From central to planar chirality, the first example of atropenantioselective cycloetherification
Gabriela Islas-Gonzalez, Michèle Bois-Choussy and Jieping Zhu*
Institut de Chimie des Substances Naturelles, CNRS, 91198 Gif-sur-Yvette Cedex, France. E-mail: zhu@icsn.cnrs-gif.fr; Fax: + 33 1 69 07 72 47; Tel: + 33 1 69 82 31 31
First published on 8th November 2002
Abstract
Using chiral quaternary ammonium hydroxide as base, cycloetherification of linear achiral diarylheptanoid 5, by way of an intramolecular SNAr reaction, provides enantiomerically enriched cyclophane 6 in good to excellent yield.
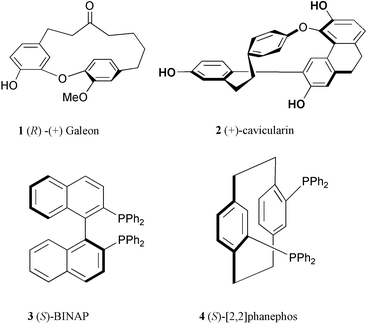
A number of optically active natural products contain planar or axial chirality as the only asymmetric element. Galeon (1)1 and cavicularin (2)2 are such examples. Thus (R)-(+)-galeon displays planar chirality, while (+)-cavicularin possesses planar as well as axial chirality; both compounds are devoid of chiral centers (Fig. 1). On the other hand, ligands with axial chirality (e.g. BINAP, 3)3 and planar chirality (e.g. [2,2]phanephos, 4)4 have found widespread applications in catalytic asymmetric processes. The asymmetric synthesis of these classes of compounds is thus of considerable importance. While palladium catalyzed enantioselective Kumada coupling5 and more recently, Suzuki coupling6 have been developed for the synthesis of axially chiral biaryls, enantioselective cyclization leading to planar chiral cyclophanes, to the best of our knowledge, remains unknown.7
 | ||
| Fig. 1 | ||
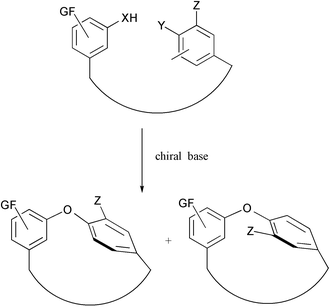
Since our original report,8 cycloetherification based on intramolecular nucleophilic aromatic substitution (SNAr) has been developed into a powerful methodology for the synthesis of macrocycles with endo aryl–aryl9 and aryl–alkyl ether bonds.10Atropdiastereoselective cycloetherification has been observed sporadically.10,11 However, no rationale could be advanced and indeed, the atropselectivity is very sensitive to subtle structural modifications and is difficult to predict. Recently, a highly atropdiastereoselective cyclization has been designed by Nicolaou and co-workers by the temporary introduction of a bulky substituent on the aromatic ring.12 We have been interested in the development of an atropenantioselective cycloetherification process as shown in Scheme 1 and wish to report herein our preliminary results.
 | ||
| Scheme 1 | ||
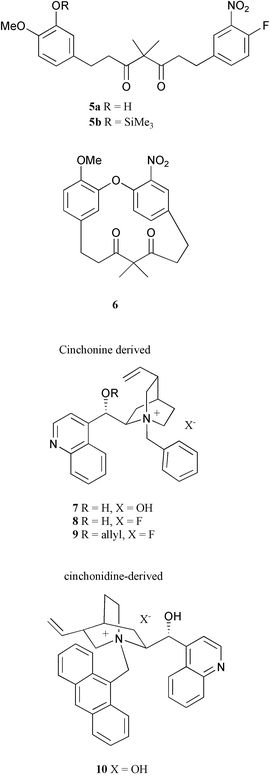
An achiral linear diarylheptanoid 5 (5a R = H, 5b R = trimethylsilyl) was selected for our studies, since its cyclization will create concomitantly a planar chirality due to the presence of a nitro group ortho to the aryl ether linkage and the constrained ring system.13 While potassium carbonate and caesium fluoride have previously been used to promote the cyclization of 5a to provide cyclophane 6 in excellent yield, we found that tetrabutylammonium fluoride (TBAF) and tetrabutylammonium hydroxide were also able to promote the cyclization of 5a. These results prompted us to investigate the projected atropenantioselective cyclization by using chiral quaternary ammonium salts. The following salts were synthesized from cinchonine and cinchonidine, respectively, according to standard procedures (Fig. 2).14 The results of the cyclization, using DMF (0.05 M) as solvent, are summarized in Table 1.
 | ||
| Fig. 2 | ||
| Entry | Substrate | Base | Yield of 6 (%) | Ee (%)b |
|---|---|---|---|---|
| a The reaction was performed in DMF (0.05 M) at room temperature in the presence of one equivalent of chiral ammonium salt.b The enantiomeric excess was measured by HPLC using a chiral column (DAICEL DO), eluent: hexane–isopropanol 85 ∶ 15, UV detection at 240 nm.c Enantioselectivity reversed. | ||||
| 1 | 5a | 7 | 60 | 20 |
| 2 | 5a | 8 | 90 | 3 |
| 3 | 5a | 9 | 68 | 0 |
| 4 | 5b | 8 | 87 | 0 |
| 5 | 5b | 9 | 83 | 0 |
| 6 | 5a | 10 | 60 | 6c |
As is seen, cyclization of 5a promoted by chiral ammonium fluoride provided the cyclophane 6† with negligible enantioselectivity (entries 2 to 5). The same is true with substrate 5b wherein the phenol was protected as a trimethylsilyl ether (entry 4). However, a notable atropenantioselectivity was observed with the ammonium hydroxide, 5 producing 6 with 20% enantiomeric excess (entry 1). The N-anthracenylmethylcinchonidinium hydroxide was also tested for cyclization.15 In this case, the opposite enantiomer was enriched under identical conditions, albeit with low ee (entry 6). The cyclophane 6 obtained was transformed to the diastereomeric mixture 11 (Fig. 3).16 The diastereomeric excess calculated from the 1H NMR spectra correlated well with the ee value determined from chiral HPLC analysis.
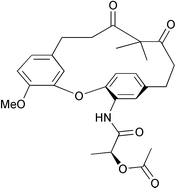 | ||
| Fig. 3 The atropisomer shown in the figure is arbitrary. | ||
We assumed that the atropenantioselectivity of the cyclization should depend on the tightness of the ion pair formed between the phenoxide and the chiral ammonium base. The latter should in turn be influenced by the solvent polarity.17 We thus examined the solvent effect using two pseudo-enantiomeric ammonium hydroxides (7, 10) and the results are summarized in Table 2.
| Entry | Solvent | Base | Time/h | Yield of 6 (%) | Ee (%)b |
|---|---|---|---|---|---|
| a The reaction was performed at room temperature in the presence of one equivalent of chiral ammonium salt (0.05 M).b The enantiomeric excess was measured by HPLC using a chiral column (DAICEL DO), eluent: hexane–isopropanol 85 ∶ 15, UV detection at 240 nm.c Enantioselectivity reversed. | |||||
| 1 | DMF | 7 | 18 | 60 | 20 |
| 2 | MeCN | 7 | 24 | 83 | 0 |
| 3 | Toluene | 7 | 6 days | 61 | 8 |
| 4 | DMF | 10 | 24 | 60 | 6 |
| 5 | MeCN | 10 | 24 | 79 | 2 |
| 6 | Toluene | 10 | 6 days | 85 | 18c |
From DMF to toluene, the presumed ion pair ArO−NR4+ should become tighter. Consequently, one may expect, at the expense of low reactivity, an increased enantioselectivity of the cyclization. However, experimental results indicated that there was no linear correlation between solvent polarity and atropenantioselectivity. Indeed, while the cyclization of 5 promoted by 10 provided a better ee in toluene (entries 6 vs. 4, 5), that promoted by 7 was best realized in DMF (entries 1 vs. 2, 3). This fact indicates that the chirality transfer from chiral ammonium salt to planar chiral cyclophane is more complex than we had envisaged.
The combination of butyllithium–sparteine18 was next examined with the idea that the ion-pair ArO−Li+-sparteine should be even tighter, thus bringing the chiral environment nearer to the reactive sites. However, no cyclization occurred under a set of conditions varying temperatures, solvents and stoichiometries and only the starting material was recovered. This result is not unexpected in view of the counterion effect we observed previously on the intramolecular SNAr reaction.19
In conclusion, we reported the first example of an enantioselective cycloetherification reaction. Although the atropenantioselectivity remained moderate, the results described here served as a proof-of-concept and laid down the foundation for future work.
Acknowledgements
Financial support from the CNRS and a SFERE-CONACYT doctoral fellowship funded jointly by the Mexican and French governments to G. Islas-Gonzalez are gratefully acknowledged.Notes and references
- K. E. Malterud, T. Anthonsen and J. Hjortas, Tetrahedron Lett., 1976, 3069–3072 CrossRef CAS.
- M. Toyota, T. Yoshida, Y. Kan, S. Takaoka and Y. Asakawa, Tetrahedron Lett., 1996, 37, 4745–4748 CrossRef CAS.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, John Wiley & Sons, Inc., New York, 1994 Search PubMed.
- P. J. Pye, K. Rossen, R. A. Reamer, N. N. Tsou, R. P. Volante and P. J. Reider, J. Am. Chem. Soc., 1997, 119, 6207–6208 CrossRef CAS.
- (a) T. Hayashi, K. Hayashizaki, T. Kiyoi and Y. Ito, J. Am. Chem. Soc., 1988, 110, 8153–8156 CrossRef CAS; (b) T. Hayashi, S. Nizuma, T. Kamikawa, N. Suzuki and Y. Uozumi, J. Am. Chem. Soc., 1995, 117, 9101–9102 CrossRef CAS; (c) T. Kamikawa and T. Hayashi, Tetrahedron, 1999, 55, 3455–3466 CrossRef CAS.
- (a) A. N. Cammidge and K. V. L. Crépy, Chem. Commun., 2000, 1723–1724 RSC; (b) J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 12051–12052 CrossRef CAS.
- (a) For recent reviews on enantioselective synthesis and application of planar chiral (arene)chromium complexes and ferrocene derivatives, see: K. Kamikawa and M. Uemura, Synlett, 2000, 938–949 Search PubMed; (b) C. Bolm and K. Muñiz, Chem. Soc. Rev., 1999, 28, 51–59 RSC; (c) C. J. Richards and A. J. Locke, Tetrahedron: Asymmetry, 1998, 9, 2377–2407 CrossRef CAS; (d) A. Togni, Angew. Chem., Int. Ed. Engl., 1996, 35, 1475–1477 CrossRef CAS.
- J. Zhu, Synlett, 1997, 133–144 CAS.
- (a) For reviews, see: K. Burgess, D. Lim and C. I. Martinez, Angew. Chem., Int. Ed. Engl., 1996, 35, 1077–1078 Search PubMed; (b) K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096–2152 CrossRef; (c) J. S. Sawyer, Tetrahedron, 2000, 56, 5045–5065 CrossRef.
- T. Temal-Laïb, J. Chastanet and J. Zhu, J. Am. Chem. Soc., 2002, 124, 583–590 CrossRef.
- (a) M. Bois-Choussy, L. Neuville, R. Beugelmans and J. Zhu, J. Org. Chem., 1996, 61, 9309–9322 CrossRef CAS; (b) R. Beugelmans, M. Bois-Choussy, C. Vergne, J. P. Bouillon and J. Zhu, Chem. Commun., 1996, 1029–1030 RSC; (c) D. A. Evans, M. R. Wood, B. W. Trotter, T. I. Richardson, J. C. Barrow and J. L. Katz, Angew. Chem.. Int. Ed., 1998, 37, 2700–2704 CrossRef CAS; (d) D. A. Evans, C. J. Dinsmore, P. S. Watson, M. R. Wood, T. I. Richardson, B. W. Trotter and J. L. Katz, Angew. Chem., Int. Ed., 1998, 37, 2704–2708 CrossRef CAS.
- K. C. Nicolaou and C. N. C. Boddy, J. Am. Chem. Soc., 2002, 124, 10451–10455 CrossRef CAS.
- G. Islas-Gonzalez and J. Zhu, J. Org. Chem., 1999, 64, 914–924 CrossRef.
- Compound 7 was prepared from cinchonine in two steps: a) BnBr, acetone, reflux, 72h. b) Amberlyst A-26, MeOH. Treatment of compound 7 in MeOH with HF (1 M) gave 8 in quantitative yield. Cinchonidine derivative 10 was prepared in a similar fashion..
- (a) B. Lygo and P. G. Wainwright, Tetrahedron Lett., 1997, 38, 8595–8598 CrossRef CAS; (b) E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414–12415 CrossRef CAS.
- Reagents and conditions: a) SnCl2, DMF, 50 °C, 7h. b) (S)-2-acetoxypropionyl chloride, Et3N, DMAP, CH2Cl2, 0 °C to room temperature, 2 h.
- A. Loupy, B. Tchoubar and D. Astruc, Chem. Rev., 1992, 92, 1141–1165 CrossRef CAS.
- D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282–2316 CrossRef CAS.
- G. Islas-Gonzalez and J. Zhu, Synlett, 2000, 412–414 CAS.
Footnote |
| † Physical and spectroscopic data of cyclophane 6. Mp: 148–151 °C; IR (CHCl3) ν 3034, 3014, 1715, 1694, 1601, 1533, 1519, 1265, 1232, 1203, 1127 cm−1; 1H NMR (CDCl3, 250 MHz) δ 1.36 (s, 3H), 1.38 (s, 3H), 2.54–2.63 (m, 4H), 3.00–3.04 (m, 4H), 3.94 (s, 3H), 5.06 (d, J = 2.0 Hz, 1H), 6.5 (dd, J = 2.0, 8.2 Hz, 1H), 6.74 (d, J = 8.2 Hz), 7.11 (d, J = 8.3 Hz, 1H), 7.48 (dd, J = 2.2, 8.3 Hz, 1H), 7.94 (d, J = 2.2 Hz, 1H); 13C NMR (CDCl3) δ 22.5, 22.6, 26.2, 28.8, 39.2, 41.2, 56.6, 62.5, 112.2, 114.3, 122.4, 126.6, 127.1, 134.3, 136.3, 140.0, 143.1, 147.0, 149.0, 150.6, 207.0, 208.3; MS (FAB) m/z 397; Anal. calcd for C22H23NO6: C, 66.49; H 5.83; N 3.52; Found: C, 66.16; H 5.85; N 3.45%. |
| This journal is © The Royal Society of Chemistry 2003 |