HIV-1 protease: mechanism and drug discovery
Ashraf
Brik
and
Chi-Huey
Wong
*
Department of Chemistry and the Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA
First published on 26th November 2002
1 Introduction
It has now been two decades since acquired immunodeficiency syndrome (AIDS) was first reported by the US Center for Diseases Control (CDC). A few years later, it was found that a retrovirus called human immunodeficiency virus (HIV) is the causative agent in AIDS.1 In a short time, AIDS increased to epidemic proportions throughout the world, affecting more than 40 million people today and killing so far more than 22 million (UNAIDS, 2001).Since the outbreak of the AIDS epidemic, tremendous efforts have been directed towards development of antiretroviral therapies that target HIV type 1 in particular (HIV-1). The identification of the HIV retrovirus and the accumulated knowledge about the role of the different elements in its life cycle led researchers around the world to develop inhibitors that target different steps in the life cycle of the virus. One of these targets is HIV-1 protease (HIV PR), an essential enzyme needed in the proper assembly and maturation of infectious virions. Understanding the chemical mechanism of this enzyme has been a basic requirement in the development of efficient inhibitors. In this review, we summarize studies conducted in the last two decades on the mechanism of HIV PR and the impact of their conclusions on the drug discovery processes.
2 The life cycle of HIV
HIV belongs to the class of viruses called retroviruses, which carry genetic information in the form of RNA. HIV infects T cells that carry the CD4 antigen on their surface. The infection of the virus requires fusion of the viral and cellular membranes, a process that is mediated by the viral envelope glycoprotein (gp120, gp41) and receptors (CD4 and coreceptors, such as CCR5 or CXCR4) on the target cell. As the virus enters a cell, its RNA is reverse-transcribed to DNA by a virally encoded enzyme, the reverse transcriptase (RT). The viral DNA enters the cell nucleus, where it is integrated into the genetic material of the cell by a second virally encoded enzyme, the integrase. Activation of the host cell results in the transcription of the viral DNA into messenger RNA, which is then translated into viral proteins. HIV protease, the third virally encoded enzyme, is required in this step to cleave a viral polyprotein precursor into individual mature proteins. The viral RNA and viral proteins assemble at the cell surface into new virions, which then bud from the cell and are released to infect another cell. The extensive cell damage from the destruction of the host's genetic system to the budding and release of virions leads to the death of the infected cells.3 HIV protease
3.1 HIV protease: a logical target for AIDS therapy
Unless the HIV life cycle is interrupted by specific treatment, the virus infection spreads rapidly throughout the body, which results in the weakness and destruction of the body's immune system. From the analysis of the HIV life cycle, one could conclude that there are several steps that might be interfered with, thus stopping the replication of the virus. For example, there are several commercially available drugs that inhibit the enzyme reverse transcriptase (RT). The first class of RT inhibitors is the nucleoside analogs such as AZT, ddI, ddC and d4T. These dideoxy compounds lack the 3′-hydroxy, causing DNA chain termination when they are incorporated into the growing DNA strand. The second class of inhibitors is the non-nucleoside inhibitors (NNIs); these inhibitors are known to bind in a pocket away from the polymerase active site, and are believed to cause a conformational change of the enzyme active site, and thus inhibit its action. Currently, there are three available non-nucleoside reverse transcriptase inhibitors (nevirapine, delavirdine, and efavirenz) for the treatment of AIDS.Another critical step in the life cycle of HIV is the proteolytic cleavage of the polypeptide precursors into mature enzymes and structural proteins catalyzed by HIV PR. It has been shown that budded immature viral particles that contain catalytically inactive protease cannot undergo maturation to an infective form.2 The necessity of this enzyme in the virus life cycle makes it a promising target for therapy of the HIV infection.3
3.2 Structure of HIV protease
Navia et al. from Merck laboratories were the first group to obtain a crystal structure of HIV PR;4 a more accurate structure was reported subsequently by Kent and coworker.5 HIV PR is a 99 amino acid aspartyl protease which functions as a homodimer with only one active site which is C2-symmetric in the free form. More than 140 structures of the HIV-1 PR, its mutants and enzymes complexed with various inhibitors have been reported so far. A database dedicated to providing structural information about HIV PR has been created at the National Cancer Institute (http://www-fbsc.ncifcrf.gov/HIVdb/). The enzyme homodimer complexed with TL-36 is shown in Fig. 1 (PDB ID: 3TLH). Each monomer contains an extended β-sheet region (a glycine-rich loop) known as the flap, that constitutes in part the substrate-binding site and plays an important role in substrate binding, and one of the two essential aspartyl residues, Asp-25 and Asp-25′ which lie on the bottom of the cavity. The substrate binds in its extended conformation, in which its interactions with the different amino acid side chains determine the specificity of the enzyme.7 Using standard nomenclature (Fig. 2), the S1 and S′1 (S2 and S′2, etc.) subsites are structurally equivalent. The two S1 subsites are very hydrophobic, the S2 subsites are mostly hydrophobic except Asp-29, Asp-29′, Asp-30 and Asp-30′. The S3 subsites are adjacent to S1 subsites and are also mostly hydrophobic.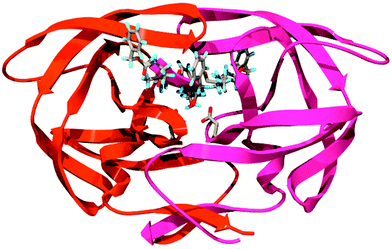 | ||
| Fig. 1 Structure of HIV PR complexed with TL-3 (PDB: 3TLH). | ||
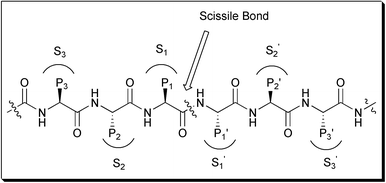 | ||
| Fig. 2 Standard nomenclature P1⋯Pn, P1′⋯Pn′ is used to designate amino acid residues of peptide substrates. The corresponding binding sites on the protease are referred to as S1⋯Sn, S1′⋯Sn′ subsites. | ||
3.3 Mechanism of the HIV protease
Proteases are known to play essential roles in many biological processes. They catalyze the hydrolysis of peptide bonds with high sequence selectivity and catalytic proficiency. These enzymes accomplish their catalysis by two different mechanisms, thus dividing them mechanistically into two broad classes of protease enzymes. The first class of enzymes uses an activated water molecule to attack the amide bond carbonyl of the substrate's scissile bond. The activation of the water molecule can be achieved either by a zinc cation (the zinc metalloproteinases) or by the two aspartyl β-carboxy groups at the active site (the aspartate proteases). In the second class of proteases, a nucleophilic atom of an amino acid side chain is used to initiate amide hydrolysis. In the first step the nucleophilic atom, which can be a hydroxy group or a thiol, is activated by another amino acid side chain. The activated nucleophile attacks the carbonyl of the scissile amide bond to form an ester or a thioester acyl intermediate. Eventually, this acyl enzyme intermediate is hydrolyzed by a water molecule to the corresponding hydrolysis products.According to several studies, HIV PR in general has been shown to belong to the class of the aspartic proteases. Examining the sequence homology of HIV PR to other cellular aspartic proteases shows that this enzyme has the sequence Asp-Thr-Gly, which is conserved among the aspartic protease enzymes.8 These results suggested that HIV PR-1 enzyme may have similar structural features to the aspartic protease enzymes as well as a similar mechanism. Indeed, mutational analysis by several groups of the highly conserved Asp 25 has shown that substituting this residue with Asn,2,9 Thr,10 or Ala,11 leads to a protein without any proteolytic activity. More support for HIV PR being a member of the aspartic protease family came from the in vitro inhibition of this enzyme by pepstatin, a natural product that selectively inhibits members of this family.9,10,12 Finally, the three-dimensional structure of this enzyme also supported the classification of HIV PR in the aspartic protease family.4,5,13 The dimeric structure, in which each monomer contributes one Asp-Thr-Gly triad to the pseudo-symmetric active site, shows an active site that is indistinguishable from those of the monomeric aspartic proteases.
The catalytic mechanism of the nonviral aspartic proteases has been extensively studied by kinetic methods, affinity labeling and X-ray crystallography. Several mechanisms of action for this family have been proposed, including a mechanism that involves the formation of a covalent intermediate.14 However, the results from most of these studies are consistent with a general acid–base mechanism, in which the two active site aspartate residues play an essential general acid–base role to activate the water molecule that acts as a nucleophile and attacks the carbonyl carbon of the scissile bond. It is generally believed that this water molecule is located between the active site aspartates, although some have suggested a different nucleophilic water molecule.15
The most widely accepted mechanism for aspartic protease has been described by Suguna et al. (Fig. 3).16 The proposed mechanism is based on the crystal structure of the Rhizopys chinensis aspartic protease complexed with a reduced peptide inhibitor. The pH–rate profile of this enzyme implies that only one of the two active site aspartic acids is unprotonated in the active pH range.17 In the proposed mechanism the Asp group that is closer to the nucleophilic water molecule was assigned the negative charge (Fig. 3). The nucleophilic water molecule held between the catalytic aspartates, after its activation by the negative aspartate side chain, attacks the carbonyl group in the substrate scissile bond to generate an oxyanion tetrahedral intermediate. Protonation of the scissile amide N atom and rearrangement result in the breakdown of the tetrahedral intermediate to the hydrolysis products. This general acid–base mechanism of the aspartic protease family precludes the use of a Lewis acid such as Zn2+ (as in the case of zinc metalloproteinases) and the formation of covalent acyl intermediates.14,18
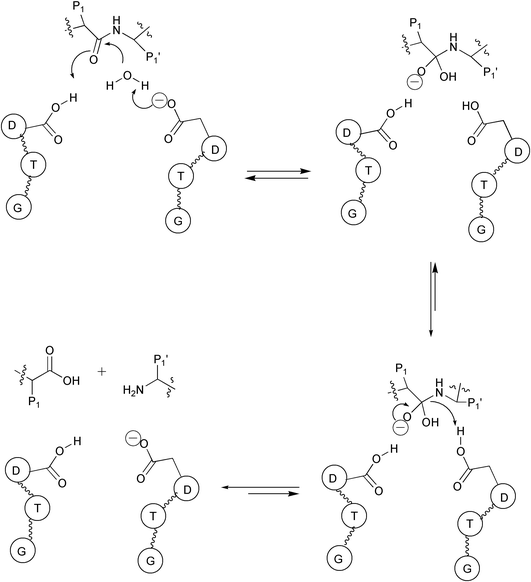 | ||
| Fig. 3 Proposed catalytic mechanism for aspartic proteases. | ||
Although the HIV PR mechanism shares many features with the rest of the aspartic protease family, the full detailed mechanism of this enzyme remains not fully understood. Jaskólski et al. have proposed a new model of the enzymatic mechanism for the HIV PR enzyme based on the crystal structure of a complex between a chemically synthesized HIV PR and an octapeptide inhibitor U-85548e (H-Val-Ser-Gln-Asn-Leu-ψ-[CH(OH)CH2]-Val-Ile-Val-OH),19 as well as other crystal structures of HIV PR complexed with different inhibitors.20 In this mechanism (Fig. 4), the hydrolysis reaction is viewed as a one-step process during which the nucleophilic water molecule and the acidic proton attack the scissile peptide bond in a concerted manner. The issue of the simultaneous attack from the nucleophile and electrophile is the major difference between this mechanism and other previously proposed mechanisms.
 | ||
| Fig. 4 Proposed concerted catalytic mechanism for HIV. | ||
As in the case of all the aspartic protease family, the possibility of covalent catalysis (e.g. Asp-25 attacks directly the carbonyl amide bond) in the chemical mechanism of HIV PR is discounted. Indeed, Hyland et al. have provided evidence against the formation of a covalent intermediate by studying the 18O incorporation into the transpeptidation product of the reaction of AcSQNYFLDG-NH2 from AcSQNYPVV-NH2 and FLDG-NH2 carried out in H218O enriched water and catalyzed by HIV PR.21 Since the incorporation of 18O into the substrate AcSQNYPVV-NH2 (Fig. 5) could be observed under the condition where the re-formation of substrate after hydrolysis was negligible, the involvement of an acyl enzyme intermediate could be excluded. From these studies, it was also found that the HIV PR-catalyzed exchange of 18O from H218O into the re-formed substrates occurs at a rate 0.01–0.012 times that of the hydrolysis rate. These results are in agreement with the formation of a kinetically competent enzyme-bound amide hydrate intermediate, the collapse of which is the rate-limiting chemical step in the reaction.
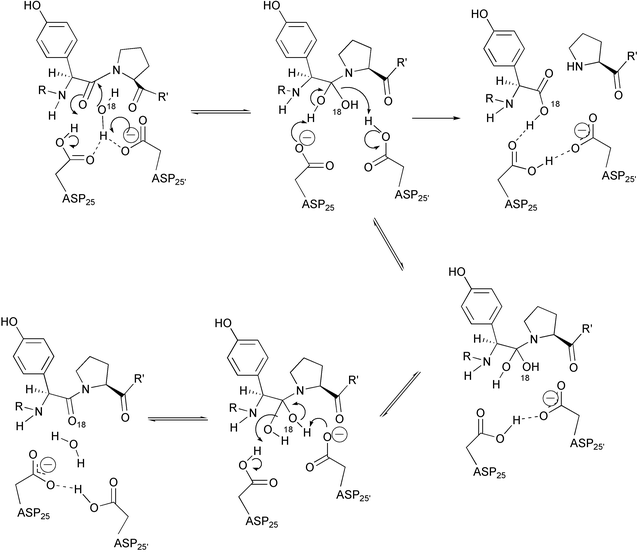 | ||
| Fig. 5 Proposed mechanism for HIV PR catalyzed incorporation of 18O from H218O into the peptide substrate. | ||
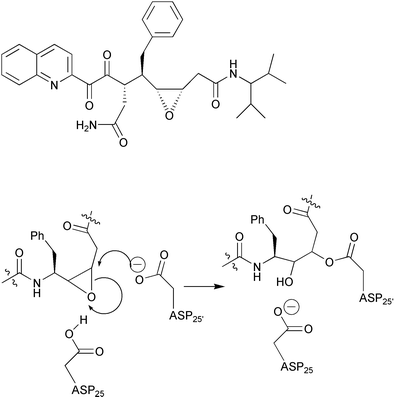
We have described a new pyrrolidine α-keto amide core structure as a mechanism-based inhibitor for HIV PR.22 The crystal structure shows that this inhibitor is bound to HIV PR in its hydrated form (Fig. 6). A 13C NMR study in DMSO-d6–D2O (5 ∶ 1) has determined that, in the presence of water, the ketone group of this inhibitor remains unhydrated even after 24 h. These results suggest that the hydration takes place within the enzyme-binding site, which means that the catalytic water is incorporated into the inhibitor. These results support the role of the nucleophilic character of the catalytic water molecule in the reaction pathway and may imply an electrophilic assistance for hydration of carbonyl with low electrophilic character.
 | ||
| Fig. 6 Proposed mechanism for the hydration of the keto amide inhibitor by HIV PR. | ||
There are four possible protonation states of the two catalytic possible states of the two catalytic aspartates: the dianionic form (−1, −1), the two monoanionic forms (−1, 0), (0, −1) in which one of the catalytic aspartate groups is anionic and the other protonated, and the diprotonated or neutral form (0, 0). It has been suggested that the protonation state of the two aspartic groups depends on the local environment near the aspartate and is different for different inhibitors.19,23 However, it has been suggested also that the difference in pKas of these residues is more a function of their proximity to each other than of their differing environments.13,24
Aspartic proteases are unique in that they function physiologically over a wide pH range (2–7.4).17 The pH–rate profile of a model substrate for HIV PR studied by Meek and coworker shows that the two aspartate groups have different pKa values of 3.1 and 5.2.25 In contrast to these results, Smith et al. have found in their NMR experiments on the 13C-enriched enzyme in the absence of inhibitor, that the two Asp side chains are equivalent and are both deprotonated at pH 6.26 However, in the presence of pepstatin inhibitor only one Asp side chain is deprotonated in the pH range 2.5–6.5. Wang et al. have also examined the protonation state of the two aspartic groups, however using the asymmetric inhibitor KNI-272.27 They also found one catalytic Asp to be protonated and the other unprotonated. In contrast to these results, Yamazaki et al. have studied the ionization state of the two aspartic groups in HIV PR complexed with the symmetric inhibitor DM323.28 This non-peptidic inhibitor contains two hydroxy groups and forms a completely symmetric complex with the enzyme, in which the side chains of the two aspartates were found to be magnetically equivalent and both protonated over the pH range 2–7. Using ab initio molecular dynamics studies of the pepstatin A–HIV PR complex, Piana et al. have proposed recently that both aspartic groups are protonated,29 in contrast to what had been reported by Smith et al.26 An answer to the question of where the acidic proton is located in the free enzyme could not be easily provided since proton locations are generally not resolvable by X-ray crystallography methods.16,19
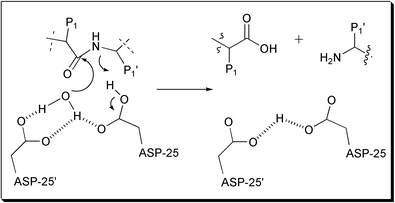
Meek et al. have shown that the known aspartic protease inactivator 1,2-epoxy-3-(4-nitrophenoxy)propane produced irreversible, time-dependent inactivation of HIV PR, through covalent modification of the enzyme's aspartyl residue.30 The pH-dependent kinetics of this inhibitor–enzyme interaction was consistent with having one protonated aspartic group in the active site of the enzyme. Following this observation, Lee and coworkers31 have designed several inhibitors based on a peptide isostere containing cis-epoxide for the irreversible inactivation of HIV (Fig. 7). These results are surprising, since one would expect the catalytic water molecule to be involved in the epoxide ring opening instead of direct attack by the aspartyl group. Perhaps the alignment of the inhibitor in the active site does not allow enough space between the epoxide ring and the two aspartyl residues to accommodate a water molecule.
 | ||
| Fig. 7 Representative structure of HIV protease inhibitor based on a peptide isostere containing cis-epoxide and mechanism of alkylation at the active site aspartic residue. | ||
Hyland et al. have proposed a detailed chemical mechanism for HIV PR32 based on kinetic data obtained from solvent kinetic isotope effects, pH–rate and 18O incorporation studies,21,25 combined with known previous structural data of HIV PR (Fig. 8). In this mechanism, Asp-25′ exists in the unprotonated state (pKa 3.1) upon binding to substrate, while the proton on Asp-25 (pKa 5.2) is positioned to hydrogen bond to the carbonyl oxygen of the substrate, and at the same time, the lytic water is positioned closer to the β-carboxylic acid of Asp-25′. This mechanism has many similar features in common with the general acid–base mechanism of aspartic proteases reported by other groups.15,16,33 It also resembles in part the concerted mechanism proposed by Jaskólski,19 in that the amine product is protonated by an active site aspartyl residue. However in Hyland's proposal, a discrete enzyme intermediate is formed while, in the concerted mechanism, there is no such intermediate.
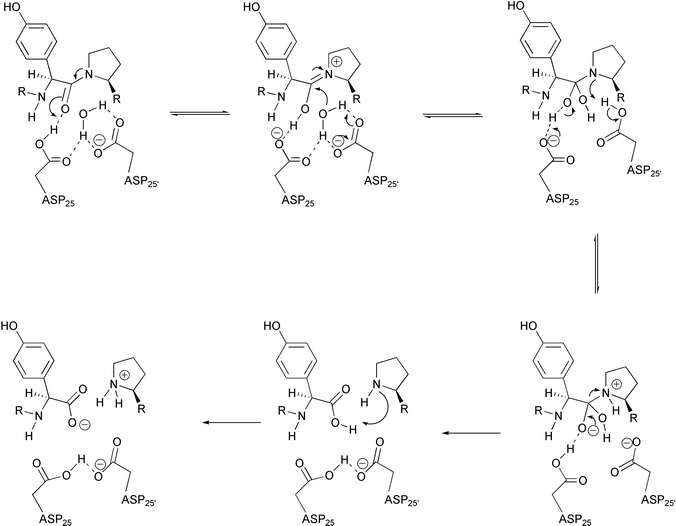 | ||
| Fig. 8 Proposed mechanism for HIV PR catalysis based on kinetic and structural data. | ||
Pearl has proposed a mechanism of aspartic proteases similar to what Sugan et al. have suggested,34 however pointing out the effect of the distortion of the scissile bond on the catalysis. In this mechanism the distortion of the scissile bond reduces the double bond character in the C–N bond and polarizes the carbonyl, rendering it more electrophilic towards the incoming nucleophilic water. The distortion of the amide bond is stabilized by interactions of the substrate with the extended binding cleft, mostly by hydrogen bond interactions between the interior side of the flap and the substrate carbonyls on either side of the scissile bond.
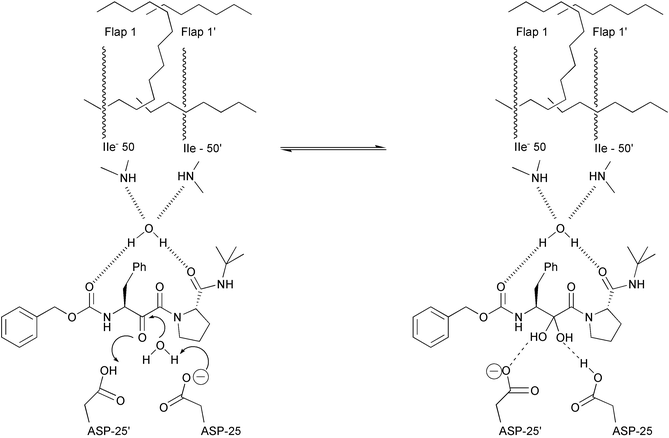
In HIV PR there are two flaps in the active dimer compared to the single flap in non-viral aspartic protease.35 Moreover, while in the pepsin-like protease there is a direct H-bonding between the flap and the substrate, in HIV PR the carbonyl group between P1 and P2 and the carbonyl between P1′ and P2′ both make hydrogen bonds to the same water molecule (Fig. 9). This water molecule makes another two hydrogen bonds to the flaps to create approximately tetrahedral geometry. The presence of this water molecule has been observed in most of the crystal structures of HIV PR bound to different inhibitors.
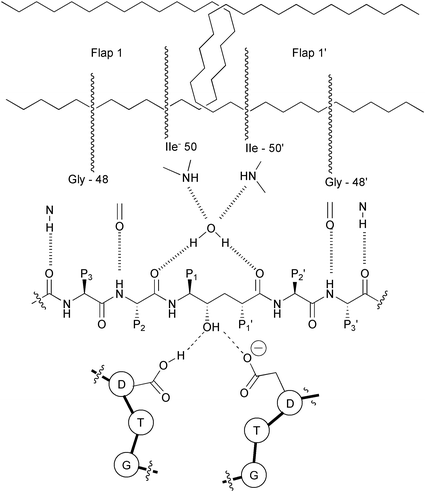 | ||
| Fig. 9 Schematic representation of two flaps of HIV PR and their hydrogen bonds with the water molecule. The same water molecule makes another two hydrogen bonds with the peptidyl inhibitor. Also showing is the interaction of the central hydroxy group of the inhibitor with the catalytic aspartates of HIV PR. | ||
It has been proposed that this water molecule is relevant for catalysis35 and a key feature in the differences of the mechanisms of the retroviral HIV PR and the corresponding cell-encoded enzymes.20 The hydrogen bonds of this water molecule apply strain on the scissile amide bond, causing it to rotate out of the plane and lose its double bond character, which enhances its vulnerability towards hydrolysis.20 Using total chemical synthesis of proteins, HIV PR was synthesized in which the Gly49–Ile50 N(H)-atom (Fig. 9) was specifically replaced by an O-atom, thus deleting one of the hydrogen bonds from one of the flaps to the water molecule. The resulting enzyme with the single flap–substrate hydrogen bonds was fully active.36 Based on these results, it has been suggested that this enzyme may make use of only one flap in the catalysis, showing a similarity to pepsin-like enzymes.
4 Mechanism based strategy for drug design
The knowledge accumulated through the last two decades about the structure and the mechanism of HIV PR has paved the way towards the development of effective drugs for this enzyme. In addition, there is a huge amount of information regarding the design of inhibitors for other aspartic proteases, such as renin, which is involved in the control of hypertension.37 Although there are no clinically useful renin inhibitors today, the lessons learned from the drug discovery process towards finding effective drugs for this enzyme have assisted researchers in finding effective drugs for HIV PR. In addition to de novo design, random screening and combination of these approaches have accelerated the drug discovery process. There are today six FDA approved drugs that function as inhibitors for HIV PR (Fig. 10).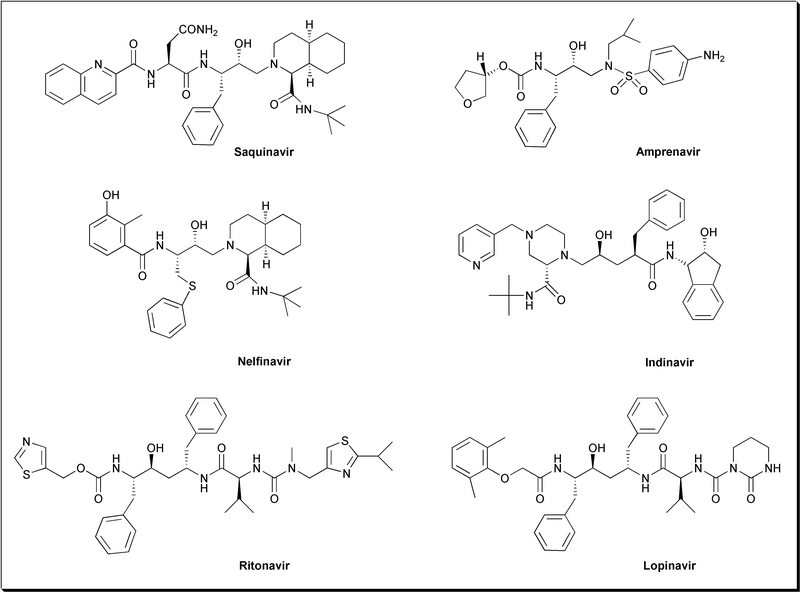 | ||
| Fig. 10 FDA approved HIV-1 protease inhibitors. | ||
In the commercially available drugs and some of the inhibitors that are currently being tested clinically, a nonhydrolyzable hydroxyethylene or hydroxyethylamine moiety is used as the basic core for the development of these inhibitors. Other noncleavable transition state isosteres have also been used, including statine, norstatine, phosphinate, reduced amide, dihydroxyethylene,38 α-keto amide39 and more recently silicon-based inhibitors40 (Fig. 11). Basically, the core is a good isostere replacement at the scissile bond that is believed to mimic the tetrahedral transition state of the proteolytic reaction. More than 2000 inhibitors have been developed and most of the effective inhibitors contain a core structure to mimic the transition state of the protease catalysis. In the crystal structures of HIV-1 PR complexed with these inhibitors, the central hydroxy group of the core is located between Asp-25 and Asp-25′ and makes favorable electrostatic contacts. It has been shown that the interaction between the two catalytic aspartates and the hydroxy group is worth more than 4 kcal mol−1.41 A review that discusses in detail the various interactions of the different inhibitors with the binding site has been published.42
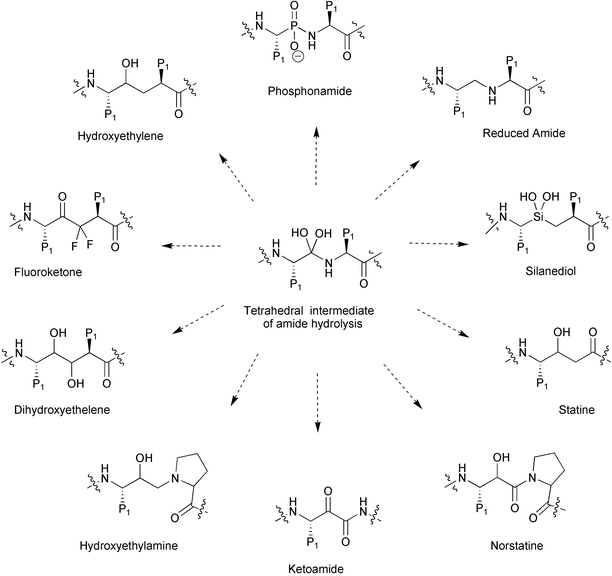 | ||
| Fig. 11 Noncleavable transition-state isostere developed for the synthesis of HIV PR inhibitors. | ||
Saquinavir (Ro 31-8959),43 which was discovered by Hoffmann-La Roche, is the first FDA approved drug (1995) for the treatment of AIDS. This potent inhibitor (Ki = 0.12 nM) has the hydroxyethylamine isostere replacement for the Phe-Pro cleavage site. The structure of this inhibitor was quite interesting since it had the R stereochemistry at the carbon bearing the central hydroxy group, which was unique among aspartic protease inhibitors that were known at that time. Both Amprenavir (Ki = 0.6 nM), which was discovered by Vertex Pharmaceuticals44 and approved by the FDA in 1999, and Nelfinavir (Ki = 2 nM), which was discovered by a collaborative effort between Lilly and Agouron45 and approved by the FDA in 1997, have similar hydroxyethylamine isostere replacement to that found in Saquinavir.
Merck group discovered a novel variation of the hydroxyethylene transition state analog in their new drug, Indinavir (Ki = 0.56 nM),46 which was approved by the FDA in 1996. Abbott Laboratories have used the (S,S,S)-aminoalcohol to prepare two of the commercially available drugs: Ritonavir (Ki = 0.01 nM),47 which was approved by the FDA in 1996, and Lopinavir (Ki = 0.003 nM),48 which is the latest approved HIV PR inhibitor (September 2000). Lopinavir is contained in a protease inhibitor formulation (Klaretra®) which includes Ritonavir. Ritonavir is known to inhibit cytochrome P-450 3A, the enzyme responsible for the metabolism of Lopinavir, therefore this combination allows for increased plasma levels of Lopinavir.
The Dupont Merck group has designed a new generation of HIV PR inhibitors based on cyclic urea cores (Fig. 12).49 The uniqueness of this class of inhibitors is the cyclic urea carbonyl oxygen that mimics the hydrogen bonding features of the key structural water molecule discussed earlier in this review (Fig. 9). The group reasoned that incorporating the cyclic urea core into an inhibitor would replace the flap water molecule, which could lead to better binding energy due to the positive entropic effect that should be provided by the cyclic urea core. These inhibitors also contained the diol functionality as a transition state mimic to interact with the catalytic aspartates. Several inhibitors based on these concepts were developed and found to be potent against HIV PR. Although some of these inhibitors were clinically tested, none of them have as yet reached the market.
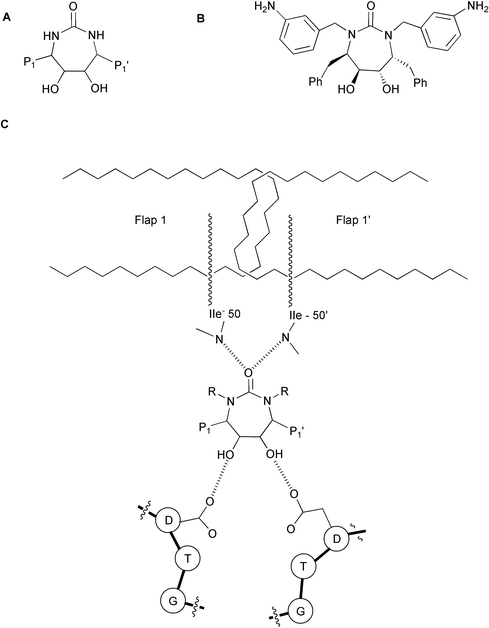 | ||
| Fig. 12 (A) General structure of cyclic urea inhibitors. (B) Structure of the inhibitor DMP4550. (C) Schematic interaction of a cyclic urea based inhibitor with HIV PR. | ||
5 The problem of drug resistance
Although there are six anti-HIV PR inhibitors, and many other anti-reverse transcriptase drugs that are commercially available, their effectiveness has been hampered by the emergence of drug-resistant and cross-resistant mutants, rendering AIDS with no definitive cure. The high rate of replication of the virus (108–109 virions/day) and the high error rate of HIV reverse transcriptase (about 1 in 10 000 bases) stand behind the rapid mutation and the selection of drug-resistant viruses.50 To date, according to the Stanford HIV reverse transcriptase and protease sequence database, 1929 isolates from patients exhibiting drug-induced protease mutations have been reported. Understanding the mechanism and structural basis of resistance in the light of the three-dimensional atomic structure of this enzyme has been reviewed.51Currently, there is no general strategy to tackle the problem of drug resistance. Modeling studies in our lab have shown that mutations that lead to drug resistance affect the hydrophobic binding site interacting with P3/P3′ and P1/P1′ groups of the inhibitors by reducing the S3 binding region.22 Thus, inhibitors with a large P3 group can no longer bind to the mutated enzyme, and drug resistance is developed.6,52 We have also found that at least six mutated residues in HIV-1 PR causing drug resistance (K20I, V32I, I50V, N88D, L90M, Q92K) are found in the structurally aligned native residues of FIV PR (Feline immunodeficiency virus protease), which is also a C2-symmetric homodimeric aspartic protease (Fig. 13) and has an identical mechanism of catalysis.53 Comparison of the structures of FIV and HIV-1 proteases and drug-resistant mutants of HIV PR further reveals a similarity between FIV PR and drug-resistant HIV PR. The FIV PR active site is more extended and contains a smaller S3 subsite, a characteristic of many drug-resistant HIV proteases.54
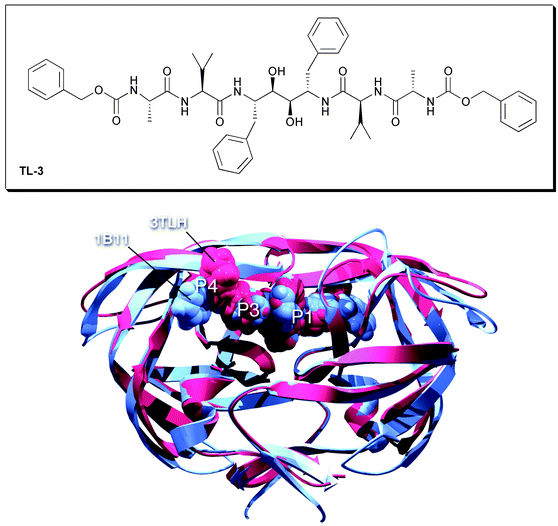 | ||
| Fig. 13 An overlay of structurally similar FIV PR (1B11) and HIV PR (3 TLH) complexed with TL-3. | ||
The above observations have suggested that FIV PR may serve as a model for drug resistance. Indeed, in a study to probe the specificity of P3–S3 interaction, it was found that inhibitors without a P3 group or a small P3 group not only exhibit a strong inhibition against the wild-type HIV protease, but also are effective against FIV protease and several drug-resistant mutants of HIV protease. One such inhibitor is TL-3 (Fig. 13), which was active in cell culture with IC50 of 0.1, 0.4 and 0.9 µM for the wild-type, the I84V and F84V mutants, respectively, and no resistance was observed over a 6-month period.
6 Conclusion and perspectives
A few questions related to the detailed mechanism of HIV PR remain to be answered: the protonation state of the two aspartic groups, the location of the acidic proton, the role of the two flaps in the catalytic mechanism. However, it is clear from all the previous studies that many researchers agree about the similarity of the mechanism to that of the other aspartic proteases: the involvement of the water molecule as nucleophile, the general role of the two active site aspartyl residues. Although the HIV PR mechanism has not been completely characterized yet, these studies have driven several academic and industrial groups to develop a huge number of inhibitors based on the transition state concept, six of which are commercially available for the treatment of AIDS.Many successes have been achieved in the war against AIDS, yet AIDS still has no definitive cure. All commercially available drugs and their combination in what is called the highly active antiretroviral therapy (HAART) improve the quality of life of the infected person. The current therapy is very expensive (95% of people who are infected with HIV/AIDS live in developing countries) and has other limitations such as the necessity of high doses, side effects and, mainly, the drug resistance problem.
Current research directed towards development of new therapies to cure AIDS is going in several directions. For example, development of new protease inhibitors that are not cross resistant to current drugs,55 inhibition of HIV-1 entry,56 and targeting conserved HIV RNA sequences with small molecules.57 Perhaps the ultimate solution is to develop a vaccine against this devastating epidemic.
Acknowledgements
This study was supported by the NIH. Many thanks to Dr Garrett M. Morris for providing the figures of HIV PR, FIV PR structures. A.B. thanks the Israel Science Foundation for financial support.References
- R. C. Gallo and L. Montagnier, Sci. Am., 1988, 259, 40 Search PubMed.
- N. E. Kohl, E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Davis, J. C. Heimbach, R. A. F. Dixon, E. M. Scolnick and I. S. Sigal, Proc. Natl. Acad. Sci. USA, 1988, 85, 4686 CAS.
- (a) R. A. Kramer, M. D. Schaber, A. M. Skalka, K. Ganguly, F. Wong-Staal and E. P. Reddy, Science, 1986, 231, 1580 CAS; (b) J. R. Huff, J. Med. Chem., 1991, 34, 2305 CrossRef CAS.
- M. A. Navia, M. D. P. Fitzgerald, B. M. Mckeever, C.-T. Leu, J. C. Heimbach, W. K. Herber, I. S. Sigal, P. L. Darke and J. P. Spronger, Nature, 1989, 615, 337.
- A. Wlodawer, M. Miller, M. Jaskolski, B. K. Sathyanarayana, E. Baldwin, I. T. Weber, L. M. Selk, L. Clawson, J. Schneider and S. B. H. Kent, Science, 1989, 245, 616 CAS.
- T. Lee, G. S. Laco, B. E. Torbett, H. S. Fox, D. L. Lerner, J. H. Elder and C.-H. Wong, Proc. Natl. Acad. Sci. USA, 1998, 95, 939 CrossRef CAS.
- S. C. Pettit, S. F. Michael and R. Swanstrom, Perspect. Drug Discovery Des., 1993, 1, 69 Search PubMed.
- (a) J. Cairns, J. Overbaugh and S. Miller, Nature, 1988, 142, 335; (b) F. W. Stahl, Nature, 1988, 142, 112 CrossRef.
- P. L. Darke, C. T. Leu, L. J. Davis, J. C. Heimbach, R. E. Diehl, W. S. Hill, R. A. Dixon and I. S. Sigal, J. Biol. Chem., 1989, 264, 2307 CAS.
- S. Seelmeir, H. Schmidt, V. Turk and K. Von Der Helm, Proc. Natl. Acad. Sci. USA, 1988, 85, 6612.
- J. Mous, E. P. Heimer and S. J. J. Le Grice, J. Virol., 1988, 62, 1433 CAS.
- J. Hansen, S. Billich, T. Schulze, S. Sukrow and K. Moelling, EMBO J., 1998, 7, 1785.
- R. Lapatto, T. Blundell, A. Hemmings, J. Overington, A. Wilderspin, S. Wood, J. R. Merson, P. J. Whittle, D. E. Danley, K. F. Geoghegan, S. J. Hawrylik, S. E. Lee, K. G. Scheld and P. M. Hobart, Nature, 1989, 342, 299 CrossRef CAS.
- (a) T. Hofmann, B. M. Dunn and A. L. Fink, Biochemistry, 1984, 23, 6956; (b) J. S. Fruton, Adv. Enzymol., 1976, 44, 1 Search PubMed.
- M. N. G. James and A. R. Sielecki, Biochemistry, 1985, 24, 1 CrossRef.
- K. Suguna, E. A. Padlan, C. W. Smith, W. D. Carlson and D. R. Davies, Proc. Natl. Acad. Sci. USA, 1987, 84, 6612.
- T. Hofmann, R. S. Hodges and M. N. G. James, Biochemistry, 1984, 23, 635 CrossRef CAS.
- (a) V. K. Atonov, L. M. Ginodman, L. D. Rumsh, Y. V. Kapitannikov, T. N. Barsheskaya, L. P. Yavashev, A. G. Gurova and L. I. Volkova, Eur. J. Biochem., 1981, 117, 195; (b) P. G. Schmidt, M. W. Holladay, F. G. Salituro and D. H. Rich, Biochem. Biophys. Res. Commun., 1985, 129, 597 CAS.
- M. Jaskólski, A. G. Tomasselli, T. K. Sawyer, D. G. Staples, R. L. Heinrikson, J. Schneider, S. B. H. Kent and A. Wlodawer, Biochemistry, 1991, 30, 1600 CrossRef CAS.
- (a) M. Miller, J. Schneider, B. K. Sathyanarayana, M. V. Toth, G. R. Marshal, L. Clawsen, L. Selk, S. B. H. Kent and A. Wlodawer, Science, 1989, 246, 1149 CAS; (b) A. L. Swain, M. Miller, J. Green, D. H. Rich, J. Schneider, S. B. H. Kent and A. Wlodawer, Proc. Natl. Acad. Sci. USA, 1990, 87, 8805 CAS.
- L. J. Hyland, T. A. Tomaszek, Jr., G. D. Roberts, S. A. Carr, V. W. Magaard, H. L. Bryan, S. A. Fakhoury, M. L. Moore, M. D. Minnich, J. S. Culp, R. L. Desjarlais and T. D. Meek, Biochemistry, 1991, 30, 8441 CrossRef CAS.
- D. H. Slee, K. L. Laslo, J. H. Elder, I. R. Ollmann, A. Gustchina, K. Kervinen, A. Zdanov, A. Wlodawer and C.-H. Wong, J. Am. Chem. Soc., 1995, 117, 11867 CrossRef CAS.
- W. E. Harte and D. L. Beveridge, Jr., J. Am. Chem. Soc., 1993, 115, 3883 CrossRef.
- L. H. Pearl and T. L. Blundell, FEBS Lett., 1984, 174, 96 CrossRef CAS.
- L. J. Hyland, T. A. Tomaszek and T. D. Meek, Biochemistry, 1991, 30, 8454 CrossRef CAS.
- R. Smith, I. M. Brereton, R. Y. Chai and S. B. H. Kent, Nat. Struct. Biol., 1996, 3, 946 CrossRef CAS.
- Y. X. Wang, D. I. Freedberg, T. Yamazaki, P. T. Wingfield, S. J. Stahl, J. D. Kaufman, Y. Kiso and D. A. Torchia, Biochemistry, 1996, 35, 9945 CrossRef CAS.
- T. Yamazaki, L. K. Nicholson, D. A. Torchia, P. T. Wingfield, S. J. Stahl, J. D. Kaufman, C. J. Eyermann, C. N. Hodge, P. Y. S. Lam, Y. Ru, P. K. Jadhav, C. H. Chang and P. C. Weber, J. Am. Chem. Soc., 1994, 116, 10791 CrossRef CAS.
- S. Piana, D. Sebastiani, P. Carloni and M. Parrinello, J. Am. Chem. Soc., 2001, 123, 8730 CrossRef CAS.
- T. D. Meek, B. D. Dayton, B. W. Metcalf, G. B. Dreyer, J. E. Strickler, J. G. Gorniak, M. Rosenberg, M. L. Moore, V. W. Magaard and C. Debouck, Proc. Natl. Acad. Sci. USA, 1989, 86, 1841 CAS.
- C. S. Lee, N. Choy, C. Park, H. Choi, Y. C. Son, S. Kim, J. H. Ok, H. Yoon and S. C. Kim, Bioorg. Med. Chem. Lett., 1996, 6, 589 CrossRef CAS.
- T. D. Meek, E. J. Rodriguez and T. S. Angeles, Methods Enzymol., 1994, 241, 127 Search PubMed.
- R. Bott, E. Subramanian and D. R. Davies, Biochemistry, 1982, 21, 6956 CrossRef CAS.
- L. Pearl, FEBS Lett., 1987, 214, 8 CrossRef CAS.
- A. Gustchina and I. T. Weber, FEBS Lett., 1990, 269, 269 CrossRef CAS.
- M. Miller, M. Baca, J. K. Mohan Rao and S. B. H. Kent, Theochem, 1998, 423, 137 CrossRef CAS.
- (a) W. J. Greenle and P. K. S. Siegl, Annu. Rep. Med. Chem, 1992, 27, 59 Search PubMed; (b) S. S. Abdel-Meguid, Med. Chem. Rev., 1993, 13, 731 Search PubMed.
- A. Wlodawer and J. W. Erickson, Annu. Rev. Biochem., 1993, 62, 543 CrossRef CAS.
- B. Munoz, C.-Z. Giam and C.-H. Wong, Bioorg. Med. Chem., 1994, 2, 1085 CrossRef CAS.
- C.-A. Chen, S. M. Sieburth, A. Glekas, G. W. Hewitt, G. L. Trainor, S. E. Viitanen, S. S. Garber, B. Cordova, S. Jeffry and R. M. Klabe, Chem. Biol., 2001, 8, 1161 CrossRef CAS.
- K. Appelt, Perspect. Drug Discovery Des., 1993, 1, 23 Search PubMed.
- R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359 CrossRef.
- N. A. Roberts, J. A. Martin, D. Kinchington, A. V. Broadhurst, J. C. Craig, I. B. Duncan, S. A. Galpin, B. K. Handa, J. Kay, A. Krohn, R. W. Lambert, J. H. Merrett, J. S. Mills, K. E. B. Parkes, S. Redshaw, A. J. Ritchie, D. L. Taylor, G. J. Thomas and P. J. Machin, Science, 1990, 248, 358 CAS.
- E. E. Kim, C. T. Baker, M. D. Dwyer, M. A. Murcko, B. G. Rao, R. D. Tung and M. A. Navia, J. Am. Chem. Soc., 1995, 117, 1181 CrossRef CAS.
- S. W. Kaldor, M. Hammond, B. A. Dressman, J. E. Fritz, T. A. Crowell and R. A. Hermann, Bioorg. Med. Chem. Lett., 1994, 4, 1385 CrossRef CAS.
- Z. Chen, Y. Li, E. Chen, D. Hall, P. Darke, C. Culberson, J. A. Shafer and L. A. Kuo, J. Biol. Chem., 1994, 269, 26344 CAS.
- D. J. Kempf, K. C. March, J. F. Denissen, E. McDonald, S. Vasavanonda, C. A. Flentge, B. E. Green, L. Fino, C. H. Park, X. P. Kong, N. E. Wideburg, A. Saldivar, L. Ruiz, W. M. Kati, H. L. Sham, T. Robins, K. D. Stweart, A. Hsu, J. J. Plattner, J. M. Leonard and D. W. Norbeck, Proc. Natl. Acad. Sci. USA, 1995, 92, 2484 CAS.
- H. L. Sham, D. J. Kempf, A. Molla, K. C. Marsh, G. N. Kumar, C. M. Chen, W. Kati, K. Stewart, R. Lal, A. Hsu, D. A. Betebenner, M. Korneyeva, S. Vasavanonda, E. McDonald, A. Saldivar, N. Wideburg, X. Chen, P. Niu, O. Park, V. Jayanti, B. Grabowski, G. R. Granneman, E. Sun, A. J. Japour, J. M. Leonard, J. J. Plattner and D. W. Norbeck, Antimicrob. Agents Chemother., 1998, 42, 3218 CAS.
- P. Y. S. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge, Y. Ru, L. T. Bacheler, O. M. J. Meek, M. M. Rayner, N. Wong, C.-H. Chang, P. C. Weber, D. V. Jackson, T. R. Sharpe and S. E. Viitanen, Science, 1994, 263, 380 CAS.
- J. M. Coffin, Science, 1995, 267, 483 CAS.
- (a) J. W. Erickson and S. K. Burt, Annu. Rev. Pharmacol. Toxicol., 1996, 36, 545 CrossRef CAS; (b) R. F. Schinazi, B. A. Lader and J. W. Mellors, Int. Antiviral News, 1997, 5, 129 Search PubMed.
- T. Lee, V.-D. Le, D. Lim, Y.-C. Lin, G. M. Morris, A. L. Wong, A. J. Olson, J. H. Elder and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 1145 CrossRef CAS.
- A. Wlodawer, A. Gustchina, L. Reshetnikova, J. Lubkowski, A. Zdanov, K. Y. Hui, E. L. Angleton, W. G. Farmerie, M. M. Goodenow, D. Bhatt, L. Zhang and B. M. Dunn, Nat. Struct. Biol., 1995, 2, 480 CrossRef CAS.
- M. Li, G. M. Morris, T. Lee, G. S. Laco, C.-H. Wong, A. J. Olson, J. H. Elder, A. Wlodawer and A. Gustachina, Proteins: Struct., Funct., Genet., 2000, 38, 29 Search PubMed.
- K. Yoshimura, R. Kato, M. F. Kavlick, A. Nguyen, V. Maroun, K. Maeda, K. A. Hussain, A. K. Ghosh, S. V. Gulnik, J. W. Erickson and H. Mitsuya, J. Virol., 2002, 76, 1349 CAS.
- D. M. Eckert and P. S. Kim, Annu. Rev. Biochem., 2001, 70, 777 CrossRef CAS.
- (a) W. K. C. Park, M. Auer, H. Jaksche and C.-H. Wong, J. Am. Chem. Soc., 1996, 118, 10150 CrossRef CAS; (b) M. R. Green, Aids Res. Rev., 1993, 3, 4 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |