Structure–reactivity relationships in the inactivation of elastase by β-sultams
Paul S.
Hinchliffe
a,
J. Matthew
Wood
a,
Andrew M.
Davis
b,
Rupert P.
Austin
b,
R. Paul
Beckett
c and
Michael I.
Page
*a
aDepartment of Chemical and Biological Sciences, University of Huddersfield, Queensgate, Huddersfield, UK HD1 3DH
bAstraZeneca R&D, Charnwood, Bakewell Road, Loughborough, UK LE11 5RH
cBritish Biotech Pharmaceuticals Limited, Wellington Road, Oxford, UK OX4 5LY
First published on 27th November 2002
Abstract
N-Acyl-β-sultams are time dependent irreversible active site directed inhibitors of elastase. The rate of inactivation is first order with respect to β-sultam concentration and the second order rate constants show a similar dependence on pH to that for the hydrolysis of a peptide substrate. Inactivation is due to the formation of a stable l ∶ l enzyme inhibitor complex as a result of the active site serine being sulfonylated by the β-sultam. Ring opening of the β-sultam occurs by S–N fission in contrast to the C–N fission observed in the acylation of elastase by N-acylsulfonamides. Structure–activity effects are compared between sulfonylation of the enzyme and alkaline hydrolysis. Variation in 4-alkyl and N-substituted β-sultams causes differences in the rates of inactivation by 4 orders of magnitude.
Proteolytic enzymes are common targets for potential therapeutic application and their inhibition by mechanism based and active site directed agents is a fruitful area of study.1 In general, this involves the covalent modification of an active site residue which is not then readily regenerated. For example, many inhibitors are acylating agents of the active site serine residue of serine proteases.2 The mechanism of inhibition involves the displacement of a leaving group from the acylating agent to generate a relatively stable acyl enzyme which only reacts slowly with nucleophiles, such as water, to regenerate the enzyme and so this process leads to effective inhibition.
Human neutrophil elastase (HNE) is a serine enzyme which is one of the most destructive proteolytic enzymes, being able to catalyse the hydrolysis of the components of connective tissue. HNE is released in response to inflammatory stimuli and has a major role in protein digestion following phagacytosis. It has been implicated in the development of diseases such as emphysema, cystic fibrosis and rheumatoid arthritis and there have been numerous studies attempting to find small molecule inhibitors of HNE.3 The enzyme belongs to the trypsin class and the structure of HNE has been determined by X-ray crystallography,4 but many structural and inhibition studies have been conducted with the related, but more readily available, porcine pancreatic elastase (PPE).5
The majority of elastase inhibitors are based an other serine protease inhibitors. They are, for example, peptidyl fluoroketones6 or ketones attached to a strongly electron withdrawing group, such as 2-benzoxazole, with suitable elements of molecular recognition.7 An alternative strategy has been to use acylating agents that generate an acyl enzyme which does not turnover, or rearranges to a more stable adduct or even generates a second electrophilic ‘trap’ which subsequently reacts with a nucleophilic group on the enzyme. For example, bicyclic [3.3.0] systems containing a γ-lactone or γ-lactam which is trans-fused to the other 5-membered ring inhibit HNE by acylating the nucleophilic hydroxy group of serine-195 in the active site of the enzyme.8 Interestingly, the classical β-lactams, traditionally used as anti-bacterial agents by inhibiting serine transpeptidases,9 have also been shown to be mechanism based inhibitors of elastase when used as neutral derivatives.10 ESI-MS and NMR studies have shown that the first step is an acylation process in which the four membered β-lactam ring is opened.11 The requirements for inhibition of elastase by monocyclic lactams have been recently described.12 They involve the initially formed acyl enzyme intermediate undergoing a conformational change to displace the hydrolytic water molecule, effectively generating a more stable complex.
In principle, sulfonation of serine proteases offers an interesting but largely unexplored strategy for inhibition as an alternative to the traditional mechanism-based acylation process. In addition to their normal acyl substrates, serine proteases are known to react with other electrophilic centres such as phosphonyl derivatives.1 The main reason why sulfonation of serine enzymes is not a well studied process is because sulfonyl derivatives are much less reactive than their acyl counterparts.13 For example, sulfonamides are extremely resistant to alkaline and acidic hydrolysis and, in general, sulfonyl transfer reactions are 102 to 104 fold slower than the corresponding acyl transfer process. However, we have recently shown that the rates of alkaline and acid hydrolysis of N-alkyl and N-aryl β-sultams are 102 to 103 fold greater than those for the corresponding β-lactams.14 β-Sultams (1) show extraordinary rate enhancements of 109 and 107, respectively, compared with the acid and base catalysed hydrolysis of the corresponding acyclic sulfonamides.15 In principle, therefore, β-sultams are excellent candidates to explore the mechanism of sulfonation and possible inhibition of serine protease enzymes. We recently demonstrated that ring opening of the β-sultam by elastase gives the sulfonate ester,16 by analogy with the acyl enzyme intermediate formed during the hydrolysis of normal substrates (Scheme 1). Herein, we report further studies on this process and the effect of structural changes in the β-sultam on the rate of inactivation of elastase.
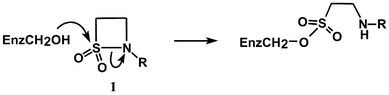 | ||
| Scheme 1 | ||
Results and discussion
One of the major requirements for successful inhibition of an enzyme by covalent modification is that the rate of the inactivation must be faster than, or at least competitive with, the turnover of the natural substrate and the inhibitor itself must be relatively stable under in vivo conditions. The rate of inactivation is controlled by two important factors:(i) the “chemical” reactivity of the inactivator
(ii) molecular recognition by the target enzyme.
Given that most modifications involve displacement of a leaving group by a nucleophile on the enzyme, chemical reactivity is usually considered in terms of the electrophilic nature of the inhibitor and the leaving group ability (nucleofugacity) of the group displaced. In addition to these electronic effects, the intrinsic chemical reactivity of a compound may be modified by steric and strain effects. Increasing intrinsic chemical reactivity may lead to a faster rate of reaction with the target enzyme but may also lead to greater hydrolytic and metabolic instability and lower specificity.
Obtaining maximum activity by molecular recognition involves using the full catalytic apparatus of the enzyme, as used for the natural substrate, and utilising the favourable non-covalent interactions at the various sub-sites of the enzyme and inhibitor.17 The most desirable inhibitor may be one where molecular recognition is maximised using these favourable non-covalent interactions and where covalent modification of the enzyme, due to the inherent reactivity of the inhibitor, is minimised.
Improving the inhibition properties of a compound may therefore be achieved by modifying the structure to enhance either chemical reactivity or molecular recognition, or a combination of both. However, analysing these separate effects on the rate of inactivation when the inhibitor is modified is not straightforward. Modification of the inhibitor structure can affect the free energies of both the initial reactant state and the transition state of the reaction with the enzyme, whereas the observed variation in rate constants for various inhibitors reacting with the enzyme only reflects the difference in energies between these two states. Different substituents can affect the ease of bond-making and -breaking by classical electronic factors such as inductive, resonance and steric effects. However, the free energy of activation of an enzyme-catalysed reaction is also affected by the favourable binding energies between the protein and substrate substituents not directly involved with the reaction site. It is, therefore, important to separate these two effects before conclusions about the efficiency of inhibition can be made. For substrates we have suggested18 that an ‘enzyme rate-enhancement factor’ (EREF) can be evaluated by dividing the second-order rate constant for the enzyme catalysed reaction, kcat/Km, by that for hydrolysis of the same substrate catalysed by hydroxide ion, kOH. For inhibitors acting by covalent modification of the enzyme, a similar ratio of ki/kOH can be used where ki is the rate constant for inactivation.
Alkaline hydrolysis of the PPelastase substrate, N-suc-(L-Ala)3-p-nitroanilide (2)
An analysis of a particular enzyme's selectivity for various substrates or covalent inhibitors requires a knowledge of their reactivity in the absence of the enzyme. Although N-suc-(L-Ala)-p-nitroanilide (2) is an established and convenient substrate for porcine pancreatic elastase (PPelastase),19 the magnitude and nature of its hydroxide ion catalysed hydrolysis has not been reported.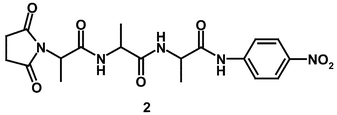
Below pH 13, N-suc-(L-Ala)3-p-nitroanilide (2) undergoes hydroxide ion catalysed hydrolysis to give p-nitroaniline, for which the second-order rate constant, kOH, is 7.19 × 10−3 M−1 s−1 at 30 °C. At pHs above ca. 13 there is a pH-independent reaction presumably due to a reversible ionisation of the aryl-NH group of (2) consistent with the pKa of simple p-nitroanilides.20 The overall pathway for the hydroxide ion hydrolysis of N-suc-(L-Ala)3p-nitroanilide (2) is shown in Scheme 2.
 | ||
| Scheme 2 | ||
Only the neutral compound undergoes hydroxide ion catalysed hydrolysis whilst the anion is relatively stable. The rate law for this reaction was determined based upon the fraction of the undissociated species present in solution [eqn. (1)], where [S] is the reactant concentration, kobs is the observed first order rate constant for hydrolysis, kOH is the second-order rate constant of hydroxide ion hydrolysis and [H+] / (Ka + [H+]) is the fraction of the anilide present in its undissociated form.
| Rate / [S] = kobs = kOH[HO−]([H+] / (Ka + [H+])) | (1) |
When the solution pH is less than 13, [H+] ≫ Ka, eqn. (1) simplifies to kobs = kOH[HO−] and a first order dependence on hydroxide ion concentration is observed. At solution pHs greater than 13, Ka ≫ [H+], eqn. (1) becomes kobs = kOH (Kw / Ka) resulting in a pH independent rate. Modelling the experimental data to this equation using the Scientist software package gave the following parameters: kOH = 7.19 × 10−3 M−1 s−1 and Ka = 9.82 × 10−14 M (pKa 13.01). Direct titration using the absorbance change at 390 nm as a function of pH, immediately after injection of (2) into sodium hydroxide solutions, gave a reasonable sigmoidal plot from which was obtained a calculated pKa, of 12.63 ± 0.07, in fairly good agreement with that determined kinetically.
PPelastase catalysed hydrolysis of substrate, N-suc-(L-Ala)3-p-nitroanilide (2)
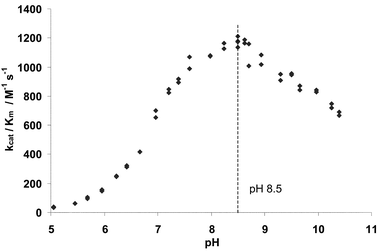 | ||
| Fig. 1 Plot of PPelastase activity (kcat/Km) towards the substrate N-suc-(L-Ala)3-p-nitroanilide (2) against pH. Initial rates measured at 1.52 × 10−6 M PPelastase, 6.0 × 10−5 M substrate, 390 nm and 30 °C in 0.1 M buffer, 6% MeCN v/v. | ||
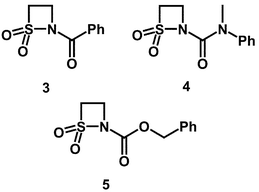
Inhibition of PPelastase by N-acyl-β-sultams
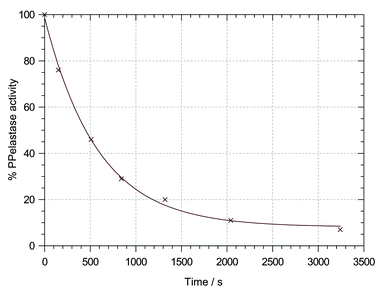 | ||
| Fig. 2 Plot of % PPelastase activity against incubation time for the inactivation of PPelastase by N-benzoyl-β-sultam (3) at pH 6.75 and 30 °C in 0.04 M buffer, 20 % ACN v/v, 8 × 10−5 M PPelastase and 1.2 × 10−3 β-sultam. | ||
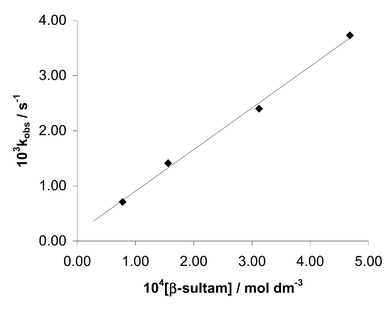 | ||
| Fig. 3 Plot of the first order rate constant for the inactivation of PPelastase by N-benzoyl-β-sultam (3) corrected for hydrolysis at pH 6.1 against β-sultam concentration. | ||
With some N-acyl β-sultams and at high pH there were problems with the accurate determination of rate constants for the inactivation process because of the competing hydrolysis of these compounds. To correct for this, the results of the inactivations were normalised with respect to the decreasing β-sultam concentration using eqn. (2).
| Normalised rate at time t = (observed rate/ [Sultam]o) × [Sultam]t | (2) |
The second-order rate constants ki for the inactivation of PPelastase by various substituted β-sultams are given in Table 1.
| β-Sultam | k i at pH 6.1/M−1 s−1 | k i at pH 7.0/M−1 s−1 | k i at pH 8.5/M−1 s−1 | k OH/M−1 s−1 | EREF at pH 8.5a |
|---|---|---|---|---|---|
| a EREF is ki (inactivation)/kOH at pH 8.5 | |||||
| N-Bz (3) | 0.85 | 2.34 | 4.06 | 1.46 × 104 | 6.1 × 10 |
| 4-iPr-N-Bz (14) | 0.23 | 0.39 | 1.63 | 300 | 1.2 × 103 |
| (E)-4-Ethylidene-N-Bz (17) | — | 3.64 | 6.33 | 2.55 × 103 | 5.4 × 102 |
| N-Carbamoyl (4) | — | 2.13 × 10−2 | 3.71 × 10−2 | 265 | 3.1 × 10 |
| 4-iPr-N-carbamoyl (15) | — | 9.14 × 10−3 | 1.59 × 10−2 | 3.80 | 9.1 × 102 |
| 4-Et-N-carbamoyl (18) | — | 0.26 | 0.46 | 26.8 | 3.8 × 103 |
| N-Cbz (5) | 8.65 | 38 | 67 | 4.48 × 104 | 3.2 × 102 |
| 4-iPr-N-Cbz (16) | 2.70 | 23.6 | 106 | 1.0 × 103 | 2.3 × 104 |
Evidence for active-site sulfonation
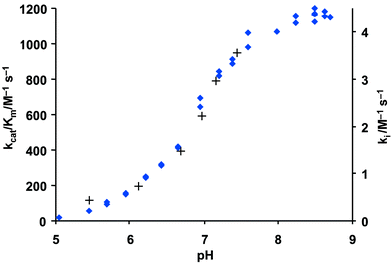 | ||
| Fig. 4 Plot of kcat/Km for the hydrolysis of N-suc-(L-Ala)3-p-nitroanilide by PPelastase (♦) and ki for N-benzoyl-β-sultam (3) with PPelastase (+) against pH. 30 °C, I = 1.0 M (KCl), 6 % MeCN v/v and 1.5% MeOH v/v. | ||
Crystals of native PPelastase were soaked for 24 h at pH 5 in a saturated solution of N-benzoyl-β-sultam (3). After soaking, X-ray crystallography revealed that the crystals were isomorphous to native PPelastase and confirmed that ring opening of the β-sultam was part of the inactivation process (Fig. 5).16 A sulfonate ester formed between Ser-195 and the inhibitor is observed along with the ethyl amino group projecting into the active site's P1/P2 region. Whilst one sulfonate oxygen atom was observed in the upper part of the S1 pocket the other was located in the oxyanion hole within hydrogen bonding distance of the amido-nitrogen atoms of Ser-195 (3.02 Å) and Gly-193 (3.23 Å). Crucially, the side chain of His-57 has been displaced by approximately 90° from its native position, which is now occupied by two water molecules (Wat-342 and Wat-355). This effect is almost identical to that observed in the inhibition of PPelastase by γ-lactams.22
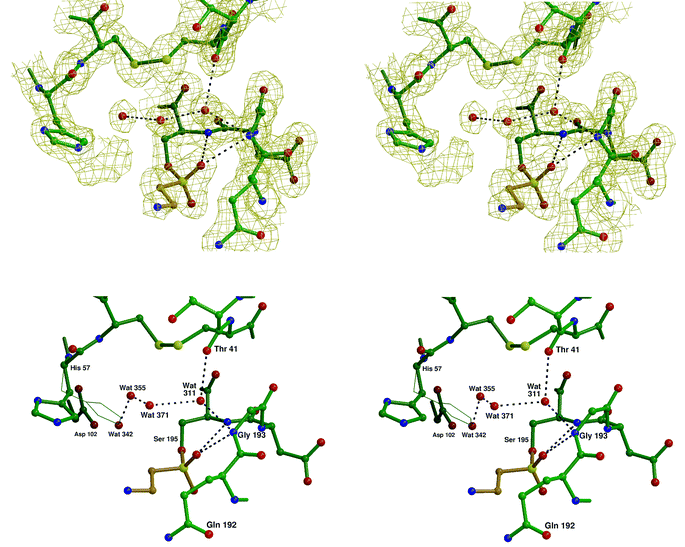 | ||
| Fig. 5 Stereo views of the active site of PPelastase (in green) showing N-benzoyl- β-sultam (3) (in beige) covalently linked via a sulfonate ester to Ser-195 and the ‘native’ position of His-57 in thin lines. The top picture shows the 2mFo − DFc electron density map contoured at 1 σ. Wat-342 has not been included so that the ‘extra’ density extending from Nε2 of His-57 can be seen more clearly. The bottom picture shows the ‘native’ position of His-57 in thin lines. | ||
N-Acylsulfonamides have been used previously to inactivate serine enzymes.8 However, the mechanism invariably involves acylation and C–N bond fission with the serine hydroxy group attacking the amide to displace the sulfonamide as the leaving group. Although the β-sultams reported here are also formally N-acylsulfonamides inactivation occurs by sulfonylation as a result of serine nucleophilic attack on the sulfonyl centre and displacement of the amide as a leaving group. This appears to be the first case of preferential S–N over C–N fission in the reaction of a N-acylsulfonamide with a serine protease.
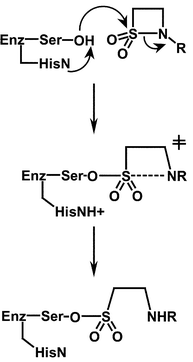 | ||
| Scheme 3 | ||
The preferred direction of nucleophilic attack at acyl centres is presumed to be at an approximately tetrahedral angle at either face. As this differs to that at sulfonyl centres, which is along the apical position bisecting the sulfonyl oxygens, attack of the serine group at the sulfonyl centre will alter the position of the other catalytic group, the histidine residue, relative to the rest of the molecule. In acyl transfer, the histidine residue has to be correctly positioned to protonate the leaving group nitrogen in the TI in its function as a general acid catalyst. The catalysis of sulfonyl transfer in N-acyl-β-sultams by elastase may therefore only be possible because ring opening occurs in a single step without general acid catalysis by the histidine residue. The relative position of this group in the active-site would therefore be less important. This apparent flexibility of enzymes has also been observed in the β-lactamase P99 which has the ability to catalyse phosphoryl transfer reactions, via trigonal bipyramidal intermediates.23
Structure–activity relationships
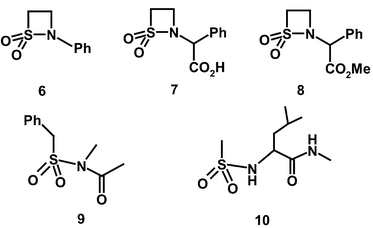
A comparison of the ki values at pH 7 with the second-order rate constants for hydroxide ion hydrolysis, kOH values (Table 1), for the N-benzoyl compound (3) and the N-carbamoyl compound (4) indicates that their relative effectiveness as PPelastase inhibitors is largely due to variations in their chemical reactivity. At pH 7, the N-benzoyl derivative (3) is a better inactivator by two orders of magnitude than the N-carbamoyl β-sultam (4) which is similar to the ratio of their kOH values, 55. This indicates that there is no significant difference in binding between these two leaving groups and the enzyme in the inactivation of PPelastase. However, replacement of the N-carbamoyl residue by the N-benzyloxycarbonyl substituent increases the rate of inactivation by 103–104. The enzyme rate enhancement factors (EREFs) for N-benzoyl (3), N-carbamoyl (4) and N-benzyloxycarbonyl-β-sultam (5) are 61, 31 and 320 respectively. This suggests that of all the N-substituents the benzyloxycarbonyl moiety binds most favourably in the active-site of PPelastase, possibly via π–π interactions with the His-57 residue in the active-site or another aromatic residue in its vicinity. Such interactions have been observed between the p-toluene ring of N-arylsulfonyl-β-lactams and His-57.30 Apparently the N-carbamoyl group of (4) does not reach sufficiently deeply into the S' sites of PPelastase's active-site and cannot interact as effectively with His-57. It both lacks one carbon atom on the side chain compared to that of (5) and free rotation is restricted by resonance of the urea moiety. There is also cis–trans isomerism of this urea moiety resulting in the N-carbamoyl compound (4) having two stereoisomers.
It is known that the α-chymotrypsin family of enzymes has a well-established binding pocket adjacent to the active serine residue, in the S1 position. In the elastase enzymes this binding pocket is relatively small and has a preference for small hydrophobic substituents. The inhibition of PPelastase by monocyclic β-lactams has also been improved by the introduction of alkyl substituents at the 3-position which are thought to bind strongly in the S1 subsite of elastase.30,31
Alkyl chains have therefore been added to the simple β-sultam ring at the 4-position with the intention of improving binding at the active-site of PPelastase. 4-Isopropylidene β-sultam (11) is unusual because the substituent causes nucleophiles to react at the acyl rather than the sulfonyl centre.32 This switch of reactive centre is accompanied by a 104-fold reduction in reactivity towards hydroxide ion. Although N-acylsulfonamides may act as inhibitors of elastase by acylation and expulsion of the sulfonamide, 4-isopropylidene-N-benzoyl-β-sultam (11) and 4-isopropylidene-N-benzyloxycarbonyl-β-sultam (12) are both inactive towards PPelastase at concentrations up to 2 mM. Interestingly, 4-isopropylidene-N-(N,N-methyl phenyl)carbamoyl-β-sultam (13) displayed a small amount of inactivation of the enzyme with an estimated ki value of 2 × 10−3 M−1 s−1 at pH 8.5. This is a reduction of approximately 10-fold compared to the parent compound (4) lacking the 4-isopropylidene substituent and is unexpected given the extremely low reactivity of this compound and the fact that it reacts with hydroxide ion at the acyl centre. It is not known whether (13) acts as a sulfonylating or acylating agent of PPelastase.
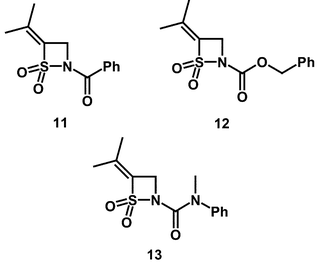
The introduction of the isopropyl substituent at the 4-position of the thiazetidine ring lowers the reactivity of β-sultams towards hydroxide ion by 50-fold—compare (14) with (3) (Table 1) and maintains the mechanism of S–N fission. In addition, unlike the 4-isopropylidene derivatives, the 4-isopropyl compounds are effective inhibitors. The second-order rate constants for inhibition obtained for 4-isopropyl-N-benzoyl-β-sultam (14), 4-isopropyl-N-(N,N-methylphenyl)carbamoyl-β-sultam (15) and 4-isopropyl-N-benzyloxycarbonyl-β-sultam (16) are summarised in Table 1.
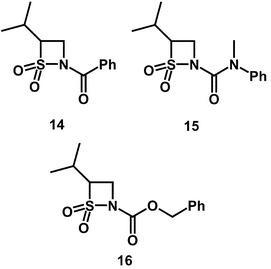
Whilst the 4-isopropyl substituent has decreased the rate of alkaline hydrolysis of N-benzoyl β-sultam (3) by 50-fold in (14), enzyme inhibitory activity was only decreased by 3-fold. If the EREF values are compared between the 4-isopropyl compound (14) and the simple N-benzoyl compound (3) then an increase of 20-fold is observed. This matches the increase in EREF of 30-fold observed between the 4-isopropyl-N-carbamoyl compound (15) and the simple N-carbamoyl compound (4). Therefore the 4-isopropyl substituent has increased the level of active site binding and made the β-sultams more reactive towards the enzyme in the presence of hydroxide ion. This is possibly a result of favourable interactions between the alkyl substituent and the hydrophobic S1 binding pocket.
The best inactivator in this series is 4-isopropyl-N-benzyloxycarbonyl-β-sultam (16) with an EREF of 2.3 × 104, 70-fold greater than that of the N-benzyloxycarbonyl compound lacking the 4-isopropyl substituent (5). The combination of the 4-isopropyl and N-benzyloxycarbonyl groups creates favourable interactions for active-site binding. If the two isopropyl substituted compounds, 4-isopropyl-N-carbamoyl-β-sultam (15) and the chemically more reactive 4 isopropyl-N-benzyloxycarbonyl-β-sultam (16) are compared, there is an increase in EREF of 25-fold on going from the carbamoyl to the benzyloxycarbonyl N-substituent. As discussed for the simple N-benzyloxycarbonyl compound (5), this again suggests that of all the N-substituents the benzyloxycarbonyl moiety binds most favourably with PPelastase. This is possibly due to its longer length permitting more favourable π–π interactions with His-57 or another aromatic residue.
The introduction of the 4-isopropyl substituent has increased the effectiveness of the lead compounds as inhibitors of PPelastase. ESIMS analysis of 4-isopropyl-N-benzoyl-β-sultam (14) incubated with PPelastase in non-buffered aqueous solution confirmed that only l ∶ l enzyme ∶ inhibitor (EI) complexes were being formed, indicative of a good degree of selectivity. Native PPelastase was observed at MW 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 890 ± 2 Da and incubation with (14) gave a time dependent emergence of a second peak consistent with the formation of a mono-sulfonylated adduct (MW 26148 ± 7 Da) up to a relative intensity of 80%. The observed mass difference of 258 is in good agreement with a calculated one of 254, consistent with sulfonylation of the enzyme. Only one equivalent of the inhibitor is observed to bind, compatible with the formation of a 1 : 1 EI complex. The conditions of the ionisation, 1 % formic acid, should result in protein unfolding as a result of the multiple side chain positive charges. As a result, bound complexes can probably be regarded as covalent because EI complexes formed from reversible inhibitors should dissociate under these conditions. The 4-isopropyl-N-benzoyl compound (14) has therefore irreversibly sulfonylated the enzyme. However, the precise location of the sulfonylation cannot be determined from these results. An X-ray crystal structure or further MS analysis of tryptic digests of the inactivated enzyme are required to determine whether or not Ser-195 is indeed sulfonylated as was observed with the simple N-benzoyl compound (3).16
890 ± 2 Da and incubation with (14) gave a time dependent emergence of a second peak consistent with the formation of a mono-sulfonylated adduct (MW 26148 ± 7 Da) up to a relative intensity of 80%. The observed mass difference of 258 is in good agreement with a calculated one of 254, consistent with sulfonylation of the enzyme. Only one equivalent of the inhibitor is observed to bind, compatible with the formation of a 1 : 1 EI complex. The conditions of the ionisation, 1 % formic acid, should result in protein unfolding as a result of the multiple side chain positive charges. As a result, bound complexes can probably be regarded as covalent because EI complexes formed from reversible inhibitors should dissociate under these conditions. The 4-isopropyl-N-benzoyl compound (14) has therefore irreversibly sulfonylated the enzyme. However, the precise location of the sulfonylation cannot be determined from these results. An X-ray crystal structure or further MS analysis of tryptic digests of the inactivated enzyme are required to determine whether or not Ser-195 is indeed sulfonylated as was observed with the simple N-benzoyl compound (3).16
In an attempt to take advantage of the smaller S1 pocket present in PPelastase the following compounds were tested as inhibitors: (E)-4-ethylidene-N-benzoyl-β-sultam (17) and 4-ethyl N-(N,N-methylphenyl)carbamoyl-β-sultam (18). The kinetic results obtained are given in Table 1.
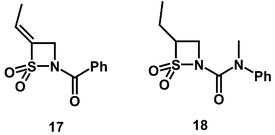
The ki value of (E)-4-ethylidene-N-benzoyl-β-sultam (17) is similar to that for the unsubstituted N-benzoyl-β-sultam (3) despite a 5-fold decrease in reactivity towards hydroxide ion catalysed hydrolysis. The EREF values for the two compounds are 540 and 61 (Table 1) respectively. These values can also be compared to the EREF value for the 4-isopropyl-N-benzoyl compound (14) of 1.2 × 103 which is the least reactive of the three N-benzoyl β-sultams.
This suggests that the 4-isopropyl substituent binds slightly more favourably into the S1 pocket of PPelastase than the 4-(E)-ethylidene substituent in the N-benzoyl series. This is surprising given the preference of the enzyme for alanine residues and may be due to the restricted rotation resulting from unsaturation.
The effectiveness of the ethyl substituent in increasing recognition was observed in 4-ethyl-N-(N,N-methylphenylcarbamoyl)-β-sultam (18). This compound shows relatively low ki values but is by far the least reactive β-sultam to display enzyme inhibition, being 10-fold less reactive than the simple N-carbamoyl compound (4). It therefore has the second highest selectivity towards PPelastase in the presence of hydroxide ion with an EREF of 3.8 × 103. This suggests that the additional methyl group in the 4-isopropyl compounds is unfavourable, possibly due to steric interactions in the active site.
In the N-carbamoyl series the presence of the alkyl substituent in the 4-position of the β-sultam ring increases the EREF value by an overall factor of 100-fold. The 4-ethyl substituted compound (18) is 4-fold more potent as an elastase inhibitor than the 4-isopropyl compound (15). This improvement is probably due to both the increased rotational mobility of the 4-ethyl substituent as a result of its lower bulk and improved binding in the S1 active-site pocket. Based on a structural comparison to an alanine-containing peptide an N-methyl-β-sultam may show even greater binding.
It is notable, despite differing N-substituents, that the EREF value for the conformationally restricted 4-(E)-ethylidene-N-benzoyl-β-sultam (17) is nearly 10-fold lower than for the 4-ethyl compound (18). This suggests that the binding of β-sultams in the S1 pocket requires the compound to have a degree of flexibility. This direct comparison has some justification despite differing leaving groups because the relative ki values of the simple N-benzoyl (3) and N-carbamoyl (4) compounds was proportional to their reactivity, which may be indicative of similar binding of the N-substituent.
The EREF value for the 4-ethyl compound (18) is still 6-fold lower than that of 4-isopropyl-N-benzyloxycarbonyl-β-sultam (16)which has the highest EREF value of the series presented here. The inhibitor EREFs have been increased by ca. 400-fold from that of the original lead compound, N-benzoyl-β-sultam (3). This increase represents a combination of changes in chemical reactivity and molecular recognition.
The structure of an enzyme : inhibitor complex formed between a 3-ethyl-N-(N-benzyl)carbamoyl-β-lactam (19)compound and HLE has been modelled.33 As expected the 4-ethyl group lies in the S1 pocket but the β-lactam ring remains intact and the resulting oxyanion formed by attack of Ser-195 at the carbonyl is stabilised by the oxyanion hole. It is notable that the binding of the benzyl urea group in the so-called hydrophobic prime site (P1′) is improved by alkylation of the benzylic carbon. This is perhaps a further possible modification to the β-sultam structure although the hydrophobic pocket exploited may not be present in PPelastase.
In conclusion, N-acyl-β-sultams are a novel class of inhibitor compounds that inactivate serine enzymes by irreversible sulfonylation of the active site serine residue. They are inactive towards enzymes lacking active serine residues, indicating that specific sulfonylation of active site residues takes place. Variation in 4-alkyl and N-substituted β-sultams causes differences in the rates of inactivation by 4 orders of magnitude. Such structure–activity relationships highlight the possibilities for further development of this compound series. Improved enzyme binding will allow the application of more stable, and hence more selective, enzyme inhibitors.
Experimental
Synthesis
Melting points were determined on a Gallenkamp melting point apparatus and are uncorrected. Infrared measurements were recorded on a Perkin Elmer 1600 series FT-IR spectrometer. 400 MHz 1H and 67 MHz 13C NMR spectra were determined on a Bruker Advance 400 MHz spectrometer. 500 MHz 1H and 100 MHz 13C NMR spectra were determined on a Bruker AMX 500 spectrometer. GC-MS were determined on a Varian GC-MS with a Finnigan MAT ion trap detector. Mass spectrometry was performed on a Fisons Quattro VG quadrupole mass spectrometer. High resolution mass spectrometry was carried out by the University of Swansea, courtesy of the EPSRC. Elemental analyses were carried out at Medac Ltd. Fluka or Merck silica gel 60 was used for all chromatographic separations, with silica gel 60 F254 TLC plates used for initial investigations. Solvents were purified according to Perrin and Armarego.34a Starting materials were purchased from Aldrich, Avocado and Lancaster and were used without further purification.1,2-Thiazetidine 1,1-dioxide (ethane β-sultam)
Taurinesulfonyl chloride (30.4 g, 169 mmol) was added to finely ground sodium carbonate (35.9 g, 339 mmol) in ethyl acetate (950 ml) and stirred at ambient temperature for 48 hours. The reaction mixture was filtered through celite and the solvent removed by reduced pressure rotary evaporation at 30 °C, giving a fine white powder (15.9 g, 89%). Mp 50–51 °C (Lit. 51–52 °C)351H NMR: δ (CDCl3 3.39 (2H, dt, J 4 and 7, CH2N), 4.32 (2H, dt, J 2 and 7, CH2SO2), 5.53 (1H, br s, NH). 13C NMR: δ (CDCl3 60.6, 26.8. IR: νmax(cm−1): 3307, 3048, 3022, 2991, 2918, 1416, 1336, 1299, 1249, 1212, 1171, 1156, 1107, 966, 803, 760, 668, 615. m/z (GC-MS) (M+H): 108, 77, 54, 42.(1,1-Dioxo-1λ6,2-thiazetidin-2-yl)(phenyl)methanone (3)
To a −5 °C stirred solution of 1,2-thiazetidine 1,1-dioxide (0.6 g, 5.6 mmol) in dry DCM (20 ml) was added DMAP (0.11 g, catalytic), and benzoyl chloride (1.18 ml, 10.2 mmol) dropwise. Triethylamine (1.88 ml, 13.5 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (10 ml) and was then washed with water (15 ml) and brine (2 × 15 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale orange solid (1.6 g) which was purified by column chromatography (75 g silica) (6 ∶ 4 diethyl ether ∶ DCM, Rf = .48) yielding a white powder (0.59 g, 49.9%). Mp 103–105 °C. 1H NMR: δ (CDCl3 8.13 (2H br d, Ph), 7.62 (1H, br tt, Ph), 7.53 (2H, br t, Ph), 4.3 (2H, t, J 7.74, CH2SO2), 3.92 (2H, t, J 7.7, CH2N). 13C NMR: δ (CDCl3) 167.4, 133.6, 132.1, 128.9, 128.4, 56.9, 30.9 IR; νmax(cm−1) 2997, 2924, 2854, 1670, 1448, 1337, 1197, 1165, 778, 715.9. HREI-MS [M+H] for C9H9SO3N calc. 212.0381 found 212.0385.1,1-Dioxo-1λ62-thiazetidine-2-carboxylic acid benzyl ester (5)
To a −5 °C stirred solution of 1,2-thiazetidine 1,1-dioxide (0.6 g, 5.6 mmol) in dry DCM (20 ml) was added DMAP (0.11 g, catalytic), and benzyl chloroformate (1.21 ml, 8.5 mmol) dropwise. Triethylamine (1.9 ml, 13.8 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (10 ml) and was then washed with water (15 ml) and brine (2 × 15 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil/solid (1.1 g) which was purified by column chromatography (50 g silica) (1 ∶ 1 DCM ∶ diethyl ether, Rf = 0.6) yielding a white powder (0.23 g, 17%). Mp 99–101 °C. 1H NMR: δ (CDCl3) 7.4 (5H, br, Ph), 5.3 (2H, s, CH2Ph), 4.2 (2H, t, J 7.21, CH2SO2), 3.75 (2H, t, J 7.33, CH2N) 13C NMR: δ (CDCl3) 151, 134.7, 128.6, 128.5, 128, 68.8, 57.9, 32. IR: νmax (cm−1) 3046, 2984, 1732, 1456, 1391, 1326, 1204, 1173, 1135, 1056, 952, 911, 780, 767, 699. HREI-MS [M+H] for C10H11SO4N calc. 259.0753 found 259.0749.1,1-Dioxo-1λ6,2-thiazetidine-2-carboxylic acid N-methyl-N-phenylamide (4)
To a −5 °C stirred solution of 1,2-thiazetidine 1,1-dioxide (0.6 g, 5.6 mmol) in dry DCM (20 ml) was added DMAP (0.11 g, catalytic), and methyl(phenyl)carbamoyl chloride (1.43 g, 8.4 mmol) dropwise. Triethylamine (1.88 ml, 13.5 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (60 ml) and was then washed with water (3 × 20 ml) and brine (2 × 20 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow solid/oil (1.5 g) which was purified by column chromatography (75 g silica) (1 ∶ DCM ∶ diethyl ether, Rf = 0.55) yielding a white powder (0.35 g, 26%). Mp 111–112 °C. 1H NMR: δ (CDCl3) 7.4 (5H, m, Ph), 4.02 (2H, t, J 7.75, CH2SO2), 3.58 (2H, dd, J 6.63 and 7.72, CH2N), 3.38 (3H, s CH3N). 13C NMR: δ (CDCl3) 152.6, 141.2, 129.6, 128.3, 128.1, 56.5, 39.3, 32.2. IR: νmax (cm−1) 2954.3, 2923.5, 2853.8, 1670.3, 1458.1, 1373.3, 1346.2, 1299.8, 1279, 1212.4, 1158, 1136. HREI-MS [M+H] for C10H12SO3N2 calc. 241.0647 found 241.0650.2,4-Bis(tert-butyldimethylsilyl)-1,2-thiazetidine 1,1-dioxide
To a −78 °C stirred solution of 1,2-thiazetidine 1,1-dioxide (2.5 g, 23.3 mmol) in dry THF (190 ml) was added n-BuLi (1.6 M, 33 ml, 52.8 mmol) dropwise over 20 minutes. The mixture was stirred for 10 minutes before the addition of TBDMSCl (17.2 g, 114 mmol) in THF (10 ml). After a period of 30 minutes, the reaction mixture was allowed to warm to ambient over a further 30 minutes and was then quenched with glacial acetic acid (3.9 g, 65 mmol) in THF (4 ml). The organics were diluted with ethyl acetate (200 ml) and saturated NH4Cl (200 ml). The aqueous layer was extracted a further 2 times with ethyl acetate (100 ml) before the combined organic extracts were washed with water (2 × 100 ml), brine (2 × 100 ml) and dried over anhydrous Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (11.1 g) which was purified by column chromatography (400 g silica) (3:2 isohexane ∶ ethyl acetate, Rf = 0.6) yielding a pale cream solid (6.14 g, 78%). Mp 89–91 °C. 1H NMR: δ (CDCl3) 4.20 (1H dd, J 9.0 and 5.5, CHSO2), 3.45 (1H dd, J 9 and 4.5, N–CH2), 3.17 (1H dd, J 4.5 and 5.3, NCH2), 1.02 (9H, s, SiC(CH3)3), 0.96 (9H, s, SiC(CH3)3), 0.33 (3H, s, SiCH3), 0.32 (3H, s, SiCH3), 0.31 (3H, s, SiCH3), 0.25 (3H, s, SiCH3).2-(tert-Butyldimethylsilyl)-4-isopropylidene-1,2-thiazetidine 1,1-dioxide
To a −78 °C stirred solution of diisopropylamine (2.79 ml, 19.9 mmol) in dry THF (130 ml) was added n-BuLi (1.6 M, 12.25 ml, 19.6 mmol) dropwise. The reaction mixture was stirred for 10 minutes before the addition of 2,4-bis(tert-butyldimethylsilyl)-12,-thiazetidine 1,1-dioxide (3.28 g, 9.79 mmol) in THF (15 ml) over 20 minutes. After stirring for a further 10 minutes, acetone (7.2 ml, 98.2 mmol) was added over 5 minutes and the mixture left to stir for 50 minutes. The reaction was quenched with saturated NH4Cl (220 ml) and then extracted with ethyl acetate (200 ml + (2 × 100 ml)) before the combined organics were washed with water (2 × 100 ml), brine (2 × 100 ml) and dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C yielded a pale yellow oil (5.3 g) which was purified by cloumn chromatography (200 g silica) (7 ∶ 3 isohexane ∶ ethyl acetate, Rf = 0.27) yielding a pale cream powder (0.5 g, 20%). Mp 100–101 °C. 1H NMR: δ (CDCl3) 3.78 (2H, s, CH2N), 2.05 (3H, s, CH3), 1.78 (3H, s, CH3), 0.98 (9H, s, SiC(CH3)3), 0.28 (6H, s, SiCH3). 13C NMR : δ (CDCl3) 142.5, 137.8, 40.7, 26.3, 21.2, 20.2, −0.55. IR: νmax (cm−1) 2965.2, 1672.7, 1672.7, 1449.6, 1314, 1282.1, 1162.8, 1037.4, 904.2, 841, 723.7, 647.8. m/z (EI-MS): (M+H) 261.8.4-Isopropylidene-1,2-thiazetidine 1,1-dioxide
To a stirred solution of 2-(tert-buty1dimethylsilyl)-4-isopropylidene-1,2-thiazetidine 1,1-dioxide (0.204 g, 0.78 mmol) in THF (20 ml) was added TBAF (1 M, 0.78 ml, 78 mmol) in one portion. The reaction mixture was stirred for 3 minutes before quenching with saturated NaCl (30 ml), extracting with ethyl acetate (15 ml) and then the combined organics were dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (0.4 g) which was purified by column chromotography (25 g silica) (2 ∶ 3 isohexane ∶ ethyl acetate, Rf = 0.32) yielding a white powder (77mg, 67%). Mp 66–68 °C. 1H NMR: δ (CDCl3)5.40 (1H, br s, NH), 3.72 (2H, s, CH2N), 2.08 (3H, s, CH3), 1.70 (3H, s, CH3). 13C NMR: δ (CDCl3)142.8, 140.8, 37.6, 21.4, 20.5. IR: νmax(cm−1) 3274.6, 1701.5, 1444.6, 1373.9, 1326.1, 1283.0, 1245.6, 1149.1, 1130.0, 1018.1, 940.7, 920.5, 869.1, 680.9.(4-Isopropylidene-1,1-dioxo-1λ6,2-thiazetidin-2-yl)phenylmethanone (11)
To a −5 °C stirred solution of 4-isopropylidene-1,2-thiazetidine 1,1-dioxide (77 mg, 0.523 mmol) in dry DCM (6 ml) was added DMAP (0.1 g, catalytic), and benzoyl chloride (0.091 ml, 0.78 mmol) dropwise. Triethylamine (0.18 ml, 1.26 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (10 ml) and was then washed with water (15 ml) and brine (2 × 15 ml)before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (0.4 g) which was purified by column chromotography (25 g silica) (3 ∶ 2 isohexane ∶ ethyl acetate, Rf = 0.36) yielding a white powder (93mg, 71%). Mp 82–84 °C. 1H NMR: δ (CDCl3) 8.01 (2H, d, J7.5, Ph), 7.61 (1H, t, J 7.4, Ph),7.51 (2H, d, J 7.6, Ph), 4.32 (2H, s, CH2), 2.10 (3H, s, CH3), 1.86 (3H, s, CH3). 13C NMR: δ (CDCl3) 168, 143.3, 140.8, 133.9, 133.2, 129.2, 128.9, 40.9, 22.1, 20.8. IR: νmax (cm−1) 2923.8, 2854, 1670.1, 1600.1, 1376.8, 1337.2, 1283.5, 1199.7, 1165.4, 1125.5, 1032.1, 778, 715.9. HREI-MS [M+H] for C12H13SO3N calc. 252.0694 found 252.0690.4-Isopropylidene-1,1-dioxo-1λ6,2-thiazetidine-2-carboxylic acid benzyl ester (12)
To a −10 °C stirred solution of 4-isopropylidene-1,2-thiazetidine 1,1-dioxide (78.5 mg, 0.533 mmol) in dry DCM (6 ml) was added DMAP (0.1 g, catalytic), and benzyl chloroformate (0.12 ml, 0.795 mmol) dropwise. Triethylamine (0.19 ml, 1.33 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (10 ml) and was then washed with water (15 ml) and brine (2 × 15 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (0.2 g) which was purified by column chromatography (25 g silica) (3 ∶ 2 isohexane ∶ ethyl acetate, Rf = 0.32) yielding a white powder (108 mg, 73%. Mp 86–88 °C. 1H NMR:δ (CDCl3) 7.35 (5H, m Ph), 5.28 (2H, s, CH2Ph), 4.10 (2H, s, CH2N), 2.09(3H, s, CH3), 1.80 (3H, s, CH3). 13C NMR: δ (CDCl3), 142.8, 138.1, 135.3, 129, 128.92, 128.9, 128.4, 69, 41.3, 22.1, 20.8. IR: νmax (cm−1) 1731.2, 1445.2, 1386.2, 1315.1, 1248.0, 1179.3, 1153.0, 699.8. HREI-MS [M+NH4] for C13H15SO3N calc. 299.1066 found 299.1069.4-Isopropylidene-1,1-dioxo-1λ6,2-thiazetidine-2-carboxylic acid N-methyl-N-phenylamide (13)
To a −5 °C stirred solution of 4-isopropylidene-1,2-thiazetidine 1,1-dioxide (120 mg 0.816 mmol) in dry DCM (10 ml) was added DMAP (0.1 g, catalytic) and N-methyl-N-phenylcarbamoyl chloride (0.21 g, 1.24 mmol) in THF (2 ml) dropwise. Triethylamine (0.27 ml, 1.89 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (10 ml) and was then washed with water (15 ml) and brine (2 × 15 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 20 °C gave a pale yellow oil (0.45 g) which was purified by column chromatography (25 g silica) (DCM→7 ∶ 3 DCM ∶ diethyl ether, gradient elution) and preparative HPLC (water ∶ ACN ∶ MeOH, gradient elution) yielding a white powder (96 mg, 42%). Mp 102–104 °C. 1H NMR: δ (CDCl3): 7.35 (5H, m, Ph), 4.08 (2H, s, NCH2), 3.39 (3H, s, NCH3), 1.95 (3H, s, CH3), 1.75 (3H, s, CH3). 13C NMR: δ (CDCl3) 153.4, 142, 141.6, 137.4, 129.4, 128.6, 128.5, 41.9, 39.8, 21.7, 20.6. IR: νmax(cm−1) 2945.8, 1670.0, 1490.4, 1442.2, 1390.9, 1315.1, 1250.0, 1201.0, 1161.6, 1128.1, 1066.4, 954.8, 883.7, 762.1, 672.1. HREI-MS [M+H] for C13H16SO3N2 calc. 281.0960 found 281.0965.2-(tert-Butyldimethylsilyl)-4-isopropyl-1,2-thiazetidine 1,1-dioxide
Palladium (0.35 g, 10% on C,) in ethyl acetate (1 ml) was added to a solution of 2-(tert-butyldimethylsilyl)-4-isopropylidene-1,2-thiazetidine 1,1-dioxide (0.45 g, 1.71 mmol) in dry ethanol (12 ml) under argon. The mixture was hydrogenated at 100 psi H2for 24 hours before being filtered through a celite plug and rotary evaporated at 30 °C, yielding a white powder (278 mg, 62%). Mp 101–104 °C. 1H NMR: δ (CDCl3) 4.05 (1H, ddd, J 6.67, 8.59 and 11.04, CHSO2), 3.29 (1H, dd, J 7.33 and 8.36, CH2N), 2.87 (1H, dd, J 6.86 and 7.17, CH2N), 2.20 (1H, d septet, J 6.55 and 10.97, CH (CH3)2), 1.07 (3H, d, J 6.43, CH3CH), 0.86 (3H, d, J 6.8, CH3CH), 0.85 (9H, s, C(CH3)3), 0.16 (6H, s, Si(CH3)2). m/z (EI-MS) (M+H): 262.4-Isopropyl-1,2-thiazetidine 1,1-dioxide
To a stirred solution of 2-(tert-butyldimethylsilyl)-4-isopropyl-1,2-thiazetidine 1,1-dioxide (0.278 g, 0.107 mmol) in THF (20 ml) was added TBAF (1 M, 1.7 ml, 1.7 mmol) in one portion, The reaction mixture was stirred for 3.5 minutes before quenching with a saturated aqueous NaCl solution (30 ml), extracting with ethyl acetate (15 ml) and then the combined organics were dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (0.8 g) which was purified by column chromatography (35 g silica) (2 ∶ 3 isohexane ∶ ethyl acetate, Rf = 0.35) yielding a white powder (123 mg, 77%). Mp 64–66 °C. 1H NMR: δ (CDCl3) 5.60 (1H, br s, NH), 4.22 (1H, ddd, J 6.79, 8.03 and 11.14, CHSO2), 3.44 (1H, m, CH2N), 3.03 (1H, dd, J 6.38 and 11.75, CH2N), 2.32 (1H, ds, J 6.6 and 11.01, CH(CH3)3), 1.16 (3H, d, J 6.63, CH3), 0.98 (3H, d, J 6.87, CH3). 13C NMR: δ (CDCl3) 81.8, 35.2, 29.4, 20.5, 19.4. IR: νmax (cm−1) 3268.7, 2969.1, 2898.6, 2872.2, 1467.2, 1366.0, 1332.9, 1292.3, 1271.1, 1239.2, 1211.8, 1153.9, 1147.6, 1126.7, 986.0, 910.4, 842.2, 733.0, 645.7.(4-Isopropyl-1,1-dioxo-1λ6,2-thiazetidin-2-yl)phenylmethanone (14)
To a −10 °C stirred solution of 4-isopropyl-1,2-thiazetidine 1,1-dioxide (117 mg 0.785 mmol) in dry DCM (15 ml) was added DMAP (0.05 g, catalytic) and benzoyl chloride (0.14 ml, 1.18 mmol) dropwise. Triethylamine (0.27 ml, 1.89 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight before the addition of DCM (5 ml) and was then washed with water (12 ml) and brine (2 × 15 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (0.4 g) which was purified by column chromatography (25 g silica) (chloroform column, eluting with 3 ∶ 2 hexane ∶ ethyl acetate, Rf = 0.3) yielding a white powder (144 mg, 72%). Mp 84–86 °C. 1H NMR: δ (CDCl3) 7.9 (2H, d, J 7.5, Ph), 7.50 (1H, t, J 7.5, Ph), 7.40 (2H, d, J 7.85, Ph), 4.06 (1H, ddd, J 6.34, 8.7 and 11.0 CHSO2), 3.84 (1H, dd, J 8.35 and 8.5, CH2N), 3.45 (1H, dd, J 6.4 and 8.0, CH2N), 2.23 (1H, ds, J 6.6 and 11.2, CH(CH3)2), 1.09 (3H, d, J 6.6, CH3), 0.93 (3H, d, J 6.7, CH3). 13C NMR: δ (CDCl3) 167.9, 133.9, 132.9, 129.2, 128.9. 37.9, 29.7, 21.6, 19.2. IR: νmax(cm−1) 2961.0, 1665.4, 1581.7, 1468.1, 1449.6, 1391.8, 1327.6, 1289.4, 1209.6, 1181.7, 1164.4, 1028.6, 844.8, 795.7, 705.7, 657.7. HREI-MS [M+NH4] for C12H15SO3N calc. 271.1116 found 271.1113.4-Isopropyl-1,1-dioxo-1λ6,2-thiazetidine-2-carboxylic acid benzyl ester (16)
To a −10 °C stirred solution of 4-isopropyl-1,2-thiazetidine 1,1-dioxide (22.2mg, 0.149 mmol) in dry DCM (2.5 ml) was added DMAP (0.02 g, catalytic) and benzyl chloroformate (0.04 ml, 0.265 mmol) dropwise. Triethylamine (0.05 ml, 0.35 mmol) was added dropwise over 5 minutes forming a pale yellow solution, The reaction mixture was stirred overnight before the addition of DCM (3 ml) and was then washed with water (2.5 ml) and brine (2 × 2.5 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (70 mg) which was purified by column chromatography (8 g silica) (3 ∶ 2 hexane ∶ ethy1 acetate, Rf = 0.33) yielding a white powder (28.4 mg, 67%). Mp 93–94 °C. 1H NMR: δ (CDCl3) 7.35 (5H, m, Ph), 5.27 (2H, s, CH2Ph), 4.09 (1H, ddd, J 6.0, 8.35 and 11.02, CHSO2), 3.75 (1H, dd, J 6.94 and 8.49, CH2N), 3.34 (1H, dd, J 6.4, CH2N), 2.31 (1H, ds, J 6.68 and 11.05, CH(CH3)2), 1.19 (3H, d, J 6.42, CH3), 1.0 (3H, d, J 6.72, CH3). 13 C NMR: δ (CDCl3) 153.2, 128.9, 128.4, 128.7, 128.4, 79.1, 69.1, 38.7, 29.4, 21.6, 19.4. IR: νmax (cm−1) 1732.0, 1455.8, 1386.3, 1341.4, 1319.7, 1207.9, 1185.5, 1170.5, 1127.9, 787.7, 769.5. HREI-MS [M+NH4] for C13H18SO4N calc. 301.1222 found 301.1221.4-Isopropyl-1,1-dioxo-1λ6,2-thiazetidine-2-carboxylic acid N-methyl-N-phenylamide (15)
To a −10° stirred solution of 4-isopropyl-1,2-thiazetidine 1,1-dioxide (62.3mg 0.418 mmol) in dry DCM (5 ml) was added DMAP (0.05 g, catalytic) and N-methyl-N-phenylcarbamoyl chloride (0.11 g, 0.65 mmol) dropwise. Triethylamine (0.14 ml, 0.98 mmol) was added dropwise over 10 minutes forming a pale purple solution. The reaction mixture was stirred overnight at ambient temperature before the addition of DCM (10 ml) and was then washed with water (12 ml) and brine (2 × 12 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale purple oil (0.12 g) which was purified by column chromatography (10 g silica) (3 ∶ 2 hexane ∶ ethy1 acetate, Rf = 0.31) yielding a white powder (87 mg, 74%). Mp 101–103 °C. 1H NMR: δ (CDCl3) 7.4 (2H, d, J 7.9, Ph), 7.36 (1H, t, J 7.37, Ph), 7.29 (2H, d, J 7.24, Ph), 3.85 (1H, ddd, J 6.67, 8.75 and 11.15, CHSO2), 3.59 (1H, dd, J 7.31 and 8.35, CH2N), 3.36 (3H, s, NCH3), 3.31 (1H, dd, J 6.86, CH2N), 2.23 (1H, ds, J 6.55 and 11.03 CH (CH3)2), 1.06 (3H, d, J 6.43, CH3), 0.93(3H, d, J 6.67, CH3). 13C NMR: δ (CDCl3) 153.2, 141.9, 129.9, 128.6, 77.1, 39.7, 38.9, 29.5, 21.5, 19.2. IR: νmax (cm−1) 2965.0, 1668.7, 1594.1, 1494.0, 1419.3, 1367.7, 1333.4, 1299.8, 1183.1, 1152.9, 1009.2, 829.0, 772.5, 697.0. HREI-MS [M+H] for C13H18SO3N2 calc. 283.1116 found 283.1113.2-(tert-Butyldimethylsilyl)-4-(E/Z)-ethylidene-1,2-thiazetidine 1,1-dioxide
To a −78 °C stirred solution of diisopropylamine (4.5 ml, 32 mmol) in dry THF (170 ml) was added n-BuLi (1.6 M, 19.4 ml, 12.1 mmol) dropwise. The reaction mixture was stirred for 10 minutes before the addition of 2,4-bis(tert-butyldimethylsilyl)-1,2-thiazetidine 1,1-dioxide (5.2 g, 15.5 mmol) in THF (20 ml) over 20 minutes. After stirring for a further 10 minutes, acetaldehyde (8.7 ml, 142 mmol) was added over 10 minutes and the mixture left to stir for 1 hour, before allowing to warm to ambient over 30 minutes. The reaction was quenched with saturated NaCl (90 ml) before the addition of sodium bisulfite (400 ml, 40%) and was then stirred for a further 1 hour. The aqueous layer was extracted with ethyl acetate (2 × 200 ml) before the combined organics were washed with water (100 ml), brine (2 × 100 ml) and dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C yielded a pale yellow oil (5.2 g) which was purified by column chromatography (200 g silica) (7 ∶ 3 isohexane ∶ ethyl acetate, Rf = 0.38 and 0.36) yielding a pale yellow oil as a mixture of the two isomers E and Z (1.9 g, 50%). 1H NMR: δ (CDCl3) 6.38 (1H, tq, J 2.34 and 7.21, CH), 3.86 (2H, dq, 1.36 and 2.7, CH2N), 1.8 (3H, dt, 1.36 and 7.2, CH3), 0.98 (9H, s, SiC(CH3)3), 0.3 (6H, s, Si(CH3)2). 13C NMR: δ (CDCl3) 149.2, 125.6, 65.3, 39.9, 25.9, 18.3, 13.6. IR: νmax (cm−1) 3518.3, 2955.5, 2931, 2859.5, 1706.6, 1471.4, 1297.4, 1255.7, 1170.8, 1126.1, 1073.3, 1005.7, 905.6, 839.9, 825.4, 785.2, 686.2, 657.9. m/z (EI-MS) (M + H) 248.4-(E/Z)-Ethylidene-1,2-thiazetidine 1,1-dioxide
To a stirred solution of 2-(tert-butyldimethylsilyl)-4-(E/Z)ethylidene-1,2-thiazetidine 1,1-dioxide (1.9 g, 7.7 mmol) in THF (80 ml) was added TBAF (1 M, 8.7 ml, 8.7 mmol) in one portion. The reaction mixture was stirred for 3 minutes, before quenching with saturated NaCl (100 ml), extracting with ethyl acetate (2 × 50 ml), washing with brine (50 ml) and then dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (2.5 g) which was purified by column chromatography (100 g silica) (2 ∶ 3 isohexane ∶ ethyl acetate, Rf = 0.3) yielding a clear colourless oil (0.92 g, 90%). 1H NMR: δ (CDCl3) 6.45 (0.5 H, qt, J 2.51 and 7.23, CH, E), 6.0 (0.5 H, qt, J 2.01 and 7.26, CH, Z), 5.66 (1H, br s NH), 3.86 (1H, m, CH2N, E),3.83 (1H, m, CH2N, Z), 2.03 (1.5 H, dt, 2.01 and 7.24, CH3, Z), 1.8 (1.5 H, dt, 1.58 and 7.23, CH3, E). 13 C NMR: δ (CDCl3) 149.148.9, 129.1, 128.4, 37.6, 36.8, 15.1, 13.9 (Z isomer in bold). IR: νmax (cm−1) 3287.6, 2957.3, 2931.6, 2862.1, 2797.1, 1703.5, 1467.1, 1377.2, 1304.4, 1167.5, 1084.8, 944.5, 872.3, 728.4-Ethyl-1,2-thiazetidine 1,1-dioxide
Palladium (0.3 g, 10% on C) in ethyl acetate (1 ml) was added to a solution of 4-(E/Z)-ethylidene-1,2-thiazetidine 1,1-dioxide (0.32 g, 2.4 mmol) in dry ethanol (25 ml) under argon. The mixture was hydrogenated for 72 hours before being filtered through a celite plug and rotary evaporated at 30 °C, yielding a pale yellow oil (98 mg, 30%).1H NMR: δ (CDCl3) 5.8 (1H, br s, NH), 4.4 (1H, m, CHSO2), 3.42 (1H, dd, J 6.27 and 8.05, CH2N), 2.94 (1H, dd, J 6.12 and 6.12, CH2N), 2.09 (1H, m, CH2CH3), 1.88 (1H, m, CH2CH3), 1.04 (3H, t, J 7.39, CH3). 13 C NMR: δ (CDCl3) 75.7, 35.2, 22.3, 11.1. IR: νmax (cm−1) 3583.8, 3291.7, 2971.7, 2880.4, 1637, 1461.4, 1383.6, 1307, 1241.2, 1196.6, 1161.4, 1040, 978.6, 850.6, 701.3.
2-(tert-Butyldimethylsilyl)-4-(E)-ethylidene-1,2-thiazetidine 1,1-dioxide
To a −78 °C stirred solution of diisopropylamine (4.28 ml, 30.5 mmol) in dry THF (180 ml) was added n-BuLi (1.6 M,19 ml, 30.4 mmol) dropwise. The reaction mixture was stirred for 10 minutes before the addition of 2,4-bis(tert-butyldimethylsilyl)-1,2-thiazetidine 1,1-dioxide (5 g, 14.9 mmol) in THF (20 ml) over 20 minutes. After stirring for a further 10 minutes, acetaldehyde (8.3 ml, mmol) was added over 10 minutes and the mixture left to stir for 1 hour before quenching with glacial acetic acid (15 ml) and stiring for a further 30 minutes. The aqueous layer was extracted with ethyl acetate (2 × 100 ml) before the combined organics were washed with brine (100 ml) and dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C yielded a pale yellow oil (8.5 g) which was purified by column chromatography (200 g silica) (7 ∶ 3→6 ∶ 4 isohexane ∶ ethyl acetate, Rf = 0.38) yielding a pale yellow oil as the product (0.4 g, 10.9%) and a second fraction determined to be starting material (Rf = 0.65, 3.8 g). Yield based upon total conversion, 45%. 1H NMR: δ (CDCl3) 6.38 (1H, tq, J 2.34 and 7.21, CH), 3.86 (2H, dq, 1.36 and 2.7, CH2N), 1.8 (3H, dt, 1.36 and 7.2, CH3), 0.98 (9H, s, SiC(CH3)3), 0.3 (6H, s, Si(CH3)2). 13C NMR: δ (CDCl3) 149.2, 125.6, 65.3, 39.9, 25.9, 18.3, 13.6. IR: νmax (cm−1) 3518.3, 2955.5, 2931, 2859.5, 1706.6, 1471.4, 1297.4, 1255.7, 1170.8, 1126.1, 1073.3, 1005.7, 905.6, 839.9, 825.4, 785.2, 686.2, 657.9.4-(E)-Ethylidene-1,2-thiazetidine 1,1-dioxide
To a stirred solution of 2-(tert-butyldimethylsilyl)-4-(E)-ethylidene-1,2-thiazetidine 1,1-dioxide (0.4 g, 1.6 mmol) in THF (20 ml) was added TBAF (1 M, 1.7 ml, 1.7 mmol) in one portion. The reaction mixture was stirred for 3 minutes, before quenching with saturated NaCl (40 ml), extracting with ethyl acetate (2 × 25 ml), washing with brine (25 ml) and then dried over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (0.55 g) which was purified by column chromatography (20 g silica) (2 ∶ 3 isohexane ∶ ethyl acetate, Rf = 0.3) yielding a clear colourless oil (0.15 g, 71%). 1H NMR: δ (CDCl3) 6.44 (0.5 H, qt, J 2.52 and 7.23, CH), 5.65 (1H, br s, NH), 3.86 (1H, m, CH2N,), 1.8 (1.5 H, dt, 1.59 and 7.22, CH3). 13C NMR: δ (CDCl3) 149.1, 128.5, 36.8, 13.9. IR: νmax (cm−1) 328.4, 2957.2, 2931.1, 2860.7, 2797.0, 1703.4, 1467, 1377.1, 1304.4, 1166.9, 1084, 872.3, 728.2.[4-(E/Z)-Ethylidene-1,1-dioxo-1λ6,2-thiazetidin-2-yl](phenyl)methanone (17)
To a −10 °C stirred solution of 4-(E/Z)-ethylidene-1,2-thiazetidine 1,1-dioxide (0.32 g 2.4 mmol) in dry DCM (35 ml) was added DMAP (0.1 g, catalytic) and benzoyl chloride (0.63 g, 3.7 mmol) dropwise. Triethylamine (0.85 ml, 6.1 mmol) was added dropwise over 10 minutes forming a pale yellow solution. The reaction mixture was stirred overnight at ambient temperature before the addition of DCM (25 ml) and was then washed with water (25 ml) and brine (2 × 25 ml) before drying over Na2SO4. Reduced pressure rotary evaporation at 30 °C gave a pale yellow oil (1 g) which was purified by column chromatography (35 g silica) (3 ∶ 2 hexane ∶ ethyl acetate, Rf = 0.28 and 0.22) yielding two fractions of white powders, E and Z isomers respectively [120 mg (E). Mp 92–94 °C] and [160 mg (Z). Mp 63–65 °C]. Overall 49% yield. E Isomer:1H NMR: δ (CDCl3) 8.03 (2H, m, Ph), 7.62 (1H, m, Ph), 7.52 (2H, m, Ph), 6.65 (1H, qt, J 2.54 and 7.22 CH), 4.43 (2H, dq, 1.62 and 2.53, CH2N), 1.93 (3H, dt, J 1.65 and 7.25, CH3). 13C NMR: δ (CDCl3) 167.5, 143.8, 133.6, 132.6, 130.7, 128.9, 128.4, 40.1, 14.4. IR: νmax (cm−1) 2954.7, 2924.2, 2854, 1670.4, 1464.5, 1449.3, 1376.9, 1334.4, 1311.8, 1267.1, 1187, 1158.1, 946.6. HREI-MS [M+NH4] for C11H11SO3N calc. 255.0803 found 255.0799.Z Isomer: 1H NMR: δ (CDCl3) 8.03 (2H, m, Ph), 7.65 (1 H, m, Ph), 7.55 (2H, m, Ph), 6.27 (1H, qt, J 2.02 and 7.24, CH), 4.39 (2H, dq, 2.1 and 2.03, CH2N), 2.15 (3H, dt, J 2.14 and 7.26, CH3). 13C NMR: 167.5, 143.2, 133.6, 132.6, 131.5, 128.9, 128.4, 40.9, 159. IR: νmax (cm−1) 3060.2, 2962.6, 1679.9, 1599.8, 1581.5, 1451.8, 1425.6, 1378.5, 1324.9 1256.2, 1216.6, 1162.3, 1136.2, 1126.7, 1028, 1017.6, 992.1, 840.8, 755.8, 710.9, 691.6, 635.4. HREI-MS [M+NH4] for C11H11SO3N calc. 255.0803 found 255.0801.
Kinetics
Standard UV spectroscopy was carried out on a Cary 1E UV-visible spectrophotometer (Varian, Australia) equipped with a twelve compartment cell block. The instrument was used in double beam mode, allowing six reaction cells to be followed in a single run. The cell block was thermostatted using a peltier system. Stopped flow experiments used an SX 18 MV Spectrakinetic monochromator (Applied Photophysics, Leatherhead, England) equipped with an absorbance photomultiplier. The reagent syringes were thermostatted with a Grant thermostatted water circulator. pH-stat experiments were performed on a ABU 91 Autoburette (Radiometer, Copenhagen, Denmark), controlled by a VIT 90 video titrator. The SAM 90 sample station incorporated a machined aluminium E2000 sample block rotor thermostatted by a MGW Lauda M3 water circulator. pH was measured by a pHG200–8 Glass pH electrode and a REF200 ‘Red Rod’ reference electrode (Radiometer). Temperature was monitored by a T101 temperature sensor.pH measurements; were made with either a ϕ40 (Beckman, Fullerton, USA) or 3020 (Jenway, Dunmow, England) pH meter. Electrodes were semi-micro Ag/AgCl and calomel (Russel, Fife, Scotland and Beckman respectively). A calibration of the pH meter was carried out at 30 °C using pH 7.00 ± 0.01, pH 4.01 ± 0.02 or pH 10.00 ± 0.02 calibration buffers.
ESIMS experiments were carried out on a VG Quattro SQ II (Micromass, Altrincham, England) and NMR experiments on a 400 MHz instrument (Bruker, Germany).
AnalaR grade reagents and deionised water were used throughout. Sodium hydroxide solutions were titrated prior to use against a 1.00 M ± 0.1% hydrochloric acid volumetric reagent (D.H. Scientific, Huddersfield, England) using phenolphthalein as an indicator.
Organic solvents were glass distilled prior to use and stored under nitrogen. For solution pHs ≥3 and ≤11 the pH was controlled by the use of ≤0.2 M buffer solutions of formate (pKa 3.75), ethanoate (pKa 4.72), 2-(N-morpholino)ethanesulfonic acid (MES) (pKa 6.1), 3-(N-morpholino)propanesulfonic acid (MOPS) (pKa 7.2), N-[tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid (TAPS) (pKa 8.4), 3-(cyclohexylamino)-2-hydroxypropane-1-sulfonic acid (CAPSO) (pKa 9.6), and 3-(cyclohexylamino)propane-1-sulfonic acid (CAPS) (pKa 10.4). For general pH work, buffers were prepared by partial neutralisation of solutions of their sodium salts to the required pH. For the alcoholysis reactions, buffers were prepared by the addition of 0.25, 0.50 or 0.75 aliquots of 1 M NaOH to solutions of the alcohol. In all experiments temperatures were maintained at 30 °C and ionic strength at 1.0 M with AnalaR grade KCl unless otherwise stated. Reaction concentrations were generally within the range ≥2×10−5 M to ≤2 × 10−4M to ensure pseudo-first order conditions.
Hydroxide ion concentrations were calculated using pKw (H2O) = 13.883 at 30 °C.34b Reactions studied by UV spectrophotometry were usually commenced by injections of acetonitrile or dioxan stock solutions of the substrate (5–50 µl) into the cells containing pre-incubated buffer (2.5 ml). Final reaction cells contained ≤5% acetonitrile or dioxan v/v. The pH of the reaction cells was measured before and after each kinetic run at 30 °C, kinetic runs experiencing a change >0.05 units were rejected. Reactant disappearance or product appearance were followed at absorbance change maxima for individual compounds. The solubility of compounds was ensured by working within the linear range of absorbance in corresponding Beer–Lambert plots. If required, greater than 1% MeCN v/v was used to aid solubility. Pseudo-first order rate constants from exponential plots of absorbance against time or gradients of initial slopes were obtained using the Enzfitter package (Elsevier Biosoft, Cambridge, England) or the CaryBio software (Varian). pH-rate profiles were modelled to theoretical equations using the Scientist program (V2.02, Micromath Software Ltd, USA).
Reactions studied by stopped flow UV spectrophotometry used stock solutions prepared at twice the standard UV concentration in 1 M KCl. Hydroxide solutions, buffer solutions or solutions of nucleophilic reagents were prepared at twice the required concentration. The substrate solution and the reaction mixture were placed in separate syringes and thermostatted at 30 °C before pneumatic injection into the reaction into the reaction cell. Where applicable, the pH of solutions was measured prior to use. If greater than 1% acetonitrile v/v was required for solubility, then organic solvent concentration of all solutions used was fixed at the required reaction cell amount. The photomultiplier voltage was set to a maximum on deionised water and absorbance wavelengths common to the standard UV experiments were used. Pseudo-first order rate constants from exponential plots of absorbance against time were obtained using the supplied fitting software (Applied Photophysics).
For reactions studied by pH-stat standardised NaOH was delivered to a stirred sample solution (10 ml) held within the thermostatted sample station. All reactions were performed under nitrogen to prevent CO2 absorption. Data were exported to a Windows PC via an RS232 interface and the terminal program (Microsoft Corp, USA). Conversion into an appropriate format was by means of an Excel (Microsoft Corp, USA) Macro and results were fitted to first order equations via the Enzfitter program (Elsevier Software). The titrant used was 0.01–0.1 M NaOH standardised prior to use by means of phenolphthalein titration against 1.00 M HCl (Volumetric reagent, D.H. Scientific). Reactions were performed in 1 M KCl, 5% MeCN v/v, with a pH set point of 6–7. Concentrations of sample were in the range of 1–2 mM with expected titrant added volumes of up to 1.0 ml.
Enzyme inhibition studies
Assays were performed at 30 °C in 0.1 M pH 8.5 TAPS buffer with I = 1.0 M (KCl). The substrate used was N-suc-(L-Ala)3-p-nitroanilide (2) and stock solutions were made up in distilled methanol. Enzyme activity was monitored by following the appearance of the p-nitroaniline product at 390 nm. During inhibition studies a fixed assay substrate concentration of 6.0 × 10−5 M was used resulting in a constant methanol concentration of 1.5% v/v. Incubations of enzyme and inhibitor (500 µl) were carried out at 30 °C in 0.04 M buffer, 20% MeCN v/v, 8 × 10−5 M PPelastase and up to 5 × 10−3 M β-sultam. These conditions were created in a separate UV cell containing stock PPelastase suspension (200 µl), 0.1 M buffer (200 µ) and inhibitor stock solution in MeCN or MeCN alone (100 µ). Aliquots of this solution (50 µl) were assayed for PPelastase activity at various time intervals by injection into 0.1 M pH 8.5 TAPS (2.36 ml) containing MeCN (150 µl) and substrate solution (40 µl). This gave final assay conditions of 6.0 × 10−5 M substrate, 1.5 × 10−6 M PPelastase, 6 % MeCN v/v and 1.5 % MeOH v/v. In all cases assays of control incubations were performed at the same time as inhibitor incubations. These incubations substituted pure MeCN for the β-sultam solution but then matched the remaining methodology in all respects. Initial rates were monitored over up to eight minutes and the gradients of the slopes obtained used as a measure of enzyme activity. Gradients from inhibitor runs were normalised with respect to the first order exponential decrease in β-sultam concentration due to hydroxide ion catalysed hydrolysis at the pH of the incubation. This was done by dividing the observed gradient by the β-sultam concentration at the start of the experiments, time = 0, and multiplying by the β-sultam concentration at the time of the assay, time = t:Normalised gradient at time t = (observed gradient/[Sultam]o) × [Sultam]t
The average control gradient was taken as 100% activity. This value and the normalised gradients were used to calculate % activity:
% Activity = (Normalised gradient/Average control gradient) × 100
Pseudo-first order rate constants of inactivation, Kobs values, were obtained from plots of % activity against time using the Enzfitter program (Elsevier Biosoft). The second order rate constants of inactivation, Ki values, were obtained by dividing the first order Kobs values by the concentration of β-sultam in the inactivation.
pKa determinations
A solution of the compound was placed in a UV cell at 30 °C and typically titrated by means of additions of NaOH or HCl (≤50 µl). Absorbance values at the wavelength of maximum change and the cell solution pH were measured after each injection of the 0.01, 0.05, 0.1 or 1.0 M standardised NaOH or HCl. Near the end point injections of the 0.01 M solution (5 µl) were made to ensure that sufficient pH values were covered in the proximity of the pKa.The results were analysed using the Enzfitter software package (Elsevier Biosoft), pKa1 determination equation, A = [(Amin + Amax) × (10(pH − pKa))]/[(10(pH − pKa)) + 1] where A = absorbance of solution at a particular pH and wavelength, Amin = minimum absorbance where 100% of dissociating species is in form with lowest absorption, Amax = maximum absorbance where 100% of dissociating species is in form with highest absorption, pH = solution pH and pKa = pKa of dissociating species.
1H-NMR experiments
The β-sultam (15 mg) was dissolved in 50 ∶ 50 v/v CD3CN ∶ D2O (ca. 3 ml) and the 1H-NMR spectra acquired at 400 MHz and 300 K. One drop additions of 4% v/v NaOD in D2O were made to the NMR tube until complete conversion of reactants had occurred with spectra being acquired after each addition. In buffer solution, the β-sultam (15 mg) was dissolved in 50 ∶ 50 v/v CD3CN ∶ 0.4 M buffer solution in D2O (ca. 3 ml), the pD of which was measured before and after the experiment. The solutions were also routinely submitted for both positive and negative mode ESIMS-MS.X-Ray crystal preparation, data collection, processing and structure refinement
PPE crystals were prepared as previously described.36 A saturated solution of the N-benzoyl β-sultam (3) in sodium acetate buffer (pH 5.0, 50 mM), sodium sulfate (25 mM) and DMSO (10% v/v) was prepared. The mother liquor containing the crystals was exchanged with this solution over a 1 h period. After 24 h soaking in the inhibitor solution, the crystals were placed in a cryoprotectant solution containing 20% (v/v) glycerol and flash-cooled in liquid nitrogen.Data were collected at 100 K using a Rigaku rotating anode generator (CuKα radiation) and 345 mm Mar Research image plate detector to a resolution limit of 1.67 Å. The crystal belonged to the space group P212121 with a = 50.77 Å, b = 57.61 Å, c = 74.48 Å. Raw data were processed with the programs MOSFLM and SCALA (CCP4 suite.37Rmerge was 4.3% for all data (19.3% in the highest resolution shell, 1.76–1.67 Å). Data were 99.3% complete (96.6% in the highest resolution shell) with 92 216 reflections of which 25 847 were unique after reduction. The overall I/σ1 was 20.1 and 4.8 in the highest resolution shell (1.68–1.67 Å).
The PPE:BCM7 structure (excluding the inhibitor) (PDB entry 1QIX) was used the starting model. The structure was refined using REFMAC (CCP4 suite) with a total of 4% of the reflections in the entire dataset randomly selected in order provide a test set for the Rfree calculations. Models and electron density maps were displayed using the program O.38 At the stage in refinement with the Fo − Fc map showed clear and unambiguous density for the β-sultam derived atoms, they were included in the model and an appropriate PROTIN dictionary file created. The sulfonate ester bond was simulated solely with bond length restraints. Only the six atoms derived from the β-sultam ring were visible in the structure with the benzoyl group apparently disordered or missing. The side chains of three residues of PPE (Arg-61, Arg-125, Arg-223) were also disordered and were included as alanines in the model. During the latter stages of refinement, 141 water molecules were added along with the bound calcium and sulfate ions,36 giving a final R-factor of 19.0% and free R-factor of 21.3%. The average B-factor for the atoms in the β-sultam inhibitor molecule was 20.3 Å2.
Acknowledgements
For CASE awards we thank the EPSRC and AstraZeneca (JMW) and British Biotech (PSH). We also thank C J Schofield and R C Wilmouth for the X-ray crystal structure.References
- M. I. Page, Enzyme Inhibition in Comprehensive Medicinal Chemistry, ed. P. G. Sammes, Pergamon Oxford, 1990, Vol. 2, pp. 61–87 Search PubMed.
- (a) C. W. Wharton, The Serine Proteinases in Comprehensive Biological Catalysis, ed. M. Sinnott, Academic Press, London, 1997, Vol. 1, Ch. 11, pp. 345–379 Search PubMed; (b) D. J. Underwood, B. G. Green, R. Chabin, S. Mills, J. B. Doherty, P. E. Finke, M. MacCoss, S. K. Shah, C. S. Burgey, T. A. Dickinson, P. R. Griffin, T. E. Lee, K. M. Swiderek, T. Covey, W. M. Westler and W, B. Knight, Biochemistry, 1995, 34, 14344–14355 CrossRef CAS; (c) J. A. Katzenellenbogen, R. Rai and W. Dai W, Bioorg. Med. Chem. Lett. 2, 1992, 11, 1399–1404 Search PubMed; (d) W. B. Knight, A. L. Maycock, B. G. Green, B. M, Ashe, P. Gale, H. Weston, P. E. Finke, W. K. Hagmann, S. K. Shah and J. B. Doherty, Biochemistry, 1992, 31, 4980–4986 CrossRef CAS; (e) C-M Kam, K. Fujikawa and J. C. Powers, Biochemistry, 1988, 27, 2547–2557 CrossRef CAS; (f) M. A. Navia, J. P. Springer, T. Y. Lin, H. R. Williams, R. A. Firestone, J. M. Pisano, J. B. Doherty, P. E. Finke and K. Hoogsteen, Nature, 1982, 327, 79–82 CrossRef CAS; (g) M. H. Gelb and R. H. Abeles, J. Med. Chem., 1986, 29, 585–589 CrossRef CAS; (h) M. H. Gelb and R. H. Abeles, Biochemistry, 1984, 23, 6569–6604.
- W. A. Metz and N. P. Peet, Prog. Inflammation Res., 1999, 9, 853–868 Search PubMed.
- (a) W, Bode, A. Z. Wei, R. Huber, E. Meyer, J. Travis and S. Neuman, EMBO J., 1986, 5, 2453–2458 CAS; (b) M. A. Navia, B. M. McKeever, J. P. Springer, T. Y. Lin, H. R. Williams, E. M. Fluder, C. P. Dorn and K. Hoogsteen, Proc. Natl. Acad. Sci. USA, 1989, 86, 7–11 CAS.
- W. Bode, E. Meyer Jr. and J. C. Powers, Biochemistry, 1989, 28, 1951–1962 CrossRef CAS.
- (a) L. H. Takahashi, R. Radhakrishan, R. E. Rosenfeld Jr., E. F. Meyer Jr. and D. A. Trainer, J. Am. Chem. Soc., 1989, 111, 3368–3374 CrossRef CAS; (b) A. Renaud, P. Lestienne, D. L. Hughes, J. G. Bieth and J-L. Dimicoli, J. Biol. Chem., 1983, 258, 8312–8316 CAS.
- (a) P. D. Edwards, E. F. Meyer Jr., J. Vijayalakshmi, P. A. Tuthill, D. A. Andisk, B. Gomes and A. Strimpler, J Am. Chem. Soc., 1992, 114, 1854–1863 CrossRef CAS; (b) M. J. Costanzo, B. E. Maryanoff, L. R. Hecker, M. R. Schott, S. C. Yabut, H-C Zhang, P. Andrade-Gordon, J. A. Kauffman, J. M. Lewis, R. Krishnan and A. Tulinsky, J. Med. Chem., 1996, 39, 3039–3043 CrossRef CAS; (c) P. R. Bernstein, B. C. Gomes, B. J. Kosmider, E. P. Vacek and J. C. Williams, J. Med. Chem., 1995, 38, 212–215 CrossRef CAS; (d) P. D. Edwards, D. W. Andisik, A. M. Strimpler, B. C. Gomes and P. A. Tuthill, J. Med. Chem., 1996, 39, 1112–1124 CrossRef CAS; (e) M. R. Angelastro, P. Bey, J. P. Burhart, T-M Chen, R. J. Cregge, S. L. Durham, R. A. Farr, S. L. Gallion, C. M. Hare, R. V. Hoffmann, E. W. Huber, M. L. Janusz, H-O. Kim, J. R. Koehl, A. L. Marquart, S. Mehdi, W. A. Metz, N. P. Peet, H. A. Schreuder, S. Sunder and C. Tardif, J. Med. Chem., 1998, 41, 2461–2480 CrossRef.
- (a) S. J. F. Macdonald, D. B. Belton, D. M. Buckley, J. E. Spooner, M. S. Anson, L. A. Harrison, K. Mills, R. J. Upton, M. D. Dowle, R. A. Smith, C. Molloy and C. Risley, J. Med. Chem., 1998, 41, 3919–3922 CrossRef CAS; (b) S. J. F. Macdonald, M. D. Dowle, L. A. Harrison, J. E. Spooner, P. Shah, M. R. Johnson, G. G. A. Inglis, G. D. E. Clarke, D. J. Belton, R. A. Smith, C. R. Molloy, M. Dixon, G. Murkitt, R. E. Godward, T. Skarzynski, O. M. P. Singh, K. A. Kumar, S. T. Hodgson, E. McDonald, G. W. Hardy, H. Finch, G. Fleetwood and D. C. Humphreys, Bioorg. Med. Chem. Lett., 2001, 11, 243–6 CrossRef CAS; (c) S. J. F. Macdonald, M. D. Dowle, L. A. Harrison, P. Shah, M. R. Johnson, G. G. A. Inglis, G. D. E. Clarke, R. A. Smith, D. Humphreys, C. R. Molloy, A. Amour, M. Dixon, G. Murkitt, R. E. Godward, T. Padfield, T. Skarzynski, O. M. P. Singh, K. A. Kumar, G. Fleetwood, S. T. Hodgson, G. W. Hardy and H. Finch, Bioorg. Med. Chem. Lett., 2001, 11, 895–8 CrossRef CAS.
- The Chemistry of β-Lactam Antibiotics, ed. M. I. Page, Blackie, Glasgow, 1992 Search PubMed.
- (a) R. Chabin, B. G. Green, P. Gale, A. L. Maycock, H. Weston, C. P. Dorn, P. E. Finke, W. K. Hagmann, J. J. Hale, M. MacCross, S. K. Shah, D. J. Underwood, J. B. Doherty and W. B. Knight, Biochemistry, 1993, 32, 8970 CrossRef CAS; (b) C. Beauve, M. Bouchet, R. Touillaux, J. Fastrez and J- Marchand-Brynaert, Tetrahedron, 1999, 55, 13301 CrossRef CAS.
- D. J. Underwood, B. G. Green, R. Chabin, S. Mills, J. B. Doherty, P. E. Finke, M. MacCoss, S. K. Shah, C. S. Burgey, T. A. Dickinson, P. R. Griffin, T. E. Lee, K. M. Swiderek, T. Covey, W. M. Westler and W. B. Knight, Biochemistry, 1995, 34, 143344 CrossRef CAS.
- R. C. Wilmouth, S. Kassamally, N. J. Westwood, R. J. Sheppard, T. D. W. Claridge, R. T. Aplin, P. A. Wright, G. J. Pritchard and C. J. Schofield, Biochemistry, 1999, 38, 7989 CrossRef CAS.
- (a) J. F. King, R. Rathore, J. Y. L. Lam, L. E. R. Gao and D. F. Klassen, J. Am. Chem. Soc., 1992, 114, 3028 CrossRef CAS; (b) J. M. Wood and M. I. Page, Trends Heterocycl. Chem., 2002, 8, 19 Search PubMed.
- N. J. Baxter, A. P. Laws, L. J. M. Rigoreau and M. I. Page, J. Am. Chem. Soc., 2000, 112, 3375 CrossRef CAS.
- N. J. Baxter, A. P. Laws, L. J. M. Rigoreau and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 1996, 2245 RSC.
- M. Beardsell, P. S. Hinchliffe, J. M. Wood, R. C. Wilmouth, C. J. Schofield and M. I. Page, β-Sultams – a novel class of serine protease inhibitors, Chem. Commun., 2001, 497–498 RSC.
- M. I. Page, Enzyme Catalysis in Comprehensive Medicinal Chemistry, 1990, Vol. 2, ed. P. G. Sammes, Pergamon, Oxford, pp. 45–60 Search PubMed.
- (a) A. P. Laws, and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 1989, 1577–1581 RSC; (b) A. P. Laws, N. J. Layland, D. G. Proctor and M. I. Page, J. Chem. Soc., Perkin Trans 2., 1993, 17–21 RSC; (c) N. O. Sykes, S. J. F. McDonald and M. I. Page, J. Med. Chem., 2002, 45, 2850 CrossRef CAS.
- J. Bieth, B. Spiess and C. G. Wermuth, Biochem. Med., 1974, 11, 350 Search PubMed.
- I. B. Blagoeva, A. J. Kirby, I. I. Kochiyashki, A. H. Koedjikov, I. G. Pojarlieff and M. M. Toteva, J. Chem. Soc., Perkin Trans. 2, 2000, 1953 RSC.
- A. R. Fersht, Enzyme Structure, Mechanism, W. H. Freeman, USA, 2nd edn., 1985, p. 405 Search PubMed.
- R. C. Wilmouth, S. Kassamally, N. J. Westwood, R. J. Sheppard, T. D. W. Claridge, R. T. Aplin, P. A. Wright, G. J. Pritchard and C. J. Schofield, Biochemistry, 1999, 38, 7989 CrossRef CAS.
- M. J. Slater, A. P. Laws and M. I. Page, Bioorg. Chem., 2001, 29, 7795 CrossRef CAS.
- A. M. Gold and D. E. Fahrney, Biochemistry, 1964, 3, 783 CrossRef CAS.
- R. A. Firestone, P. L. Barker, J. M. Pisano, B. M. Ashe and M. E. Dahlgren, Tetrahedron, 1990, 46, 2255 CrossRef CAS.
- W. B. Knight, A. L. Maycock, B. G. Green, B. M. Ashe, P. Gale, H. Weston, P. E. Finke, W. K. Hagmann, S. K. Shah, D. Underwood and J. B. Doherty, Biochemistry, 1992, 31, 4980 CrossRef CAS.
- (a) M. O. Lively and J. C. Powers, Biochim. Biophys. Acta, 1978, 525, 171 CAS; (b) T. Yoshimura, L. N. Barker and J. C. Powers, J. Biol. Chem., 1982, 257, 5077 CAS; (c) D. E. Fahrney and A. M. Gold, J. Am. Chem. Soc., 1963, 85, 997 CrossRef CAS.
- (a) O. R. Zaborsky and E. T. Kaiser, J. Am. Chem. Soc., 1970, 92, 860 CrossRef CAS; (b) E. T. Kaiser, Intra-Sci. Chem. Rep., 1972, 6, 5 Search PubMed.
- J. L. Kice, Adv. Phys. Org. Chem., 1980, 17, 65 Search PubMed.
- R. C. Wilmouth, N. J. Westwood, K. Anderson, W. Brownlee, T. D. W. Claridge, I. J. Clifton, G. J. Pritchard, R. T. Aplin and C. J. Schofield, Biochemistry, 1998, 37, 17506 CrossRef CAS.
- R. A. Firestone, P. L. Barker, J. M. Pisano, B. M. Ashe and M. Dahlgren, Tetrahedron, 1990, 46, 2255 CrossRef CAS.
- P. S. Hinchcliffe, J. M. Wood, A. M. Davis, R. P. Austin, R. P. Beckett and M. I. Page, J. Chem. Soc., Perkin Trans. 2, 2001, 1503 RSC.
- P. E. Finke, S. K. Shah, D. S. Fletcher, B. M. Ashe, K. A. Brause, G. O. Chandler, P. S. Dellea, K. M. Hand, A. L. Maycock, D. G. Osinga, D. J. Underwood, H. Weston, P. Davies and J. B. Doherty, J. Med. Chem., 1995, 38, 2449 CrossRef CAS.
- (a) D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 3rd edn., Pergamon, Oxford, 1988 Search PubMed; (b) Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw-Hill, 11th edn., 1973, pp. 5–7 Search PubMed.
- A. Champseix, J. Chanet-Ray, A. Etienne, A. Le Berre, J. Masson, C. Napierale and R. Vessiere, Bull. Soc. Chim. Fr., 1985, 483 Search PubMed.
- E. Meyer, G. Cole, R. Radhakrishnan and O. Epp, Acta. Crystallogr., Sect. B, 1985, B44, 26–38.
- S. Bailey, Acta Crystallogr., Sect D, 1994, D50, 760–3 CAS.
- A. T. Jones, J. Y. Zou, S. W. Cowan and M. Kjeldgaard, Acta Crystallogr., Sect A., 1991, A47, 110–9 CrossRef.
| This journal is © The Royal Society of Chemistry 2003 |