A donor–acceptor substituted molecular motor: unidirectional rotation driven by visible light†
Richard A.
van Delden
,
Nagatoshi
Koumura
,
Annemarie
Schoevaars
,
Auke
Meetsma
and
Ben L.
Feringa
*
Department of Organic and Molecular Inorganic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@chem.rug.nl; Fax: (+31) 50-3634296; Tel: (+31) 50-3634278
First published on 8th November 2002
Abstract
A newly designed donor–acceptor substituted molecular motor 1 allows unidirectional rotation driven by visible light and shows some unique photophysical properties.
In the nanotechnological endeavour towards molecular devices, different functions have to be addressed at a molecular level.1 Molecular switches,2 brakes,3 gears,4 turnstiles,5 and muscles6 have been developed over the years. Controlled molecular motion is an essential feature of these systems.7 Other examples of the control of molecular movement in the pursuit of true molecular motors8 involve molecular machines based on redox-driven metal ion translocation9 and molecular shuttles based on linear pseudo-rotaxane or rotaxane or catenane systems.10 A unidirectional rotating molecular motor will be one of the most prominent members of the future nanotechnology's toolbox. Recently, the first examples of unidirectional rotating molecular motors based on simple organic molecules were reported.11,12 Following our initial design, a series of second-generation motors was developed,13 where we combined the design versatility of chiroptical molecular switches2 with the unique rotational behavior of a chiral 2-methyl-2,3-dihydrothiopyran propeller. For these motors the rotation speed can be tuned very precisely.14
Here, we report a novel donor–acceptor functionalized molecular motor 1, which allows repetitive 360° unidirectional rotation driven by visible light. Furthermore, a remarkable enhancement of the isomerization process upon protonation is observed. The molecular design is based on a helical sterically overcrowded alkene with a rotor upper half combined with a donor–acceptor substituted 7-dimethylamino-2-nitro-9H-thioxanthene15 lower half (Scheme 1). Asymmetric donor–acceptor substitution allows excitation of 1 by visible light.
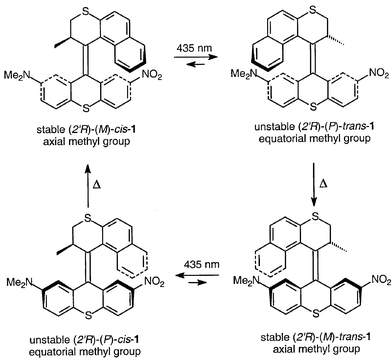 | ||
| Scheme 1 Unidirectional rotation of a donor–acceptor substituted molecular motor consisting of four distinct stages. | ||
The key step in the synthesis of 1 is the formation of the central double bond. Analogous to previously reported systems, this was achieved via a diazothioketone coupling of the upper and lower halves, followed by desulfurization. Compound 1 is formed as a mixture of diastereoisomers, which was resolved by HPLC.16 All stereoisomers with a (2′R)-configuration at the stereogenic center could be assigned by comparison of their circular dichroism (CD) spectra with the spectra of related compounds,14 taking into account the preferred (pseudo)axial orientation of the methyl substituent. This preferred axial orientation of the methyl substituent was underlined by an X-ray crystal structure of (2′R)-(M)-cis-1 (Fig. 1).17‡
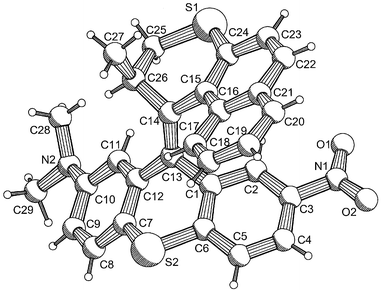 | ||
| Fig. 1 X-Ray crystal structure of energetically stable (2′R)-(M)-cis-1 with the methyl substituent in a (pseudo)axial orientation. | ||
Irradiation of (2′R)-(M)-cis-1, with an axial orientation of the methyl group, in CHCl3 (3.309 × 10−5 M) at 5 °C with visible light at 435 nm resulted in the formation of (2′R)-(P)-trans-1, with the methyl substituent in an equatorial orientation, as was confirmed by 1H NMR. The NMe2-protons shift from δ 3.05 to 2.25 ppm, as a result of increased shielding due to the proximity of the upper arene part, indicating cis to trans isomerization. The upper half methyl protons shift from δ 0.86 to 1.06 ppm, indicative of the forced equatorial orientation, where the methyl group is shielded by the lower arene moiety. The (M) to (P) reversal of helicity is readily observed by CD spectroscopy (Fig. 2), where the major band shifted from 281 to 276 nm and changed sign from Δε −121.8 to + 138.3 (for the photostationary state at 435 nm (PSS435)). A PSS with a ratio (2′R)-(M)-cis-1 ∶ (2′R)-(P)-trans-1 of 1 ∶ 9 was observed, as indicated both by HPLC and NMR analyses.
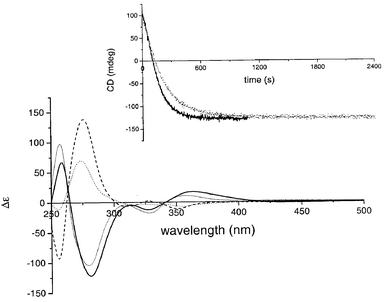 | ||
Fig. 2 CD spectra of all stages of the unidirectional molecular motor. ![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) (2′R)-(M)-cis-1;
(2′R)-(M)-cis-1; ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) (2′R)-(P)-trans-1
(PSS435); —
(2′R)-(M)-trans-1; ⋯
(2′R)-(P)-cis-1
(PSS435). The inset shows the change in CD signal at 274 nm in time upon heating at 50 °C; grey line: conversion of (2′R)-(P)-cis-1 to (2′R)-(M)-cis-1; black line: conversion of (2′R)-(P)-trans-1 to (2′R)-(M)-trans-1.
(2′R)-(P)-trans-1
(PSS435); —
(2′R)-(M)-trans-1; ⋯
(2′R)-(P)-cis-1
(PSS435). The inset shows the change in CD signal at 274 nm in time upon heating at 50 °C; grey line: conversion of (2′R)-(P)-cis-1 to (2′R)-(M)-cis-1; black line: conversion of (2′R)-(P)-trans-1 to (2′R)-(M)-trans-1. | ||
Next, this mixture was heated in the dark at 50 °C and the CD signal at 274 nm was monitored in time. An inversion of helicity was observed after approximately 20 min of heating, indicating formation of stable (2′R)-(M)-trans-1 with a major CD band (Δε −113.4) at 279 nm. From the time transient and the known CD spectra of both (2′R)-(P)-trans-1 and (2′R)-(M)-trans-1, the rate constant of this thermal process was determined to be 6.90 × 10−3 s−1. From this value both the half-life of the process (t½ = 1.01 × 102 s) and the Gibbs free energy of activation (ΔG≠ = 92.7 kJ mol−1) could be calculated.18 The inversion of CD signal was accompanied by a change in 1H NMR absorption, clearly indicating the trans geometry of the compound (–NMe2δ 2.20 ppm) and the axial orientation of the upper half methyl substituent (δ 0.74 ppm). Subsequent irradiation, again with 435 nm light, induced a second (trans to cis) isomerization resulting in the formation of (2′R)-(P)-cis-1, with the methyl substituent in an equatorial orientation as indicated by 1H NMR. The NMe2-protons shift from δ 2.20 to 3.07 ppm, the upper half Me-protons shift from δ 0.74 to 1.25 ppm. Again inversion of the CD absorption was observed (to Δε +75.9 at 274 nm). A PSS with a ratio (2′R)-(M)-trans-1 ∶ (2′R)-(P)-cis-1 of 3 ∶ 7 was observed (HPLC, NMR). Upon heating to 50 °C in the dark again, a clear helix inversion was visible in the CD spectrum and 1H NMR analysis confirmed the expected formation of (2′R)-(M)-cis-1. Here, the rate constant, half life, and Gibbs free energy of activation were determined to be 4.95 × 10−3 s−1, 1.40 × 102 s, and 93.6 kJ mol−1, respectively, similar to the values found for the trans helix inversion. The CD spectra corresponding to the four different stages of the molecular motor are depicted in Fig. 2; the inset shows the change in CD signal at 274 nm with time upon heating during the two thermal helix inversion steps.
Analogous to the previously reported second-generation motors, here four stages of the molecular process combine to a full 360° rotation of the upper (rotor) half of the molecule relative to the other (stator) half in a counterclockwise fashion, dictated by the configuration at the stereogenic center and the accompanying helicity of the molecule. The process is driven by two energetically uphill photoisomerization steps induced by visible light, forcing the methyl substituent in an energetically unfavorable equatorial conformation. The release of internal energy is accomplished by helix inversion, where the methyl substituent adopts the favorable axial conformation again. Two of these energetically downhill helix inversions ensure the unidirectionality of rotation.
Compound 1 can be envisioned as a basic element for a molecular solar cell,19 which directly converts solar energy into mechanical unidirectional rotary motion. The use of visible light as a driving force, or fuel, is more convenient than the use of UV light and it corresponds to lower energy consumption since photon energy is inversely proportional to the wavelength of light. The thermal barrier for helix inversion is remarkably low and close to the lowest barrier found thus far for the second generation motors.14 Since the two thermal helix inversions are the rate determining steps in the rotation process, this is one of the fastest second-generation motors. This is surprising since the barrier height is so far assumed to be caused by steric effects, mainly determined by the size of the bridging (hetero-)atoms in the upper and lower halves of the molecule. Relatively small atoms like carbon and oxygen give the lowest barriers, whereas larger sulfur atoms result in strongly increased barriers and slower rotation. For steric reasons, for the S,S-bridged overcrowded alkene 1, a considerably higher barrier was expected and electronic factors appear to play a decisive role here. Next to this unprecedented fast rotation speed, additional advantages of the presented system can be envisioned. The dipole moment of the molecule, caused by asymmetric electron donor–acceptor substitution, might offer the possibility of using an electric field to align this compound.
Another advantage of the substitution pattern of compound 1 is the possibility to influence its behavior by a second stimulus. For a donor–acceptor substituted chiroptical molecular switch, such a feature resulted in a gated switching system.20 In that case, protonation was readily achieved using trifluoroacetic acid (TFA) and the protonated switch did not show any photoisomerization whereas its fluorescence was completely quenched. This allows locking of the switching process; a highly desired property for information storage. As far as the fluorescence behavior of the donor–acceptor motor 1 is concerned, protonation with TFA has a similar effect. The unprotonated (M)-trans-1 shows green fluorescence (λmax = 527 nm) in n-hexane (where the cis-form is insoluble) and both (M)-trans-1 and (M)-cis-1 show orange fluorescence in chloroform (λmax(trans) = 710 nm and λmax(cis) = 705 nm). Both in n-hexane as well as chloroform solution, the fluorescence is completely quenched upon protonation with TFA. Unexpected and completely different from the related chiroptical molecular switch, 435 nm irradiation of the protonated motor, starting from (2′R)-(M)-cis-1, leads to a faster and more efficient isomerization process. UV-Vis spectroscopy (Fig. 3) shows that only 420 s of irradiation of a 5.16 × 10−5 M chloroform solution are sufficient to fully reach the PSS with unstable (P)-trans in excess. For the unprotonated compound 1, under identical conditions, an irradiation time of 720 s is necessary to reach the PSS. Furthermore, the selectivity of the photoisomerization has slightly increased; where the unprotonated motor gives a PSS435 with (M)-cis-1 ∶ (P)-trans-1 10 ∶ 90, in the protonated case this ratio ((M)-cis ∶ (P)-trans) is 5 ∶ 95. These preliminary results clearly illustrate that the mechanism in this energetically uphill photoisomerization process is completely different from that of a donor–acceptor substituted molecular switch bearing identical substituents.
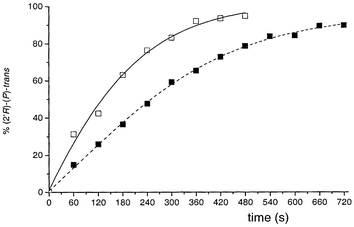 | ||
| Fig. 3 Conversion vs. time for the photoisomerization of protonated (solid) and unprotonated (dashed) motor 1. | ||
The second step of the rotation cycle for the protonated structure, the thermal helix inversion going from unstable (P)-trans-1 to stable (M)-trans-1 was also monitored by UV-Vis spectroscopy. It was shown that this helix inversion indeed takes place and the Gibbs free energy was established to be 101.1 kJ mol−1, a value substantially higher than for the unprotonated compound and more or less comparable to the value found for the parent molecular motor without donor–acceptor substituents. These observations clearly illustrate that electronic effects, caused by the donor–acceptor substitution pattern in the lower half of the molecule, play an important role in both the photoisomerization as well as the thermal helix inversion steps in these motors. Detailed analysis of these processes is in progress.
References
- (a) Sci. Amer., special issue: Nanotech: The Science of Small Gets Down to Business, September 2001 Search PubMed; (b) R.P. Feynman in Miniturization, ed. H. D. Gilbert, New York, 1971 Search PubMed; (c) K. E. Drexler, Nanosystems: Molecular Machinery, Manufacturing and Computation, Wiley, New York, 1992 Search PubMed.
- (a) B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS; (b) Molecular Switches, ed. B.L. Feringa, Wiley, Weinheim, 2001 Search PubMed.
- T. R. Kelly, M. C. Bowyer, K. V. Bhaskar, D. Bebbington, A. Garcia, F. R. Lang, M. H. Kim and M. P. Jette, J. Am. Chem. Soc., 1994, 116, 3657–3658 CrossRef CAS.
- (a) A. M. Stevens and C. J. Richards, Tetrahedron. Lett., 1997, 38, 7805–7808 CrossRef CAS; (b) J. Clayden and J. H. Pink, Angew. Chem., Int. Ed. Engl., 1998, 37, 1937–1939 CrossRef CAS.
- T. C. Bedard and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 10662–10671 CrossRef CAS.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef CAS.
- B. L. Feringa, N. Koumura, R. A. van Delden and M. K. J. ter Wiel, Appl. Phys. A, 2002, 75, 301–308 Search PubMed.
- (a) J. P. Abrahams, A. G. W. Leslie, R. Lutter and J. E. Walker, Nature, 1994, 370, 621–628 CrossRef CAS; (b) H. Noji, M. Yasuda and K. Kinosita Jr., Nature, 1997, 386, 299–302 CrossRef CAS; (c) Special issue: Science, Movement: Molecular to Robotic, 2000, 288, 79–106 Search PubMed.
- V. Amendola, L. Fabbrizzi, C. Mangano and P. Pallavicini, Acc. Chem. Res., 2001, 34, 488–493 CrossRef CAS.
- For reviews see for example: (a) V. Balzani, A. Credi, M. Raymo and J. F. Stoddard, Angew. Chem., Int. Ed., 2000, 39, 3349–3391 CrossRef; (b) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS; (c) R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445–455 CrossRef CAS; (d) C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS; (e) J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jiminez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS.
- N. Koumura, E. M. Geertsema, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2000, 122, 12005–12006 CrossRef CAS.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037–5051 CrossRef CAS.
- W. F. Jager, J. C. de Jong, B. de Lange, N. P. M. Huck, A. Meetsma and B. L. Feringa, Angew. Chem., Int. Ed. Engl., 1995, 34, 348–350 CrossRef CAS.
- See supplementary material for details on synthesis, spectroscopic data and resolution.
- Crystal data for cis- 1, C29H24N2O2S2, Mr = 496.64, orthorhombic, P212121, a = 7.7426(3), b = 12.4048(5), c = 24.867(1) Å, V = 2388.36(16) Å3, Z = 4, Dx = 1.381 g cm−3, λ(MoKα) = 0.71073 Å, µ = 2.54 cm−1, F(000) = 1040, T = 100 K, GooF = 1.037, Rint = 0.0160, wR(F2) = 0.0769 for 6379 reflections and 412 parameters and R(F) = 0.0285 for 6253 reflections obeying Fo ≥ 4.0 σ(Fo) criterion of observability. The absolute configuration of (cis)-1 could not be determined via X-ray methods.
- For a unimolecular reaction: k = t½−1 ln 2 and ΔG≠ = −RT ln (kh/kb).
- The monochromatic efficiency, considering the energy differences between the unstable and stable forms and the UV-Vis absorption of the isomers, can be estimated to be about 0.6%.
- N. P. M. Huck and B. L. Feringa, J. Chem. Soc., Chem. Commun., 1995, 1095–1096 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR details, and HPLC resolution of compound 1. Details of UV-Vis absorption spectra of all four isomers of compound 1 and the photochemical experiments. See http://www.rsc.org/suppdata/ob/b2/b209378b/ |
| ‡ CCDC reference number 194360. See http://www.rsc.org/suppdata/ob/b2/b209378b/ for crystallographic files in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |