Development of β-keto 1,3-dithianes as versatile intermediates for organic synthesis
Matthew J.
Gaunt
,
Helen F.
Sneddon
,
Peter R.
Hewitt
,
Paolo
Orsini
,
David F.
Hook
and
Steven V.
Ley.
*
University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: svl1000@cam.ac.uk; Fax: +44 (0) 1223 336442; Tel: +44 (0) 1223 336398
First published on 3rd December 2002
Abstract
β-Keto 1,3-dithianes can be generated by the double conjugate addition of dithiols to propargylic ketones, esters and aldehydes in excellent yields. As masked 1,3-dicarbonyl systems these substrates can be converted to a range of functionalised oxygen-containing heterocycles that can be used in natural product synthesis.
The generation of 1,3-oxygenated functionality in organic molecules is an important process of modern synthetic chemistry.1 While there have been many advances in this area, and especially for the assembly of polyketide natural products, there remains a constant need to develop new methods for the introduction of 1,3-oxidation patterns with enantio- and diastereocontrol.2
We were interested in developing a general functionalised molecular unit from which we could generate a range of 1,3-substituted molecules. Here we report our initial studies on the facile double conjugate addition of dithiols to propargylic carbonyls to form a β-carbonyl dithiane. This intermediate provides a versatile platform for the preparation of heterocycles containing 1,3-oxidation arrangements.
 | ||
| Scheme 1 | ||
There are a number of methods available for the generation of β-keto 1,3-dithianes, although in some cases their synthesis is complicated and reported applications have therefore been limited.3 Potentially, however, orthogonally protected dicarbonyl intermediates have wide ranging applications. We conceived that a dithiol would undergo a base mediated double conjugate addition to a propargylic ketone to generate the desired β-keto 1,3-dithiane (Scheme 1, 2).4
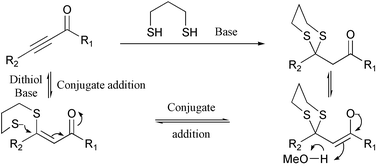 | ||
| Scheme 2 Conjugate addition of a dithiol to propargylic ketones. | ||
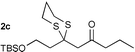
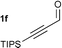
Initial investigations proceeded on the addition of propane-1,3-dithiol to alkyne 1a to generate desired β-keto dithiane 2a in an excellent yield. The optimal system involved the use of NaOMe in MeOH with either THF or CH2Cl2 as co-solvent. The scope of the reaction was investigated using a range of propargylic compounds (1a–f). Pleasingly, the dithiol additions worked in good to excellent yields, Table 1.† Ketones (entries 1, 3, and 4) and esters (entry 2) with a range of substituents were good substrates for this process.5 In general, we found that the addition of the base to a solution of dithiol and the propargylic carbonyl at −10 °C and allowing the temperature to rise to 0 °C overnight gave optimal yield of product. Particularly noteworthy is the formation of dithiane 2e from aldehyde 1e (entry 5). This versatile aldehyde derivative was generated in good yield. The dithiol addition is amenable to large-scale production of dithiane adducts. For example, ethyl propiolate 1b is converted to dithiane 2b in 84% yield on a 0.1 mole scale. Dithiane 2f is formed in modest yield from 1f (entry 6). This product is probably formed by dithiol addition6 to the alkyne followed by attack of methoxide on the resulting aldehyde and subsequent 1,4-Brook rearrangement.7
| Entry | Substratea | Time/h | Yield (%) | Product |
|---|---|---|---|---|
| a Reagents and conditions: NaOMe (1.3 equiv.), propane-1,3-dithiol (1.1–1.3 equiv.), MeOH–CH2Cl2 (4 : 1, 0.05 M), −10 °C to 0 °C. b NaOEt, EtOH–CH2Cl2 are used. c NBu4OH (1.3 equiv.), propane-1,3-dithiol, THF–MeOH, 0 °C. | ||||
| 1 |

|
16 | 94 |

|
| 2b |

|
4 | 84 |

|
| 3 |

|
24 | 88 |
|
| 4 |

|
24 | 82 |

|
| 5 |

|
18 | 88 |

|
| 6c |
|
18 | 48 |

|
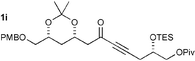
More complex substrates are also amenable to this reaction. As part of our natural product synthesis programmes we required a number of poly-oxygenated 1,3-dicarbonyl compounds. The propargylic ketones were readily prepared by a two step procedure involving acetylide addition to an aldehyde and oxidation of the resulting propargylic alcohols. Table 2 shows the addition of dithiols to more complex substrates (1g–i), highlighting the compatibility of this reaction with a range of common protecting groups. Moreover, the stereochemical integrity of ketone 1g is conserved in the formation of 2g implying that this methodology is adaptable to molecules with base sensitive asymmetric centres.
| Entry | Substratea | Time/h | Yield (%) | Product |
|---|---|---|---|---|
| a Reagents and conditions: NaOMe (1.3 equiv.), propane-1,3-dithiol (1.1 equiv.), MeOH–CH2Cl2 (4 : 1, 0.05 M), −10 to 0 °C. | ||||
| 1 |

|
16 | 80 |

|
| 2 |

|
16 | 90 |

|
| 3 |
|
16 | 95 |

|
We also investigated the addition to 1j, a substrate that contains a second electrophilic site. Dithiol addition proceeded smoothly and was followed by intramolecular aldol reaction to form 2j as a 3 ∶ 1 (syn–anti) mixture of diastereomers in 65% yield. This tandem process generates cyclic systems with high levels of functionality (Scheme 3).
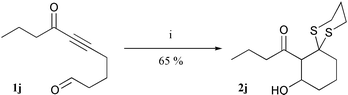 | ||
| Scheme 3 Tandem dithiol-intramolecular aldol reaction. Reagents and conditions: (i) NaOMe (1.3 equiv.), propane-1,3-dithiol (1.1 equiv.), MeOH–CH2Cl2 (3 : 1), 0 °C to rt, 14 h. | ||
The conversion of the propargylic ketone unit to a β-keto 1,3-dithiane removes the rigid acetylene unit from the molecule permitting a range of cyclisation reactions. For example, treatment of 2h with p-TsOH in MeCN–H2O for 16 hours facilitated removal of the two silicon groups and subsequent cyclisation afforded spiroketal 3. This sequence of reactions provided a model compound for the synthesis of the AB spiroketal unit of spongistatin 1 (Scheme 4).8 Interestingly, the 1,3-dithiane unit seems to have a significant effect on the cyclisation of 2h. Cyclisation of the corresponding dione proved capricious,9however, in the presence of the dithiane unit the cyclisation cleanly produced the desired spiroketal in excellent yield (95%).
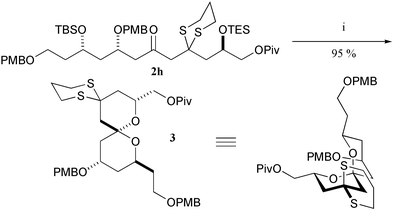 | ||
| Scheme 4 Application to the synthesis of spiroketal 3. Reagents and conditions: (i) p-TsOH, MeCN–H2O (4 : 1), 30 °C. | ||
In summary, we have developed an efficient method for the conjugate addition of dithiols to propargylic ketones, esters and aldehydes. The resulting β-carbonyl-dithianes are isolated in good to excellent yields across a range of substrates. The potential of these units as versatile platforms for the generation of 1,3-oxygenated structures is highlighted through the synthesis of spiroketal 3. Functionalised dithianes (2f) can be accessed from a silyl substituted propargylic aldehyde (1f) and we have also reported a new tandem process that forms highly functionalised cyclic systems. We are currently exploring the application of these species in a number of synthesis programmes and these results will be reported in due course.
Acknowledgements
We would like to thank the British Ramsay Memorial Trust and Magdalene College for Fellowship (to M.J.G.), the EPSRC (to H.F.S. and D.F.H.), Pharmacia S.p.A., Viale Pasteur, 10-20014 Nerviano (MI) – Italy. (to P.O) and the Novartis Research Fellowship (to S.V.L).Notes and references
- (a) C. Schneider, Angew. Chem., Int. Ed., 1998, 37, 1375 CrossRef CAS; (b) R. D. Norcross and I. Paterson, Chem. Rev., 1995, 95, 2041 CrossRef CAS.
- (a) For some recent examples see: A. B. Smith and A. M. Boldi, J. Am. Chem. Soc., 1997, 119, 6925 Search PubMed; (b) S. D. Rychnovsky, U. R. Khire and G. Yang, J. Am. Chem. Soc., 1997, 119, 2058 CrossRef CAS; (c) D. A. Evans, D. M. Fitch, T. E. Smith and V. J. Cee, J. Am. Chem. Soc., 2000, 122, 10033 CrossRef CAS; (d) I. Paterson, G. J. Florence, K. Gerlach, J. P. Scott and N. Sereinig, J. Am. Chem. Soc., 2001, 123, 9535 CrossRef CAS; (e) M. J. Zacuto, S. J. O'Malley and J. L. Leighton, J. Am. Chem. Soc., 2002, 124, 7890 CrossRef CAS; (f) S. A. Burova and F. E. McDonald, J. Am. Chem. Soc., 2002, 124, 8188 CrossRef CAS.
- (a) E. C. Taylor and J. L. LaMattina, Tetrahedron Lett., 1977, 2077 CrossRef CAS; (b) I. Paterson and L. G. Price, Tetrahedron Lett., 1981, 2829 CrossRef CAS; (c) K. Hatanaka, S. Tanimoto, T. Sugimoto and M. Okano, Tetrahedron Lett., 1981, 3243 CrossRef CAS.
- (a) B. C. Ranu, S. Bhar and R. Chakraborti, J. Org. Chem., 1992, 57, 7349 CrossRef CAS; (b) H. Kuroda, I. Tomita and T. Endo, Synth. Commun., 1996, 26, 1539 CAS. For example of a diol addition to a propargylic ketone see (c) J. Aiguade, J. L. Hao and C. J. Forsyth, Org. Lett., 2001, 3, 979 CrossRef CAS.
- All compounds were characterised by 1H and 13C NMR, HRMS, I.R. and optical rotation where applicable.
- Although we could isolate the initial dithiol adduct (39% yield) the reaction proved capricious.
- A. Brook, Acc. Chem. Res., 1974, 7, 77 CrossRef CAS.
- J. Pietruszka, Angew. Chem., Int. Ed., 1998, 37, 2629 CrossRef CAS.
- Dr A. J. Hazelwood, Ph. D. Thesis, 2002, University of Cambridge.
Footnote |
| † General procedure. NaOMe (1.3 equiv.) was added in one portion to a stirred solution of propargylic carbonyl compounds and propane-1,3-dithiol (1.1 equiv.) in MeOH–CH2Cl2 (4 ∶ 1, 0.05 M) at approximately −10 °C (ice–acetone bath). The reaction mixture was stirred for 14 hours, allowing the temperature to rise to 0 °C. On completion the reaction was quenched by the addition of NH4Cl solution and extracted with Et2O. The organic fractions were washed (water and brine), dried (MgSO4), concentrated under reduced pressure and purified by flash column chromatography. |
| This journal is © The Royal Society of Chemistry 2003 |