 °C using a Lambda 40 UV spectrophotometer equipped with a peltier temperature programmer with the heating rate of 1 °C per minute. Prior to the measurements samples (1 µM of AON and 1 µM RNA mixture) were preannealed by heating to 80 °C for 5 min followed by slow cooling till 3 °C and 30 min equilibration at this temperature.32P Labeling of oligonucleotides
The oligoribonucleotides, oligodeoxyribonucleotides as well as phosphorothioates were 5′-end labeled with 32P using T4 polynucleotide kinase, [γ-32P]ATP and standard procedure 23. Labeled AONs and RNA were purified by 20% denaturing PAGE and specific activities were measured using a Beckman LS 3801 counter.Kinetics of RNase H hydrolysis
Values of Km and Vmax for this method were determined directly from v0versus [S0] plots by use of the correlation SigmaPlot 2000 Program, where the correlation equation was: y = ax/(b + x) where a = Vmax and b = Km. Since Vmax = E0 × kcat (E0 = initial enzyme concentration), and for all of our RNA concentration dependent kinetics, the E0 was identical, hence Vmax is proportional to kcat. In other words, kcat can be understood by comparing simply the Vmax.
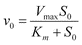
Exonuclease degradation studies
Stability of the AONs towards 3′-exonucleases was tested using snake venom phosphodiesterase from Crotalus adamanteus. All reactions were performed at 3 µM DNA concentration (5′-end 32P labeled with specific activity 50000 cpm) in 56 mM Tris-HCl (pH 7.9) and 4.4 mM MgCl2 at 22 °C. Exonuclease concentration of 17.5 ng µl−1 was used for digestion of oligonucleotides.Total reaction volume was 14 µl. Aliquots were quenched by addition of the same volume of 50 mM EDTA in 95% formamide. Reaction progress was monitored by 20% 7 M urea PAGE and autoradiography.
Endonuclease degradation studies
Stability of AONs towards endonuclease was tested using DNase 1 from bovine pancreas. Reactions were carried out at 0.9 µM DNA concentration (5′-end 32P labeled with specific activity 50000 cpm) in 100mM Tris-HCl (pH 7.5) and 10 mM MgCl2 at 37 °C using 30 unit of DNase 1. Total reaction volume was 22 µl. Aliquots were taken at 60, 120, 180 and 240 min and quenched with the same volume of 50 mM EDTA in 95% formamide. They were resolved in 20% polyacrylamide denaturing gel electrophoresis and visualised by autoradiography.
Stability studies in human serum
AONs (6 µl) at 2 µM concentration (5′-end 32P labeled with specific activity 90000 cpm) were incubated in 30 µl of Human Serum (male AB) at 37 °C (total reaction volume was 36 µl). Aliquots (3 µl) were taken at 5, 15, 30, 60 and 120 min, and quenched with 6 µl of solution containing 8 M urea and 50 mM EDTA, resolved in 20% polyacrylamide denaturing gel electrophoresis and visualized by autoradiography.
Acknowledgements
Authors thank the Swedish Research Council (Vetenskapsrådet) and Philip Morris Inc (USA) for generous financial support. We also thank Dr Suresh Gohil for recording the MALDI-TOF MS. P. I. Pradeepkumar and N.V. Amirkhanov have contributed equally in this project.References
- (a) N. Dias and C. A. Stein, Mol. Cancer Ther., 2002, 1, 347 Search PubMed; (b) S. T. Crooke, Methods Enzymol., 2000, 313, 3 Search PubMed; (c) S. T. Crooke, Biochem. Biophys. Acta, 1999, 1489, 31 Search PubMed.
- (a) S. Kanaya, M. IkeharaSubcell Biochemistry, in Proteins: Structure, Function, and Engineering, ed. B. B. Biswas and Siddhartha Roy, Plenum Press, New York, 1995, p. 24, p. 377–422 Search PubMed; (b) E. Zamaratski, P. I. Pradeepkumar and J. Chattopadhyaya, J. Biochem. Biophys. Methods, 2001, 48, 189 CrossRef CAS.
- (a) C. A. Stein, J. Clin. Invest., 2001, 108, 641 Search PubMed; (b) I. Lebedeva and C.A. Stein, Annu. Rev. Pharmacol., 2001, 41, 403–419 Search PubMed.
- M. Manoharan, Biochem. Biophys. Acta, 1999, 1489, 117 Search PubMed.
- (a) S. T. Crooke, K. M. Lemonidis, L. Neilson, R. Griffey, E. A. Lesnik and B. P. Monia, Biochem. J., 1995, 312, 599 CAS; (b) W. F. Lima and S. T. Crooke, Biochemistry, 1997, 36, 390 CrossRef CAS.
- J. Kurreck, E. Wyszko, C. Gillen and V. A. Erdmann, Nucleic Acids Res., 2002, 30, 1911 CrossRef CAS.
- (a) E. Uhlmann, A. Peyman, A. Ryte, A. Schmidt and E. Buddecke, Methods Enzymol., 2000, 313, 268 Search PubMed; (b) A. Peyman, M. Helsberg, G. Kretzschmar, M. Mag, A. Ryte and E. Uhlmann, Antiviral Res., 1997, 33, 135 CrossRef CAS.
- (a) R. V. Giles and D. M. Tidd, Anti-Cancer Drug Des., 1992, 7, 37 Search PubMed; (b) D. M. Tidd, Perspect. Drug Discovery Des., 1996, 4, 51 Search PubMed.
- V. K. Rait and B. R. Shaw, Antisense Nucleic Acid Drug Dev., 1999, 9, 53 CAS.
- (a) K.-H. Altman, R. Kesselring, E. Francotte and G. Rihs, Tetrahedron Lett., 1994, 35, 2331 CrossRef CAS; (b) M. A. Siddiqui, H. Ford, C. George and V. E. Marquez, Nucleosides Nucleotides, 1996, 15, 235 Search PubMed; (c) V. E. Marquez, M. A. Siddiqui, A. Ezzitouni, P. Russ, J. Wang, W. R. Wanger and D. M. Matteucci, J. Med. Chem., 1996, 39, 3739 CrossRef CAS; (d) S. Obika, D. Nanbu, Y. Hari, K. Morio, Y. In, T. Ishida and T. Imanishi, Tetrahedron Lett, 1997, 38(50), 8735 CrossRef CAS; (e) A. A. Koshkin, S. K. Singh, P. Nielson, V. K. Rajwanshi, R. Kumar, M. Meldgaard and J. Wengel, Tetrahedron, 1998, 54, 3607 CrossRef CAS; (f) D. G. Schultz and S. M. Grynazov, Nucleic Acids Res., 1996, 24, 2966 CrossRef CAS; (g) G. Wang, J.–L. Giradet and E. Gunic, Tetrahedron, 1999, 55, 7707 CrossRef CAS; (h) J. Wengel, Acc. Chem. Res., 1999, 32, 301 CrossRef CAS; (i) M. Sekine, O. Kurasawa, K. Shohda, K. Seio and T. Wada, J. Org. Chem., 2000, 65(12), 3571 CrossRef CAS.
- (a) P. Herdewijn, Biochem. Biophys. Acta, 1999, 1489, 167 Search PubMed; (b) L. Kvaerno and J. Wengel, Chem. Commun., 2001, 1419–1424 RSC.
- (a) P. I. Pradeepkumar, E. Zamaratski, A. Foldesi and J. Chattopadhyaya, Tetrahedron Lett., 2000, 41, 8601 CrossRef CAS; (b) P. I. Pradeepkumar, E. Zamaratski, A. Foldesi and J. Chattopadhyaya, J. Chem. Soc., Perkin Trans. 2, 2001, 402 RSC; (c) P. I. Pradeepkumar and J. Chattopadhyaya, J. Chem. Soc., Perkin Trans. 2, 2001, 2074 RSC.
- N. V. Amirkhanov, P. I. Pradeepkumar and J. Chattopadhyaya, J. Chem. Soc., Perkin Trans. 2, 2002, 976 RSC.
- (a) D. Ossipov, E. Zamaratski and J. Chattopadhyaya, Helv. Chim. Acta, 1999, 82, 2186 CrossRef CAS; (b) E. Zamaratski, D. Ossipov, P. I. Pradeepkumar, N. V. Amirkhanov and J. Chattopadhyaya, Tetrahedron, 2001, 57, 593 CrossRef CAS.
- N. V. Amirkhanov and J. Chattopadhyaya, J. Chem. Soc., Perkin Trans. 2, 2002, 271 RSC.
- (a) G. Godard, A. S. Boutorin, E. Saison-Behmoaras and C. Helene, FEBS, 1995, 232, 404 Search PubMed; (b) S. T. Crooke, M. J. Graham, J. E. Zuckerman, D. Brooks, B. S. Conklin, L. L. Cummins, M. J. Greig, C. J. Guinosso, D. Kornbrust, M. Manoharan, H. M. Sasmor, T. Schleich, K. L. Tivel and R. H. Griffey, J. Pharmacol. Exp. Ther., 1996, 277, 923 Search PubMed; (c) M. Manoharan, L. K. Johnson, C. F. Bennet, T. A. Vickers, D. J. Ecker, L. Cowsert, M. M. Frier and P. D. Cook, Bioorg. Med. Chem. Lett., 1994, 4, 1053 CrossRef CAS; (d) A. S. Boutorin, L. V. Gus'kova, E. M. Ivanova, N. D. Kobetz, V. F. Zarytova, A. S. Ryte, L. V. Yurchenko and V. V. Vlassov, FEBS Lett., 1989, 254, 129 CrossRef CAS; (e) A. S. Ryte, V. N. Karamyshev, M. V. Nechaeva, Z. V. Guskova, E. M. Ivanova, V. F. Zarytova and V. V. Vlassov, FEBS Lett., 1992, 299, 124 CrossRef CAS.
- (a) C. F. Bleczinski and C. Richert, J. Am. Chem. Soc., 1999, 121, 10889 CrossRef CAS; (b) H. Gao and J. R. Dias, Synth. Commun., 1997, 27, 757 CAS; (c) W. Kramer and G. Wess, Eur. J. Clin. Invest., 1996, 26, 715 CAS; (d) T. J. Lehmann and J. W. Engels, Bioorg. Med. Chem., 2001, 9, 1827 Search PubMed; (e) D. Opsenica, G. Pocsfalvi, Z. Juranic, B. Tinant, J. P. Declercq, D. E. Kyle, W. K. Milhous and B. A. Solaja, J. Med. Chem., 2000, 43, 3274 CrossRef CAS.
- Oxetane and oxetane modified mixmer AON with DPPZ at the 3′-end were found to be non-toxic from 1 µM to 20 µM in K562 Cell culture assays. A. P. Gewirtz, University of Pennsylvania, USA; personal communication.
- M. Dixon, E. C. Webb, in Enzymes, 3rd edn., Longman Group Ltd, London, 1979, p. 60 Search PubMed.
- M. D. Sorensen, L. Kvaerno, T. Bryld, A. E. Hakansson, B. Verbeure, G. Gaubert, P. Herdewijn and J. Wengel, J. Am. Chem. Soc., 2002, 124, 2164 CrossRef CAS.
- C. Wahlestedt, L. Salmi, J. K. Good, T. Johnsen, T. Hokfelt, C. Broberger, F. Porreca, A. Koshkin, M. H. Jacobson and J. Wengel, Proc. Natl. Acad. Sci. USA, 2000, 97, 5633 CrossRef CAS.
- D. D. Perrin, W. L. F. Armarego, D. R. Perrin, Purification of Laboratory Chemicals, 1985, Pergamon Press, Oxford Search PubMed.
- F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith and K. Struhl, Current Protocols in Molecular Biology, 1995, 3.10.3, John Wiley & Sons, New York Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: RNA cleavage kinetics. See http://www.rsc.org/suppdata/ob/b2/b210163g/ |
| This journal is © The Royal Society of Chemistry 2003 |