Concise synthesis of (±)-horsfiline and (±)-coerulescine by tandem cyclisation of iodoaryl alkenyl azides†
Dimitrios E.
Lizos
and
John A.
Murphy
*
Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, Scotland, UK G1 1XL. E-mail: John.murphy@strath.ac.uk
First published on 29th November 2002
Abstract
A brief, efficient and economical synthesis of the spiropyrrolidinyloxindoles, horsfiline and coerulescine, has been achieved, starting from itaconic acid and, respectively, p-anisidine or o-iodoaniline. Tandem radical cyclisation of iodoaryl alkenyl azides is the key step in both syntheses.
Introduction
The recent isolation of the natural products1 spirotryprostatin A 1 and spirotryprostatin B 2 (Scheme 1), and the discovery of their activity (albeit mild activity) as cell cycle inhibitors has added considerable impetus to research on the spiropyrrolidinyloxindole alkaloids. Total syntheses2 of the spirotryprostatins A and B have been achieved, but it transpires that synthetic intermediates prepared during Danishefsky's synthetic studies3 on these compounds, may be more interesting than the spirotryprostatins themselves. Three compounds, 3–5, were identified to have potent and selective action on human breast cancer cell lines.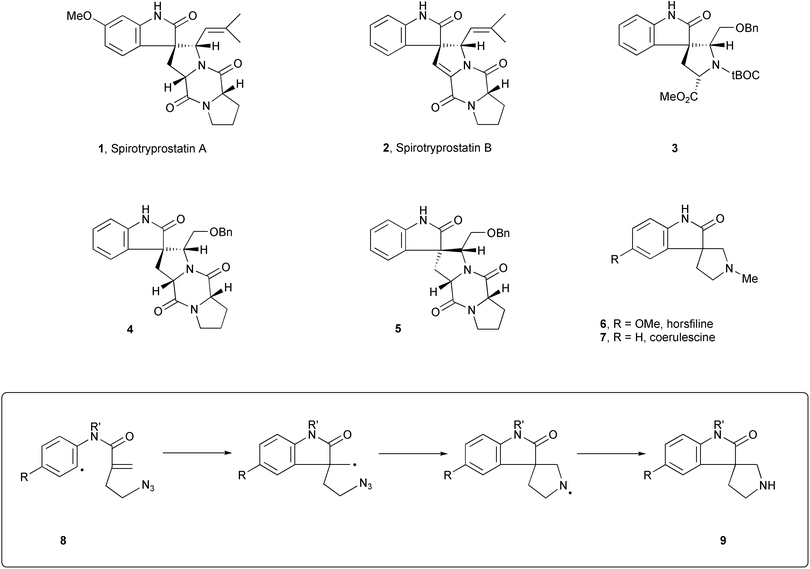 | ||
| Scheme 1 | ||
Among these, tricyclic compound 3 is exciting since it is a relatively uncomplicated potential lead for development of novel anti-tumour agents. Also highly active was the pair of diastereomers 4 and 5. The discovery of equal activity for the diastereomeric pair shows that the stereochemistry of the spiro centre in these molecules is not crucial for anti-tumour action. Although the cellular site of action of these three compounds has not yet been disclosed, if it is assumed that they act at a common locus, as a working hypothesis, then the anti-tumour activity of tricyclic spiropyrrolidinyloxindoles requires further investigation, and therefore synthetic routes that provide easy access to these classes of compounds become important.
It is not surprising therefore that there has been a significant and increasing interest in the synthesis of the tricyclic natural products, horsfiline4a–h,4j6 (isolated by Bodo and co-workers4a) and coerulescine4d,i–l7 (isolated5 by Colegate and co-workers) that bear the same tricyclic skeleton as 3. A range of synthetic approaches has been used. With respect to horsfiline, Jones and Wilkinson4b used a free radical cyclisation as the key step in their synthesis. Lévy and co-workers produced4d two routes featuring (i) oxidative rearrangement of a tetrahydrocarboline (see also ref. 4a) and (ii) oxidation and formaldehyde-treatment of an appropriately substituted tryptamine. In a synthesis of (−)-horsfiline as well as, separately, the unnatural enantiomer, Borschberg and co-workers4c studied the stereospecific conversion of tryptamine derivatives firstly to tetrahydrocarbolines and thence to spiropyrrolidinylindolones, while Palmisano et al. prepared4e (−)-6via the (chiral auxiliary-directed) cycloaddition of azomethine ylides onto 3-methyleneindolones. Meanwhile, Fuji and co-workers used4f asymmetric nitroolefination of indolones to introduce the correct stereochemistry at what would become the spiro-carbon, subsequently making the pyrrolidine ring. More recently, in preparations of (±)-6, Carreira's group4g have ring-expanded a spiro-cyclopropylindolone to the requisite spiropyrrolidine, and Kumar et al. rearranged4j an (aziridinoethylidene)indolone to a pyrrolidinylindolone.
From our perspective, the spiropyrrolidinyl-oxindole skeleton should be an ideal target for synthesis using the iodoaryl azide tandem radical cyclisation strategy that we recently exploited in the total synthesis of aspidospermidine6 and in the formal total synthesis of vindoline.7 Thus tandem cyclisation of a radical, 8, should afford the tricyclic system, 9, in a single process. This paper reports8 the realisation of that goal.
Results and discussion
To synthesise horsfiline, dilithiated t-BOC-p-anisidine 10a was iodinated9 with 1,2-diiodoethane, to give 11a (86%). Deprotection of the t-BOC group gave 12a (89%), which was reductively alkylated with benzaldehyde10 (94%). (Conversion of the primary amine 12a to a secondary amine such as 13a is necessary since the radical cyclisations about to be undertaken on the amide substrates, e.g.17 in Scheme 2 do not succeed4b with secondary amides. We chose benzylation for this conversion, because of the later easy removal of the benzyl group). Condensation of the benzylamine 13a with the acid chloride1114 of the monomethyl ester of itaconic‡ acid (in turn readily available11 from commercially available itaconic acid) afforded the amide 15a (94%). It was now necessary to reduce the ester group in amide 15a to an alcohol, 16a, before conversion to azide 17a and cyclisation. Initial attempts to reduce the ester to alcohol 16a with DIBAL-H led to the desired product 16a as the major product, but it was contaminated with a minor compound that, based on examination of the NMR spectrum, we propose to be 18, where unexpected reduction of the alkene has occurred. Unfortunately the two compounds had similar physical properties, so separation was not possible. To overcome this problem, protection of the alkene double bond in 15a was undertaken. Reaction of 15a with PhSeNa, formed in situ by reduction of diphenyl diselenide with sodium borohydride, surprisingly led to no reaction, and we attribute this to steric hindrance to attack on the alkene by the large phenylseleno group. To decrease the steric problems, PhSH was instead reacted with ester 15a in the presence of base to give an excellent yield of product 19 (96%). Reduction of 19 afforded the desired alcohol 20 (55%) together with a small amount of unexpected amine 21 (10%). Alcohol 20 was purified and oxidized to the corresponding sulfoxide, which was directly subjected to thermal elimination affording the alcohol 16a (72%). | ||
| Scheme 2 Syntheses of horsfiline and coerulescine. Reagents and conditions: (a) t-BuLi, Et2O, −20 °C then ICH2CH2I, 86% for 11a; (b) TFA, DCM, 0 °C, 89% for 12a; (c) ZnCl2, PhCHO, MeOH, NaCNBH3, 94% for 13a, 92% for 13b; (d) Et3N, PhH, 94% for 15a; 94% for 15b; (e) PhSH, DBU, THF, Δ, 96% for 19; (f) DIBAL-H, PhMe, −78 °C, 55% for 20; (g) m-CPBA, DCM, then PhMe, Δ, 72 h, 72% for 16a; (h) LiCI, NaBH4, EtOH, THF, 83% for 16a, 84% for 16b; (i) PPh3, (PhO)2P(O)N3, DEAD, THF, 68% for 17a; 70% for 17b; (j) (Me3Si)3SiH, AIBN, PhH; (k) CH2O, NaCNBH3, MeCN, yield over 2 steps: 60% for 23a, 68% for 23b; (1) Na, NH3, 86% for 6, 83% for 7. | ||
Although 16a had now been produced, the process of protecting and later deprotecting the alkene detracted from the directness of the synthetic route. With this in mind, we returned to investigate further whether a clean reduction of 15a to 16a could be found. Gratifyingly, success was achieved with LiBH4, generated in situ from lithium chloride and sodium borohydride.12 This afforded the desired product cleanly and in high yield (83%). Conversion to the azide 17a (68%) was accomplished using diphenylphosphoryl azide, and this was followed by tandem cyclisation to afford the tricyclic product 22a; this was subjected to in situ methylation to yield 23a (60% over two steps). Finally deprotection under Birch conditions afforded horsfiline 6 (87%).
Significantly, the tandem radical cyclisation afforded solely the desired tricyclic skeleton; no product arising from apparent 6-endo-trig cyclisation of the aryl radical onto the alkene was seen. In studies related to the synthesis of these same alkaloids, Jones et al. had shown13 how the balance of 5-exo to apparent 6-endo cyclisation is determined by kinetic and thermodynamic factors. At lower temperatures, the 5-exo predominates,14 but at higher temperatures, rearrangement to 6-endo can occur through a neophyl rearrangement. In our case, the product of the initial 5-exo cyclisation is likely to be trapped by the azide group, preventing side-products from forming.
The same protocol was then followed for the synthesis of coerulescine and with generally excellent yields, producing intermediates 13b (92%), 15b (94%), 16b (84%), 17b (70%) and 23b (68%). We were a little concerned about the final deprotection step to afford coerulescine. Debenzylation of 23a in the horsfiline case involves selective addition of solvated electrons to the aromatic ring of the benzyl protecting group. It is entirely reasonable that this should be a very selective process, since the aromatic ring in the horsfiline skeleton is likely to be far more electron-rich, and hence more difficult to reduce, than that of the benzyl group, due to the electron-donating properties of the methoxy and amide groups. With coerulescine, the absence of the methoxy group should make the difference between the two rings less pronounced, but the benzyl group should still bear the more electron-poor ring. In practice, the selectivity was maintained and the reaction proceeded in an excellent (83%) yield.
It is seen that the iodoaryl alkenyl azide tandem radical cyclisation reaction affords a rapid route to (±)-horsfiline in 20% yield from t-BOC-p-anisidine [18% from anisidine], and (±)-coerulescine in 29% yield from 2-iodoaniline. An acid chloride derived from the economical itaconic acid is the key partner reagent for both syntheses. The efficiency of this route ranks with the highest reported.
In mechanistic terms, this route shows greatest similarities with the route of Jones and Wilkinson,4b since both use radical cyclisations as the key steps (Scheme 3). However, the two routes differ considerably in all other facets, (e.g. single cyclisation versus tandem cyclisation) and most particularly in the nature and reactivity of the final radicals, carbon radical 24 in their case, versus nitrogen radical 25 in this case. Importantly, in designing the synthesis of more complex spiropyrrolidinylindolones, the diversity of efficient routes should now allow access to a wide variety of substitution patterns of analogues for anti-tumor testing.
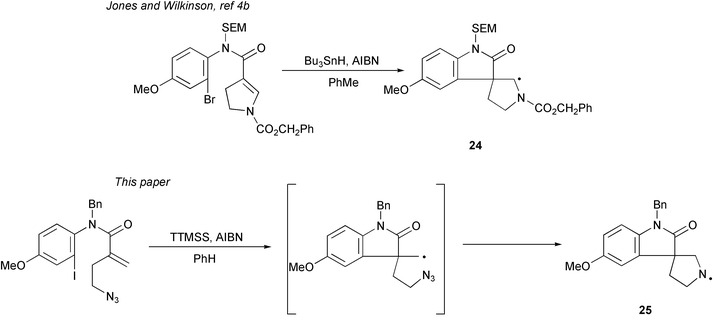 | ||
| Scheme 3 The different regiochemical outcomes of the two radical cyclisation approaches to spiropyrrolidinylindolones. | ||
Experimental section
Melting points were carried out on a Reichert hot stage apparatus and are uncorrected. Infra red spectra were obtained on a Perkin Elmer 1720-X FTIR. Mass spectra were recorded on a JEOL JMS-AX505 HA instrument using, electron impact ionisation (EI+), chemical ionisation (CI), electrospray (ES) or fast atom bombardment (FAB). Some low and high resolution mass spectra were obtained on JLSX 102 and VG ZAB-E instruments using peak-matching techniques at the EPSRC Mass Spectrometry Centre, Swansea. 1H NMR spectra were recorded at 400 MHz on a Bruker DPX 400 spectrometer. 13C{H} NMR spectra were similarly recorded at 100.61 MHz on a Bruker DPX400 machine. NMR experiments were carried out in deuterochloroform with tetramethylsilane as internal reference unless otherwise stated. 13C NMR spectra were acquired in a broadband decoupled mode with the multiplicities obtained using a DEPT or Jmod sequence. Chemical shifts (δ) are reported in parts per million (ppm) from tetramethylsilane. The following abbreviations are used for multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet, br = broad. Coupling constants (J) are reported in Hertz (Hz).tert-Butyl (2-iodo-4-methoxyphenyl)carbamate 11a9
Addition of tert-butyllithium in pentane (1.7 M, 39.6 cm−3, 67.2 mmol, 2.5 equiv.) to a solution of tert-butyl (4-methoxyphenyl)carbamate (6.0 g, 26.9 mmol, 1.00 equiv.), in dry diethyl ether (60 cm−3), was performed under a nitrogen atmosphere at −20 °C, followed by stirring for 3 h at the same temperature. To this mixture was added 1,2-diiodoethane (11.4 g, 40.3 mmol, 1.5 equiv.) in dry diethyl ether (80 cm−3) at −78 °C, and the reaction was gradually warmed to room temperature and stirred for 16 h. After addition of a saturated aqueous solution of sodium thiosulfate (200 cm−3), the mixture was extracted with diethyl ether. The organic extracts were then washed with brine, dried over anhydrous sodium sulfate and evaporated under reduced pressure. The residue was purified by column chromatography (10 ∶ 1 petroleum ether–ethyl acetate) to yield tert-butyl (2-iodo-4-methoxyphenyl)carbamate 11a as colourless prisms (8.1 g, 86%); mp 48–49 °C (lit.9 mp 49–50 °C); [Found (EI) M+ 349.0172. C12 H16INO3 requires M, 349.0175]. νmax (film)/cm−1 3399, 2976, 2933, 2834, 1725, 1511, 1367, 1250, 1161, 1037; δH 1.51 (9H, s, 3 × CH3), 3.72 (3H, s, CH3), 6.55 (1H, br s, NH), 6.84–6.87 (1H, dd, J 9.0, 2.8 ArH), 7.28 (1H, d, J 2.8, ArH) 7.76 (1H, d, J 9.0, ArH); δC 28.5 (q), 55.9 (q), 80.9 (s), 90.5 (s), 115.1 (d), 122.3 (d), 123.9 (d), 132.6 (s), 153.3 (s), 156.3 (s). m/z (EI) 349 (29%, M+), 293 (96), 249 (46), 166 (34), 106 (22) and 57 (100).4-Amino-3-iodoanisole 12a
Trifluoroacetic acid (21.2 cm−3, 274.9 mmol, 12.00 equiv.) was added to a solution of tert-butyI (2-iodo-4-methoxyphenyl)carbamate (8.0 g, 22.9 mmol, 1.00 equiv.) in dry DCM (120 cm−3) at 0 °C and the reaction was stirred at room temperature for 2 h. Sodium hydroxide (2 M) was added followed by hydrochloric acid (2 M) until neutral pH had been attained. The two layers were separated and the aqueous one was extracted with DCM. The organic layers were washed with brine, dried over anhydrous sodium sulfate and the solvent was removed in vacuo. The crude product was purified by column chromatography (4 ∶ 1 petroleum ether–ethyl acetate) giving 4-amino-3-iodoanisole12a as a colourless oil (5.1 g, 89%); [Found (ES): MH+ 249.9732 C7H9INO, requires MH, 249.9729]; νmax (film)/cm−1 3431, 3348, 2939, 2830, 1596, 1493, 1232, 1037; δH 3.62 (2H, br s, NH2), 3.77 (3H, s, CH3), 6.74 (1H, d, J, 8.7, ArH), 6.82 (1H, dd, J 8.7, 2.7, ArH), 7.26 (1H, d, J 2.7, ArH); δC 56.4 (q), 84.7 (s), 115.9 (d), 116.5 (d), 124.0 (d), 141.3 (s), 153.1 (s); m/z (CI) 267 (MNH4+, 63%), 250 (M+, 100).Benzyl(2-iodo-4-methoxyphenyl)amine 13a
A stirred mixture of 4-amino-3-iodoanisole 12 (3 g, 12.00 mmol, 1.00 equiv.), zinc(II) chloride (1.97 g, 14.45 mmol, 1.20 equiv.) and benzaldehyde (1.47 cm−3, 14.45 mmol, 1.20 equiv.) in methanol, (50 cm−3) was treated with sodium cyanoborohydride (0.91 g, 14.45 mmol, 1.20 equiv.) and warmed at reflux under nitrogen for 2 h. The cooled reaction mixture was diluted with sodium hydroxide (1 M, 50 cm−3) and extracted with diethyl ether. The combined ether layers were washed with water, dried over anhydrous sodium sulfate and concentrated in vacuo. Benzyl(2-iodo-4-methoxyphenyl)amine13a was purified by column chromatography on silica gel eluting with petroleum ether–ethyl acetate 5 ∶ 1 to give pure title compound as a yellow oil (3.85 g, 94%); Found: C, 49.87; H, 4.15; N, 4.06. C14H14INO requires C, 49.56; H, 4.16; N, 4.13%; (Found (ES): MH+ 340.0196, C14H15INO requires MH 340.0198); νmax (film)/cm−1 3401, 3028, 2998, 2830, 1603, 1504, 1282, 1039; δH 3.75 (3H, s, CH3), 4.33 (1H, br s, NH), 4.39 (2H, d, J 4.2, CH2), 6.54 (1H, d, J 8.9, ArH), 6.84, (1H, dd, J 2.9, 8.9, ArH), 7.30–7.42 (6H, m, ArH); δC 49.2 (t), 56.1 (q), 85.5 (s), 111.7 (d), 115.7 (d), 124.8 (d), 127.4 (d), 128.8 (d), 139.1 (s), 142.0 (s), 152.2 (s); m/z (CI) 340.1 ([MH]+, 100%).3-Chlorocarbonylbut-3-enoic acid methyl ester 1411
Thionyl chloride (7.1 cm−3, 97.2 mmol, 1.40 equiv.) was added to a solution of 2-methylenesuccinic acid 4-methyl ester (10 g, 69.4 mmol, 1.00 equiv.) in dry diethyl ether (8 cm−3) and the reaction mixture was refluxed gently until gas evolution was essentially complete (15 min). Distillation gave 3-chlorocarbonylbut-3-enoic acid methyl ester 14 as a colourless oil (10.8 g, 96%) bp 84–87 °C (11 mmHg) (Found: M+ 162.0080, C6H7ClO3 requires M 162.0084); νmax (film)/cm−1 3004, 2955, 2847, 1740, 1638, 1437, 1343, 1206; δH 3.39 (2H, d, J 0.8, CH2), 3.72 (3H, s, CH3), 6.17 (1H, t, J 0.8, CH2), 6.70 (1H, s, CH2); δC 37.8 (t), 52.8 (q), 136.5 (t), 138.7 (s), 168.6 (s), 170.3 (s); m/z (EI) 163 ([MH]+, 5%), 127 (60), 113 (95), 99 (50), 85 (100).3-[Benzyl(2-iodo-4-methoxyphenyl)carbamoyl]but-3-enoic acid methyl ester 15a
Triethylamine (1.6 cm−3, 11.4 mmol, 1.10 equiv.) was added to a stirred solution of benzyl(2-iodo-4-methoxyphenyl)amine (3.5 g, 10.3 mmol, 1.00 equiv.) in dry benzene (10 cm−3) at room temperature. 3-Chlorocarbonylbut-3-enoic acid methyl ester (2.18 g, 13.4 mmol, 1.30 equiv.) in dry benzene (10 cm−3) was added dropwise. The resulting solution was stirred at rt for 30 min, then heated under reflux for 90 min. After cooling, the solvent was removed under reduced pressure and the residue dissolved in diethyl ether (50 cm−3). The ether was washed with water, dried over anhydrous sodium sulfate and concentrated. Column chromatography eluting with petroleum ether–ethyl acetate 4 ∶ 1 gave pure 3-[benzyl(2-iodo-4-methoxyphenyl)carbamoyl]but-3-enoic acid methyl ester15a as a white solid mp 107–110 °C (4.5 g, 94%); Found C, 51.68; H, 4.36; N, 3.03. C20H20INO4 requires C, 51.61; H, 4.33; N, 3.01%; [Found: (ES) MH+ 466.0521, C20H21INO4 requires MH 466.0515]; νmax (film)/cm−1 3065, 2998, 2841, 1736, 1651, 1622, 1491, 1287, 1175; δH 3.06 (1H, d, J 16.5, CH2), 3.64–3.69 (4H, CH3 + CH2), 3.78 (3H, s, CH3), 4.17 (1H, d, J 14.3, CH2), 5.16 (1H, s, CH2), 5.27 (1H, s, CH2), 5.77 (1H, d, J 14.3, CH2), 6.70 (1H, dd, J 2.8, 8.7, ArH), 6.81 (1H, d, J 8.7, ArH), 7.23–7.31 (5H, m, ArH), 7.42 (1H, d, J 2.8, ArH); δC 40.1 (t), 52.0 (q), 52.1 (t), 55.7 (q), 100.1 (s), 114.4 (d), 123.2 (t), 125.0 (d), 127.6 (d), 128.4 (d), 129.6 (d), 132.0 (d), 136.5 (s), 136.8 (s), 137.5 (s), 159.0 (s), 169.2 (s), 171.3 (s); m/z (ES) 466 [(MH)+, 60%], 340 (100).Reduction of 3-[benzyl(2-iodo-4-methoxyphenyl)carbamoyl]but-3-enoic acid methyl ester 15a using NaBH4 and LiCl
Lithium chloride (16.2 mg, 0.43 mmol, 2.00 equiv.) was added to a solution of 3-[benzyl(2-iodo-4-methoxyphenyl)carbamoyl]but-3-enoic acid methyl ester 15a (100 mg, 0.21 mmol, 1.00 equiv.) in dry THF–EtOH (2 ∶ 1 mixture, 3 cm−3). When it had dissolved, NaBH4 (16.2 mg, 0.43 mmol, 2.00 equiv.) was added and the reaction mixture was stirred at room temperature for 16 h. Then the solvent was evaporated near dryness and H2O (4 cm−3) was added. The organic phase was extracted with ethyl acetate, washed with H2O, dried over anhydrous sodium sulfate and concentrated under vacuum. The reaction mixture was subjected to column chromatography eluting with petroleum ether–ethyl acetate 1 ∶ 1 to afford N-benzyl-2-(2-hydroxyethyl)-N-(2-iodo-2-methoxyphenyl)acrylamide16a as a colourless oil (76 mg, 83%); [Found (ES): MH+ 438.0563, C19H21INO3 requires MH 438.0566]; νmax (film)/cm−1 3409, 2938, 1614, 1487, 1287, 1030; δH 2.01 (1H, br s, OH) 2.25–2.31 (1H, m, CH2), 2.48–2.54 (1H, m, CH2), 3.62–3.77 (5H, m, CH2 and CH3), 4.10 (1H, d, J 14.1, CH2), 5.10 (1H, s, CH2), 5.14 (1H, s, CH2), 5.71 (1H, d, J 14.1, CH2), 6.56 (1H, d, J 8.7 ArH), 6.68 (1H, dd, J 2.7, 8.7, ArH), 7.21–7.39 (5H, m, ArH), 7.40 (1H, d, J 2.7, ArH); δC 38.2 (t), 52.4 (t), 55.7 (q), 62.6 (t), 100.1 (s), 114.4 (d), 120.2 (t), 125.0 (d), 127.9 (d), 128.6 (d), 129.6 (d), 131.6 (d), 136.6 (s), 137.1 (s), 142.0 (s), 159.1 (s), 172.2 (s); m/z (El) 438.1 [(MH)+, 100%]2-(2-Azidoethyl)-N-benzyl-N-(2-iodo-4-methoxyphenyl)acrylamide 17a
Triphenylphosphine (363 mg, 1.38 mmol, 1.10 equiv.), DEAD (0.22 cm−3, 1.38 mmol, 1.10 equiv.) and diphenylphosphoryl azide (0.3 cm−3, 1.38 mmol, 1.10 equiv.) were added sequentially to a solution of alcohol 16a in dry THF (12 cm−3), and the solution was stirred at rt for 24 h. Then the solvent was evaporated and the residue was subjected to column chromatography using petroleum ether–ethyl acetate 5 ∶ 1 to afford pure 2-(2-azidoethyl)-N-benzyl-N-(2-iodo-4-methoxyphenyl) acrylamide17a as a slightly yellow oil (396 mg, 68%); [Found (ES): MH+ 463.0622, C19H20IN4O2 requires MH 463.0631]; νmax (film)/cm−1 3030, 2938, 2838, 2096, 1651, 1622, 1488, 1287, 1030; δH 2.34–2.41 (1H, m, CH2), 2.55–2.62 (1H, m, CH2), 3.43 (2H, t, J 6.9, CH2), 3.78 (3H, s, CH3), 4.09 (1H, d, J 14.1, CH2), 5.12 (1H, s, CH2), 5.17 (1H, s, CH2), 5.71 (1H, d, J 14.1, CH2), 6.55 (1H, d, J 8.7, ArH), 6.69 (1H, dd, J 2.7, 8.7, ArH), 7.21–7.35 (5H, m, ArH), 7.41 (1H, d, J 2.7, ArH); δC 33.9 (t), 50.1 (t), 52.3 (t), 55.8 (q), 100.4 (s), 114.5 (d), 120.3 (t), 125.1 (d), 127.8 (d), 128.6 (d), 129.7 (d), 131.7 (d), 136.9 (s), 137.1 (s), 140.7 (s), 159.2 (s), 170.5 (s); m/z (CI) 463 [(MH)+, 35%], 115 (100).Tandem radical cyclisation of 2-(2-azidoethyl)-N-benzyl-N-(2-iodo-4-methoxyphenyl)acrylamide 17a
2-(2-Azidoethyl)-N-benzyl-N-(2-iodo-4-methoxyphenyl)acrylamide 17a (295 mg, 0.64 mmol, 1.00 equiv.) was dissolved in dry benzene (20 cm−3) and the solution was purged with argon for 30 min. The solution was brought to reflux and tristrimethylsilylsilane (TTMSS) (0.22 cm−3, 0.70 mmol, 1.10 equiv.) and AIBN (21 mg, 0.13 mmol, 0.2 equiv.) were added and the mixture was refluxed for 5 h. The reaction mixture was allowed to cool down to room temperature and the solvent was removed in vacuo. Ethyl acetate (50 cm−3) was added and the mixture extracted with hydrochloric acid (2 M). The aqueous phase was basified with sodium hydroxide (2 M) and then it was extracted with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and concentrated in vacuo. The crude 1-benzyl-5-methoxy-1H-spiro[indole-3,3′-pyrrolidin]-2-one 22a (150 mg, 0.49 mmol, 1.00 equiv.) from the previous step was dissolved in acetonitrile (7 cm−3) and treated5f with formaldehyde [(0.24 cm−3, 2.92 mmol, 6.00 equiv.) 37% in water] and NaCNBH3 (61 mg, 0.97 mmol, 2.00 equiv.) at rt. The mixture was stirred for 15 min and neutralised with acetic acid. After stirring was continued for another 1 h, the mixture was basified with ammonia solution and the solvent was removed in vacuo. The residue was extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure. Column chromatography eluting with ethyl acetate: acetone 4 ∶ 1 gave pure 1-benzyl-5-methoxy-1′-methyl-1H-spiro[indole-3,3′-pyrrolidin]-2-one 23a as a colourless oil (123 mg, 60% over two steps) (Found MH+ 323.1761, C20H23N2O2 requires MH 323.1759); νmax (film)/cm−1 2941, 2836, 2787, 1707, 1601, 1491, 1177, 1035; δH 2.12–2.20 (1H, m, CH2), 2.40–2.46 (1H, m, CH2), 2.51 (3H, s, CH3), 2.78–2.85 (1H, m, CH2), 2.91 (1H, d, J 9.4, CH2), 3.01 (1H, d, J 9.4, CH2), 3.15–3.20 (1H, m, CH2), 3.77 (3H, s, CH3), 4.88 (2H, s, CH2), 6.59 (1H, d, J 8.5, ArH), 6.68 (1H, dd, J 2.6, 8.5, ArH), 7.11 (1H, d, J 2.6, ArH), 7.24–7.33 (5H, m, ArH); δC 38.4 (t), 42.0 (q), 44.1 (t), 53.9 (s), 56.1 (q), 56.8 (t), 66.2 (t), 109.3 (d), 110.6 (d), 112.5 (d), 127.4 (d), 127.8 (d), 129.0 (d), 135.6 (s), 136.2 (s), 137.0 (s), 156.6 (s), 180.2 (s); m/z (CI) 323 [(MH)+, 100%].(±)-Horsfiline 64g
Ammonia (ca. 5 cm−3) was condensed at −50 °C and sodium (31 mg) was added with vigorous stirring. 1-Benzyl-5-methoxy-1′-methyl-1H-spiro[indole-3,3′-pyrrolidin]-2-one 23a (29.4 mg, 0.09 mmol, 1.00 equiv.) was added in THF (1 cm−3). After 12 min, a small amount of NH4Cl was added and the ammonia left to evaporate. Water (25 cm−3) was added and the reaction was extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and the solvent was evaporated in vacuo. Column chromatography on silica gel with DCM–MeOH 9 ∶ 1 afforded pure (±)-horsfiline as a white solid (18.2 mg, 87%); mp 154–156 °C (lit.4g mp 156–157 °C) [Found (ES): MH+ 233.1287, C13H17N2O2 requires MH 233.1290]; νmax (film)/cm−1 3400, 2944, 2849, 1704, 1604, 1491, 1304, 1209, 1031; δH 2.05–2.18 (1H, m, CH2), 2.38–2.44 (1H, m, CH2), 2.49 (3H, s, CH3), 2.77–2.84 (1H, m, CH2), 2.90 (1H, d, J 9.4, CH2), 2.97 (1H, d, J 9.4, CH2), 3.06–3.11 (1H, m, CH2), 3.80 (3H, s, CH3), 6.72 (1H, dd, J 2.5, 8.4, ArH), 6.80 (1H, d, J 8.4, ArH), 7.06 (1H, d, J 2.5, ArH), 8.32 (1H, br s, NH); δC 38.3 (t), 41.9 (q), 54.3 (s), 56.1 (t), 56.8 (q), 66.2 (t), 110.0 (d), 110.6 (d), 112.9 (d), 133.6 (s), 137.4 (s), 156.5 (s), 182.8 (s); m/z (CI) 233 [(M + H)+), 100%].Benzyl(2-iodophenyl)amine 13b
12b (5 g, 22.80 mmol, 1.00 equiv.), was treated as for 12a and afforded benzyl(2-iodophenyl)amine 13b as a yellow oil (6.5 g, 92%) [Found (ES): MH+ 310.0095, C13H13IN requires MH 310.0093]; νmax (film)/cm−1 3400, 3060, 3027, 2854, 1589, 1502, 1318, 1004; δH 4.52 (2H, d, J 5.6, CH2), 4.74 (1H, br s, NH) 6.56 (1H, ddd, J 1.4, 7.3, 7.8, ArH), 6.64 (1H, dd, J 1.3, 8.1, ArH), 7.26 (1H, ddd, J 1.4, 7.3, 8.1, ArH), 7.39–7.48 (5H, m, ArH), 7.78–7.80 (1H, dd, J 1.4, 7.8, ArH); δC 48.5 (t), 85.5 (s), 111.1 (d), 119.0 (d), 127.4 (d), 127.5 (d), 128.9 (d), 129.6 (d), 138.8 (s), 139.2 (d), 147.3 (s); m/z (EI) 309 [(MH)+, 18%], 180 (20), 91 (100) and 65 (22).3-[Benzyl(2-iodophenyl)carbamoyl]but-3-enoic acid methyl ester 15b
Benzyl(2-iodophenyl)amine 13b (4.5 g, 14.55 mmol, 1.00 equiv.) was reacted as for 13a and gave 3-[benzyl(2-iodophenyl)carbamoyl]but-3-enoic acid methyl ester15b as a white solid mp 60–62 °C (5.96 g, 94%); [Found (EI): M+ 435.0339, C19H18INO3 requires M 435.0331]; νmax (film)/cm−1 2949, 1736, 1653, 1625, 1470, 1172; δH 3.06 (1H, d, J 16.5, CH2), 3.67–3.70 (4H, m, CH2 and CH3), 4.23 (1H, d, J 14.3, CH2), 5.15 (1H, s, CH2), 5.27 (1H, s, CH2), 5.78 (1H, d, J 14.3, CH2), 6.94–7.00 (2H, m, ArH), 7.16–7.20 (1H, m, ArH), 7.24–7.30 (5H, m, ArH), 7.91 (1H, d, J 7.9, ArH); δC 40.0 (t), 52.0 (q), 52.5 (t), 99.8 (s), 123.4 (t), 127.7 (d), 128.4 (d), 129.0 (d), 129.4 (d), 129.6 (d), 132.0 (d), 136.4 (s), 136.6 (s), 140.2 (d), 144.7 (s), 168.9 (s), 171.3 (s); m/z (EI) 435 (M+, 5%), 376 (9), 308 (18), 180 (19) and 91 (100).N-Benzyl-2-(2-hydroxyethyl)-N-(2-iodophenyl)acrylamide 16b
3-[Benzyl(2-iodophenyl)carbamoyl]but-3-enoic acid methyl ester 15b (12.1 g, 27.8 mmol, 1.00 equiv.) was treated as for 15a to afford N-benzyl-2-(2-hydroxyethyl)-N-(2-iodophenyl)acrylamide16b as a colourless oil (9.5 g, 84%) [Found (ES): MH+ 408.0461, C18H19INO2 requires MH 408.0460]; νmax (film)/cm−1 3401, 2931, 1647, 1619, 1469, 1030; δH 2.27–2.31 (1H, m, CH2), 2.50–2.57 (1H, m, CH2), 3.20 (1H, br s, OH), 3.74–3.81 (2H, m, CH2), 4.14 (1H, d, J 14.3, CH2), 5.10 (1H, s, CH2), 5.13 (1H, s, CH2), 5.70 (1H, d, J 14.3, CH2), 6.68 (1H, d, J 7.6, ArH), 6.97–7.00 (1H, m, ArH), 7.15–7.28 (6H, m, ArH), 7.92 (1H, dd, J 1.4, 7.9, ArH); δC 38.3 (t), 52.2 (t), 62.8 (t), 99.9 (s), 120.7 (t), 127.9 (d), 128.7 (d), 128.9 (d), 129.6 (d), 129.7 (d), 131.6 (d), 136.6 (s), 140.4 (d), 141.9 (s), 144.4 (s), 171.9 (s); m/z (EI) 407 (2%, M+), 376 (12), 280 (17), 180 (17), 91 (100) and 65 (25).2-(2-Azidoethyl)-N-benzyl-N-(2-iodophenyl)acrylamide 17b
Alcohol 16b (480 mg, 1.18 mmol, 1.00 equiv.) was treated as for alcohol 16a to afford pure 2-(2-azidoethyl)-N-benzyl-N-(2-iodophenyl)acrylamide17b as a slightly yellow oil (357 mg, 70%); [Found (EI): M+ 432.0446, C18H17IN4O requires M 432.0447]; νmax (film)/cm−1 3031, 2934, 2875, 2095, 1651, 1624, 1469, 1385; δH 2.37–2.41 (1H, m, CH2), 2.57–2.64 (1H, m, CH2), 3.44 (2H, t, J 6.7, CH2), 4.16 (1H, d, J 14.2, CH2), 5.12 (1H, s, CH2), 5.17 (1H, s, CH2), 5.70 (1H, d, J 14.2, CH2), 6.70 (1H, d, J 7.5, ArH), 6.96–7.00 (1H, m, ArH), 7.16–7.38 (6H, m, ArH), 7.90 (1H, dd, J 1.4, 7.8, ArH); δC 33.9 (t), 50.1 (t), 52.2 (t), 99.9 (s), 120.3 (d), 120.7 (t), 125.8 (d), 127.9 (d), 128.6 (d), 129.0 (d), 129.6 (d), 130.1 (d), 131.7 (d), 136.9 (s), 140.5 (d), 140.5 (s), 145.4 (s) 170.5 (s); m/z (CI) 432 (100%, M+).Tandem radical cyclisation of 2-(2-azidoethyl)-N-benzyl-N-(2-iodophenyl)acrylamide 17b
2-(2-Azidoethyl)-N-benzyl-N-(2-iodophenyl)acrylamide 17b (160 mg, 0.37 mmol, 1.00 equiv.) when treated as for azide 17a gave pure 1-benzyl-1′-methyl-1H-spiro[indole-3,3′-pyrrolidin]-2-one 23b as a colourless oil (73.6 mg, 68% over two steps) [Found (ES): MH+ 293.1652, C19H21N2O requires MH 293.1654]; νmax (film)/cm−1 2940, 2838, 2786, 1711, 1611, 1486, 1467, 1359, 1183; δH 2.11–2.18 (1H, m, CH2), 2.41–2.46 (1H, m, CH2), 2.50 (3H, s, CH3), 2.77–2.83 (1H, m, CH2), 2.89 (1H, d, J 9.4, CH2), 2.95 (1H, d, J 9.4, CH2), 3.09–3.14 (1H, m, CH2), 4.9 (2H, s, CH2), 6.71 (1H, d, J 7.7, ArH), 7.04 (1H, ddd, J 1.5, 7.3, 7.7, ArH), 7.14 (1H, ddd, J 0.8, 7.7, 7.7, ArH), 7.24–7.33 (5H, m, ArH), 7.44 (1H, dd, J 1.4, 7.8, ArH); δC 38.3 (t), 42.0 (q), 44.0 (t), 53.5 (s), 56.9 (t), 66.6 (t), 108.9 (d), 123.1 (d), 123.2 (d), 127.4 (d), 127.7 (d), 127.8 (d), 129.0 (d), 136.0 (s), 136.2 (s), 142.1 (s), 180.6 (s); m/z (EI) 292 [(M)+, 30%], 235 (100), 91 (80).(±)-Coerulescine 7
1-Benzyl-1′-methyl-1H-spiro[indole-3,3′-pyrrolidin]-2-one 23b (22 mg, 0.075 mmol, 1.00 equiv.) was treated as for 23a and afforded pure (±)-coerulescine 7 as a pale yellow gum (12.7 mg, 83%); [Found (ES): MH+ 203.1182, C12H14N2O requires MH 203.1184]; νmax (film)/cm−1 3385, 2969, 2793, 1711, 1619, 1471, 1336, 1196, 1047; δH 2.10–2.18 (1H, m, CH2), 2.39–2.45 (1H, m, CH2), 2.50 (3H, s, CH3), 2.80–2.86 (1H, m, CH2), 2.89–2.96 (2H, m, CH2), 3.05–3.11 (1H, m, CH2), 6.88 (1H, d, J 7.7, ArH), 7.07 (1H, ddd, J 0.9, 7.2, 7.7, ArH), 7.20 (1H, ddd, J 1.2, 7.7, 7.7, ArH), 7.42 (1H, d, J 7.2, ArH), 8.16 (1H, br s, NH); δC 38.1 (t), 41.9 (q), 53.9 (s), 56.9 (t), 66.2 (t), 109.8 (d), 123.1 (d), 123.5 (d), 128.0 (d), 136.1 (s), 140.4 (s), 183.3 (s); m/z (EI) 202 (40%, M+), 145 (25), 130 (28), 57 (100) and 51 (12).Acknowledgements
We thank: the Royal Society for a Leverhulme Senior Research Fellowship to JAM; University of Strathclyde for a studentship to D. L. and EPSRC Mass Spectrometry Service Swansea for mass spectra.References
- (a) C.-B. Cui, H. Kakeya, G. Okada, R. Onose and H. Osada, J. Antibiot., 1996, 49, 527 Search PubMed; (b) C.-B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 832 Search PubMed; (c) C.-B. Cui, H. Kakeya and H. Osada, Tetrahedron, 1996, 52, 12651 CrossRef CAS.
- (a) P. R. Sebahar and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 5666 CrossRef CAS; (b) H. Wang and A. Ganesan, J. Org. Chem., 2000, 65, 4685 CrossRef CAS; (c) L. E. Overman and M. D. Rosen, Angew. Chem., Int. Ed., 2000, 39, 4596 CrossRef CAS; (d) T. D. Bagul, G. Lakshmaiah, T. Kawabata and K. Fuji, Org. Lett., 2002, 4, 249 CrossRef CAS; (e) T. Lindel, Nachr. Chem., 2000, 48, 1498 Search PubMed; (f) H. S. Wang and A. Ganesan, J. Org. Chem., 2000, 65, 4685 CrossRef CAS; (g) F. von Nussbaum and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 2175 CrossRef CAS.
- S. Edmondson, S. J. Danishefsky, L. Sepp-Lorenzino and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147 CrossRef.
- (a) A. Jossang, P. Jossang, H. A. Hadi, T. Sévenet and B. Bodo, J. Org. Chem., 1991, 56, 6527 CrossRef CAS; (b) K. Jones and J. Wilkinson, J. Chem. Soc., Chem. Commun., 1992, 1767 RSC; (c) C. Pellegrini, C. Strässler, M. Weber and H.-J. Borschberg, Tetrahedron: Asymmetry, 1994, 5, 1979 CrossRef CAS; (d) S.-I Bascop, J. Sapi, J.-Y. Laronze and J. Lévy, Heterocycles, 1994, 38, 725 Search PubMed; (e) G. Palmisano, R. Annunziata, G. Papeo and M. Sisti, Tetrahedron: Asymmetry, 1996, 7, 1 CrossRef CAS; (f) G. Lakshmaiah, T. Kawabata, M. Shang and K. Fuji, J. Org. Chem., 1999, 64, 1699 CrossRef CAS; (g) C. Fischer, C. Meyers and E. M. Carreira, Helv. Chim. Acta, 2000, 83, 1175 CrossRef CAS; (h) G. Cravotto, G. B. Giovenzana, T. Pilati, M. Sisti and G. Palmisano, J. Org. Chem., 2001, 66, 8447 CrossRef CAS; (i) M. E. Kuehne, D. M. Roland and R. Hafter, J. Org. Chem., 1978, 43, 3705 CrossRef CAS; (j) U. K. Syam Kumar, H. Ila and H. Junjappa, Org. Lett., 2001, 3, 4193 CrossRef CAS; (k) M. Sonei, K. Noguchi, R. Yamagami, Y. Kawada, K. Yamada and F. Yamada, Heterocycles, 2000, 53, 7 Search PubMed; (l) S. E. V. Bell, R. F. C. Brown, F. W. Eastwood and J. M. Horvath, Aust. J. Chem., 2000, 53, 183 CrossRef CAS.
- N. Anderton, P. A. Cockrum, S. M. Colegate, J. A. Edgar, K. Flower, I. Vit and R. I. Willing, Phytochemistry, 1998, 48, 437 CrossRef CAS.
- B. Patro and J. A. Murphy, Org. Lett., 2000, 2, 3599 CrossRef CAS.
- S.-Z. Zhou, S. Bommezijn and J. A. Murphy, Org. Lett., 2002, 4, 443 CrossRef CAS.
- For preliminary report, see: D. Lizos, R. Tripoli and J. A. Murphy, Chem. Commun., 2001, 2732 Search PubMed.
- Y. Kondo, S. Kojima and T. Sakamoto, J. Org. Chem., 1997, 62, 6507 CrossRef CAS.
- L. F. Tietze and T. Grote, Chem. Ber., 1993, 126, 2733 CAS.
- B. R. Baker, R. E. Schaub and J. H. Williams, J. Org. Chem., 1952, 17, 116 CrossRef.
- S. E. Webber, K. Okano, T. L. Little, S. H. Reich, Y. Xin, S. A. Fuhrman, D. A. Matthews, R. A. Love, T. F. Hendrickson, A. K. Patick, J. W. Meador, R. A. Ferre, E. L. Brown, C. E. Ford, S. L. Binford and S. T. Worland, J. Med. Chem., 1998, 41, 2786 CrossRef CAS note that commercial lithium borohydride was not tried in this experiment.
- K. Jones, S. A. Brunton and R. Gosain, Tetrahedron Lett., 1999, 40, 8935 CrossRef CAS.
- A. R. Lee, W. H. Huang, T. L. Lin, K. M. Shih and H. F. Lee, J. Heterocycl. Chem., 1995, 32, 1 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra are provided for the compounds; the preparations of compounds 19, 20 and 21 and the conversion of 20 to 16 are also included. See http://www.rsc.org/suppdata/ob/b2/b208114h/ |
| ‡ The IUPAC name for itaconic acid is methylenesuccinic acid. |
| This journal is © The Royal Society of Chemistry 2003 |