Promoting laccase activity towards non-phenolic substrates: a mechanistic investigation with some laccase–mediator systems
Paola
Baiocco
,
Anna Maria
Barreca
,
Maura
Fabbrini
,
Carlo
Galli
* and
Patrizia
Gentili
*
Dipartimento di Chimica and Centro CNR Meccanismi di Reazione, Università
‘La Sapienza’, P. le A. Moro 5, 00185 Roma, Italy
First published on 29th November 2002
Abstract
The oxidation of benzyl alcohols with the enzyme laccase, under mediation by appropriate mediator compounds, yields carbonylic products, whereas laccase can not oxidise these non-phenolic substrates directly. The oxidation step is performed by the oxidised form of the mediator (Medox), generated on its interaction with laccase. The Medox can follow either an electron transfer (ET) or a radical hydrogen atom transfer (HAT) route of oxidation of the substrates. Experimental evidence is reported that enables unambiguous assessment of the occurrence of either one the oxidation routes with each of the investigated mediators, namely, ABTS, HBT, HPI and VLA. Support to the conclusions is provided by (i) investigating the intermolecular selectivity of oxidation with appropriate substrates, (ii) attempting Hammett correlations for the oxidation of a series of 4-X-substituted benzyl alcohols, (iii) measuring the kinetic isotope effect, (iv) investigating the product pattern with suitable probe precursors. Based on these points, a HAT mechanism results to be followed by the laccase–HBT, laccase–HPI and laccase–VLA systems, whereas an ET route appears feasible in the case of the laccase–ABTS system.
Introduction
Laccase is a family of ‘blue-copper’ oxidase proteins containing four copper ions, and excreted by white-rot fungi under ligninolytic conditions. Laccase cooperates with other enzymes in the degradation of lignin in wood. With respect to lignin peroxidase (LiP)1 and manganese peroxidase (MnP),2 that are more powerful oxidants, laccase has a lower redox potential (ca. 0.7–0.8 V/NHE)3,4 and therefore is able to catalyse single-electron oxidation steps only with the easy-to-oxidise phenolic constituents of lignin, with the concomitant reduction of O2 to water. However, the activity of laccase can be fostered and expanded towards more difficult to oxidise non-phenolic substrates by the use of appropriate mediators.5–7 The role of mediators in laccase oxidations is outlined in Scheme 1.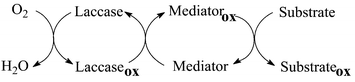 | ||
| Scheme 1 The role of a mediator on laccase activity. | ||
In general, a mediator could be a sort of ‘electron shuttle’ that, after being oxidised by the enzyme, diffuses away from the active site to oxidise any substrate that, for its size, could not enter the enzymatic pocket directly.8,9 In addition, the oxidised form of the mediator (Medox), being structurally ‘diverse’ from the enzyme, might rely on a different mechanism of oxidation, thereby extending the range of substrates susceptible to the enzymatic action.7,10 For example, 3-hydroxyanthranilic acid (HAA) has been reported to promote the laccase activity of the fungus Pycnoporus cinnabarinus towards non-phenolic lignin structures.11 Suitable enzyme–mediator systems could also enable the environmentally benign (i.e., chlorine free) bleaching process of kraft pulps for the paper industry.5b,12 Understanding the role and mechanism of action of these mediators is a practical issue, that however requires a knowledge of fundamental reactivity features; the latter can be addressed by the typical experimental approach of physical organic chemistry.
We have undertaken a systematic investigation of the mediation phenomenon with laccase, and have already reported on a comparative evaluation of the efficiency of a number of mediators in the oxidation of non-phenolic lignin model compounds, such as the benzylic alcohols.7 The mediators ABTS, HBT, HPI, VLA (structures and abbreviated names in Fig. 1) and TEMPO, for various reasons, presented the more promising features as catalysts in this oxidation. A simple and synthetically-attractive oxidation of alcohols to carbonyl compounds by oxygen, induced by the laccase–TEMPO system, has been developed from the previous investigation.13
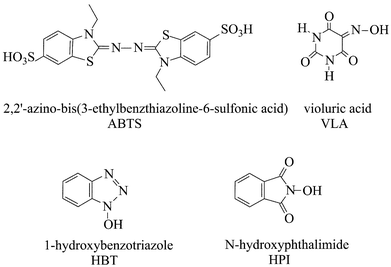 | ||
| Fig. 1 Structure and abbreviated name of the mediators employed. | ||
In the present paper we report on new advances in the interpretation of the reactivity features of laccase–mediator systems, and will particularly focus on the mechanism that the oxidised form of the mediator can employ in the oxidation of non-phenolic substrates. The possibility exists that the Medox species abstracts either one-electron (ET route) or a H-atom (HAT route) from the substrate, depending on the reactivity propensity of the latter.10 In order to clarify this important point, the following experimental evidence has been acquired: (i) reactivity trends or intermolecular selectivity with significant substrates (key features: redox potential, stereoelectronic effects); (ii) product patterns with suitable probe precursors, which unambiguously indicate either an electron-transfer or a radical mechanism of oxidation; (iii) effect of substituents in the oxidation of benzyl alcohols (Hammett correlation); (iv) determination of the kinetic isotope effect. The results obtained allow us to come to conclusions about the oxidation mechanism adopted by the various laccase–mediator systems. The case of mediator TEMPO, in view of its peculiar and ionic oxidation route, is the object of a separate investigation.14 Our previous experience in the case of competing electron-transfer vs. radical routes of oxidation was certainly helpful in the approach to the present problem.15
Results and discussion
Purified laccase from Trametes villosa (viz. Poliporus pinsitus)7 was employed in the experiments. These were performed at room temperature in a stirred water solution, buffered at pH 5 (0.1 M in sodium citrate) and purged with O2 for 30 min prior to the addition of the reagents.7 A first set of substrates was investigated, that could provide information about the oxidation mechanism, as well as hints about any possible substrate-specialisation of the mediator. The yield of oxidation (Table 1) was determined by GC analysis, and calculated with respect to the molar amount of the substrate. It must be stressed that, in contrast to similar reports in the literature, a deficiency of mediator is used in our experiments, in combination with a very small amount of laccase, so that the overall oxidation of the substrate by oxygen (eqn. (1) and Scheme 1) is truly catalysed by the laccase–mediator system adopted.7,16 In fact, yields in excess of 100% are obtained if calculated vs. the molar amount of the mediator, thereby implying an oxidation process with turnover.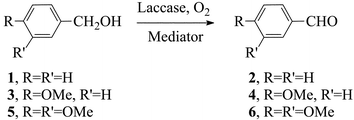 | (1) |
| Substrate (E°, in V/NHE, in H2O)a | With ABTS | Product yield (% vs. substrate) | ||
|---|---|---|---|---|
| With HBT | With HPI | With VLA | ||
| a The source of the E° data,22 and a few experimental details, were already given.7 b In competition experiments. | ||||
| Benzyl alcohol, 1 (2.4) | 2, 2 | 2, 30 | 2, 54 | 2, 42 |
| 4-MeO-benzyl alcohol, 3 (1.8) | 4, 22 | 4, 76 | 4, 65 | 4, 65 |
| Veratryl alcohol, 5 (1.4) | 6, 37 | 6, 92 | 6, 74 | 6, 61 |
| 1-(4-MeO-phenyl)ethanol, 7 (1.4) | 8, 15 | 8, 49 | — | 8, 82 |
| Methyl veratryl ether, 9 (ca. 1.7) | 0 | 6, 57; 10, 25 | 6, 38; 10, 15 | 6, 24; 10, 1 |
| 4-MeO-ethylbenzene, 11 (ca. 1.7) | 0 | 7, 8; 8, 20 | 7, 4; 8, 11 | 7, 1; 8, 1 |
| k 3 /k1 relative rateb | 30 | 4.1 | 10 | 2.2 |
| k 3 /k7 relative rateb | 1.5 | 0.7 | 0.8 | 0.4 |
Reactivity of substrate
The concentration of the reagents was: [substrate], 20 mM; [mediator], 6 mM, with 10 units of laccase. The reaction time was 24 h, even though a reaction time of 7–8 h was previously found sufficient in most cases.7 Three benzylic alcohols (1, 3 and 5) were considered as significant structural models of lignin (eqn. (1)). It must be stressed that, in the absence of mediators, laccase alone does not oxidise these non-phenolic precursors, as expected, nor can the mediators alone do it, in the absence of the enzyme.3,7,17 The three benzyl alcohols are mechanistically significant in that they differ in redox potential (E° in V/NHE in H2O given in Table 1),7 as brought about by the different number of electron–donor methoxy substituents in the aromatic ring.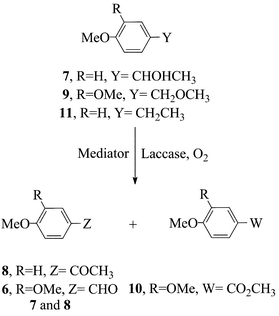 | (2) |
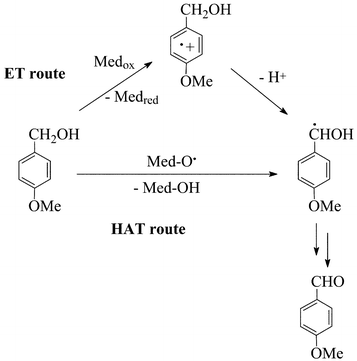 | ||
| Scheme 2 Mechanisms of oxidation. | ||
Competition of a primary (3) vs. secondary (7) benzylic alcohol (eqn. 2) was additionally carried out, and a k3/k7 relative rate of 3 obtained. This result is also consistent with the ET route of Scheme 2, because stereoelectronic effects are known to retard deprotonation from the radical cation of an encumbered (secondary) alkylaromatic substrate,15 such as 7. As for the nature of the Medox species deriving from ABTS on interaction with laccase,20,21 either ABTS˙+ or ABTS++ (E° = 0.69 and 1.1 V/NHE, respectively)7 could be generated by the enzyme, whose redox potential is around 0.7–0.8 V.4 Clearly, if the Medox were the ABTS++ species, the subsequent monoelectronic oxidation of appropriate substrates, such as 3, 5 and 7 would occur in a moderately endoergonic ET step, whereas it had to be much more endoergonic if the Medox were the weaker ABTS˙+.16 It appears therefore likely that the Medox form of ABTS is the dication.10,20 It is finally worth emphasizing the lack of reactivity of the laccase–ABTS system with a benzylic ether (9) or with an alkylbenzene (11) (eqn. (2)), despite the fact that the redox potential of 9 and 10 is close to that of 322 which reacted satisfactorily. A specificity of this mediator towards benzylic alcohols would therefore emerge.16
In the HAT radical route (Scheme 2), the redox potential of the substrate ought to have a negligible relevance on reactivity. However, effects arising from the polarity of the N-oxyl radical, an electrophilic radical,24–26 and from the electron-richness of the substrate, could provide a dipolar stabilisation to the radical transition state of H-abstraction, due to the important contribution from a charge-separated resonance structure,28,29 thereby explaining the slightly higher oxidation yields obtained with substrates 3 and 5, that bear electron-donor groups. Anyhow, such polar effects are small, as the k3/k1 relative rates of 10, 4.1 and 2.2 with HPI, HBT and VLA, respectively, do confirm (compare with the value of 30 for ABTS). Support to the HAT route also comes from the value of the primary vs. secondary (k3/k7) selectivity, which is lowerthan 1 with HBT, HPI and VLA (0.7, 0.8 and 0.4, respectively), rather than 1.5 as obtained with ABTS. An easier cleavage of a secondary vs. primary benzylic C–H bond is indeed expected in a radical route, based on the pertinent BDE(C–H) values of 88.0 and 85.4 kcal mol−1, respectively.15 Finally, the conversion into oxidised products from both the ether 9 and alkylbenzene 11 is appreciable, and opens up synthetically interesting possibilities.16 Peculiar is the additional formation of the methyl ester 10 from 9, besides the aldehyde 6. Once again, a hint to a possible mediator-to-substrate specialisation emerges, as HBT proves more competent towards the ether moiety, whereas VLA proves more competent towards a secondary alcohol.
A final comment on these sections is that the observed variety of results not only provides support to the operation of two different mechanisms of oxidation (ET vs. HAT, in Scheme 2), but also confirms the dominant role of the Medox species (rather than that of laccase; see Scheme 1) in the interaction with the substrate. Clearly, the ‘natural’ phenol-oxidase activity of laccase is profoundly affected from its interaction with the mediators.16
Hammett correlation
A more extensive and quantitative treatment of the effect of substituents upon reactivity in the oxidations induced by laccase–mediator systems was sought through a Hammett-type correlation. Five X-substituted benzyl alcohols, i.e., 4-NO2- (p-12), 3-NO2- (m-12), 4-Cl- (14), 4-Me- (16), 4-MeO–C6H4CH2OH (3), were oxidised pair-wise in competition experiments vs. the unsubstituted precursor (1) as the relay compound (eqn. (3)),25 and the kX/kH ratios determined by determining the relative amounts of the corresponding aldehydes (p-13, m-13, 15, 17, 4, and 2, respectively) by GC analysis (Table 2). The log kX/kH ratios gave better fits when plotted vs. the σ+ rather than the σ of the substituents,30 and the resulting ρ correlation parameters of the four laccase–mediator systems are given in Table 2; the example of the laccase–ABTS correlation is presented in Fig. 2.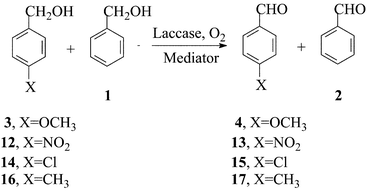 | (3) |
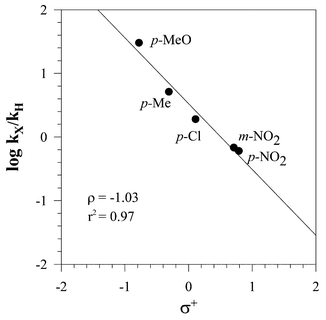 | ||
| Fig. 2 Hammett correlation for the laccase–ABTS oxidation of substituted benzylic alcohols: competition experiments. | ||
| Mediator | kp-NO2/kH | km-NO2/kH | k Cl/kH | k Me/kH | k MeO/kH | ρ |
|---|---|---|---|---|---|---|
| a Typical error of the GC determinations: ±3%. Runs in triplicate. | ||||||
| ABTS | 0.60 | 0.67 | 1.9 | 5.1 | 30 | −1.03 |
| HBT | 0.68 | 0.59 | 1.7 | 2.6 | 6.6 | −0.64 |
| HPI | 0.47 | 0.44 | 1.5 | 3.8 | 11 | −0.89 |
| VLA | 0.57 | — | 0.84 | 1.4 | 2.5 | −0.41 |
These ρ parameters are admittedly rather small and similar. Nevertheless, it cannot be denied that the ρ values of HBT, HPI and VLA are the smaller ones, as it ought to be for a HAT mechanism.25 In fact, small ρ values are obtained in unambiguous radical processes,29 such as the reactions of substituted toluenes with bromine or NBS28 or with Cl3C˙,31 with ρ values in the range of −0.3 to −1.5. Indeed, the relevance24–26 of polar effects on the Med–O˙ electrophilic radicals, resulting from these N–OH mediators, would be responsible for making the substituent effects of these laccase-mediated oxidations more or less sensible,25 and making the absolute values of the ρ parameters of VLA < HBT < HPI ‘significant’. The smaller ρ value determined for VLA is consistent with the smaller k3/k1 and k3/k7 values (Table 1) obtained with this particular mediator, and would suggest that the corresponding Med–O˙ intermediate is the one with the lower polar character. In the contest of polar radicals it is indeed common to find better correlations of the log kX/kH data with the σ+, rather than with the σ of the substituent.28,29,31 On the other hand, in the ET route of mediator ABTS (Scheme 2), the effect of the substituents on the reactivity of oxidation plays a divergent role in the two steps of the reaction.19 In fact, removal of electrons from the substrate by Medox to form a radical cation is favoured by electron-donor substituents, whereas the ensuing deprotonation of the substrate radical cation is disfavoured by electron-donor substituents. The two effects tend to compensate each other, and this might explain the small ρ value obtained here with ABTS; anyway, small ρ values with ET processes are not always the case.32,33 Support to the present mechanistic conclusions comes also from the following section.
Probe substrates
Attention was turned towards the benzylic alcohol 18 (and 20) (Scheme 3). Besides being very comparable in structure with other widely-employed lignin model compounds,5c,3418 presents the distinct feature of giving rise to two diverse end-products depending on the oxidation mechanism.10 In fact, under genuine ET conditions with chemical oxidants,3318 gives the transient 18˙+ intermediate that cleaves at the Cα–Cβ bond, to produce p-anysaldehyde 4 and tert-butyl radical.22a This cleavage is driven by steric and stereoelectronic factors,35 that make the loss of the tertiary radical fragment easy.22a,36 Conversely, under bona fide radical HAT conditions, 18 undergoes fragmentation of the Cα–H benzylic bond,22a and produces ketone 19 exclusively.33,37 This clear-cut behaviour makes 18 a useful probe, capable of providing evidence for the oxidation mechanism from product analysis.10,33 Use of probe 20 was additionally made whenever the conversion to products was low, as in the case of mediator ABTS (or, with some other mediator, for consistency); in fact 20 is structurally analogous to 18 but more viable to monoelectronic oxidation (to veratryl aldehyde 6), in view of its additional methoxy-substituent.38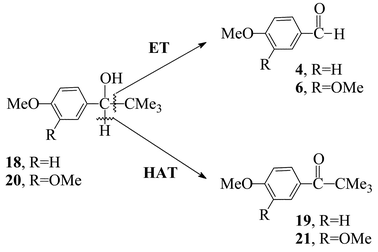 | ||
| Scheme 3 Oxidation of probe substrates 18 and 20 by laccase–mediator systems: Cα–Cβvs. Cα–H cleavage. | ||
Test results (Table 3) show that reaction of 18 with a bona fide one-electron oxidant, such as K5CoIIIW12O40 (viz. Co(III)W; E° 1.4 V),10,39 causes Cα–Cβ bond cleavage and yields aldehyde 4exclusively. Analogous results are obtained on independent oxidation of 20 with ABTS++ (E° 1.1 V),7 unambiguously generated from ABTS (in the absence of laccase) by use of the strong oxidant (NH4)2CeIV(NO3)6 (viz., CAN; E° 1.5 V).10,33 Consistent with this evidence for the ET route, the laccase–ABTS system gave only the aldehyde 6 from precursor 20 (no appreciable conversion from 18), whereas the three laccase–NOH mediator systems gave only the ketone 19 or 21 from their corresponding precursors, in keeping with the HAT route. With respect to Table 3, it must be stressed that no other side-products are detected, and that the precursor probe is always recovered in amounts consistent with the yield of product, thereby providing a quantitative mass balance.
| Mediator | Substrate | Product (% yield vs. subst.)b | |
|---|---|---|---|
| a Reaction conditions: [Subs.] = 20 mM, [Med] = 6 mM, [Lc] = 3 U ml−1, pH 5, reaction time 24 h, at 20 °C, O2 was purged for 30 min initially. b Determined by GC. c The substrate was quantitatively recovered. d Reaction time: 3 days. e [Co(III)W]: [Sub] = 2 ∶ 1, pH 5, reaction time 24 h, at 20 °C; without laccase. f Generated in situ by using the CeIV salt, without laccase. | |||
| ABTS | 18 20 | 4, 0c | 6, 2d |
| HBT | 18 20 | 19, 20 | 21, 50 |
| HPI | 18 20 | 19, 30 | 21, 70 |
| VLA | 18 20 | 19, 5d | 21, 20 |
| Co(III)We | 18 | 4, 15 | |
| ABTS++f | 20 | 6, 4 | |
Two conclusions have been made concerning the results of Table 3. With reference to Scheme 3, the product obtained from the Cα–Cβ cleavage (i.e., 6) with the laccase–ABTS system is in keeping with the oxidation pattern of the bona fide ET reagent Co(III)W, as well with that of the ABTS++ species independently generated. Conversely, mediators HBT, HPI and VLA, that give the C–H cleavage product (19 or 21), follow the HAT pattern. Conversion is strangely low from 18 with mediators ABTS and VLA, in spite of the fact that they did catalyse the oxidation of the analogous mono-methoxylated substrate 3 (see Table 1) satisfactorily. The bulky tert-butyl group in 18 (and 20) could be responsible for lowering the yield of oxidation with ABTS and VLA, perhaps for steric reasons. This does not seem to affect the reaction of the other mediators to a similar extent.
The virtue of probe 18 (and 20) is its specificity, because it gives rise uniquely to either one (4) or the other (19) of the end-products, depending on the oxidation mechanism. Less specific probe substrates are also available,33 however, and their products pattern is consistent with these mechanistic conclusions, although being less unambiguous. In fact, probe 22, that features a benzylic residue rather than the tert-butylic residue of 18, yields both the Cα–Cβ cleavage product (i.e., p-MeO-benzaldehyde, 4) and the Cα–H fragmentation product (ketone 23, as for 19) under bona fide ET conditions (46% and 9%, respectively),37 while giving rise to modest amounts (1%) of the aldehyde 4 in reaction with laccase–ABTS system. Conversely, ketone 23predominates (30 ∶ 1) over 4 in the oxidation of 22 with the laccase–HBT system.
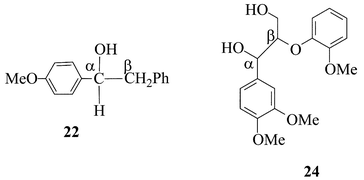
Analogously, another lignin model compound, i.e., Adlerol (24),40a yields predominantly the Cα–Cβ cleavage product 6 (i.e., veratryl aldehyde) vs. the Cα–H fragmentation product (i.e., ketone Adlerone, 25) under bona-fide ET conditions, as for example with metalloporphyrins and KHSO5,40 or in an anodic oxidation,41 whereas ketone 25 is almost exclusively obtained from laccase–mediator (i.e., HBT, VLA) oxidations that follow the HAT route.42 It can be concluded that the approach of the probe substrates, and the resulting mechanistic information, have a wide structure-generality.
Intramolecular isotope effect determination
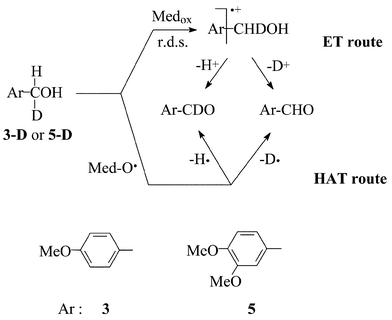
In the ET mechanism of Scheme 2, a deprotonation at the Cα–H bond of the radical cation of the substrate is implied, but this is likely to be a fast step, following the rate-determining monoelectronic oxidation of the substrate by the Medox species.15,19 Fragmentation of a Cα–H bond is instead the only and rate-determining step in the concurrent HAT mechanism of oxidation (Scheme 2).15 On mono-deuteriation of a substrate (such as 5) at the benzylic position, either 1H+ or 2H+ ought to be loss to the solvent, if the ET route operates, with the concomitant formation of either Ar-CDO or Ar-CHO, respectively (Scheme 4). Instead, in the HAT route, either 1H˙ or 2H˙ ought to be abstracted from precursor 5, and the two corresponding aldehydes Ar-CDO or Ar-CHO formed. Mass spectrometric determination of the relative amount of the two aldehydes enabled determination of the intramolecular kH/kD selectivity (Table 4) for these oxidations, on a suitably synthesised 5-D (or 3-D) precursor.10| Oxidant | k H/kDd | |
|---|---|---|
| With 3-D | With 5-D | |
| a [Co(III)W]: [Sub] = 2 ∶ 1, pH 5, reaction time 24 h, 25 °C; without laccase. b [Sub] = 20 mM, [Med] = 6 mM, [Lc] = 3 U ml−1, pH 5, reaction time 24 h, 25 °C, O2 for 30 min. c Generated in situ by the CeIV salt, without laccase. Unreacted substrate is recovered (65%). d Determined by GC-MS. | ||
| Co(III)Wa | — | 3.8 |
| Laccase + ABTSb | 3.1 | 3.6 |
| Laccase + VLAb | — | 6.4 |
| Laccase + HBTb | 6.2 | 6.4 |
| Laccase + HPIb | 6.4 | 6.2 |
| ABTS++c | 3.3 | 3.7 |
 | ||
| Scheme 4 | ||
The results obtained support the stronger kinetic relevance of the H/D loss with mediators HBT, VLA and HPI, once again in keeping with the rate-determining H-abstraction step in the radical route (Scheme 2). Large kH/kD values are indeed documented for the oxidation of benzhydrols by the N-oxyl radical of HPI.43 On the other hand, with laccase–ABTS we obtain a smaller kH/kD value, that is practically coincident with those from the bona fide ET oxidations with Co(III)W, and with ABTS++ independently generated.10 Small kH/kD values are consistently reported from enzymatic and biomimetic studies involving the radical cations of the alkylanilines.44
Conclusion
The present work reports on fundamental reactivity features of the mediation phenomenon with the enzyme laccase towards non-phenolic substrates, and offers a comprehensive mechanistic rationalisation of it. Experimental evidence is provided for the oxidation mechanism undertaken by the oxidised form (Medox) of four mediators, namely, ABTS, HBT, HPI and VLA. This evidence derives from determining the Hammett correlations and the kinetic isotope effect for each mediator. Additionally, experiments with some probe substrates, whose end-products of oxidation unambiguously indicate the occurrence of either the ET or the HAT route of oxidation, are described. Conclusive clues allowing to solve the dichotomy between ET vs. HAT oxidation routes are in this way obtained. In general, HBT and HPI prove to be the more efficient mediators in the HAT route. These laccase–mediator systems show promise for environmental friendly biotechnological applications to the delignification of kraft pulps for the paper industry. Tests of their efficiency towards samples of pulps are underway.Experimental
General
Most of the precursors and products, and the solvents were commercially available (Aldrich). Others (18, 20, 22, and their oxidation products) were available from previous investigations.7,22a,33,36,37 Monodeuteriated alcohols (3D and 5D) were also available from previous work.45 Laccase from a strain of Trametes villosa (viz. Poliporus pinsitus) (Novo Nordisk Biotech) was employed.7 It was purified by ion-exchange chromatography on Q-Sepharose by elution with phosphate buffer,46 and an activity of 9000 U mL−1 determined spectrophotometrically by the standard reaction with ABTS.47Instrumentation
A VARIAN 3400 Star gas chromatograph, fitted with a 20 m × 0.25 mm methyl silicone gum capillary column, was employed in the GC analyses. GC-MS analyses were performed on a HP 5892 GC, equipped with a 12 m × 0.2 mm methyl silicone gum capillary column, and coupled to a HP 5972 MSD instrument, operating at 70 eV. The acquisition of the MS signal was in the SIM mode for the Ar-CHO vs. Ar-CDO relative intensity determinations.Enzymatic reactions
The oxidation reactions were performed at room temperature in water solution (3 mL), buffered at pH 5 (0.1 M in sodium citrate) and purged with O2 for 30 min prior to the addition of the reagents.7 In case of sparingly soluble substrates, 10% acetonitrile was added. The concentration of the reagents was: [substrate], 20 mM; [mediator], 6 mM, with 10 units of laccase. The incubation was carried out for 24 h under constant stirring, keeping a latex balloon half-filled with oxygen on top the reaction vessel. Following a conventional work-up, the yields of oxidation were determined by GC analysis with respect to an internal standard (acetophenone or p-methoxyacetophenone), suitable response factors being determined from authentic products. The identity of the products was also confirmed by GC-MS analyses. No other products, besides those indicated in the Tables, were detected.Competition experiments
Competition experiments of two substrates, either for the Hammett's correlations (kX/kH), but also for the k3/k1 and k3/k7 relative rate determinations, were similarly run on a 40 mmol amount of each of the substrates, giving the following initial concentrations: [ArCH2OH], 20 mM; [PhCH2OH], 20 mM, [mediator], 6 mM, with 10 units of laccase. The yields of products were determined by GC, after a reaction time (4–5 h) that would ensure only a modest/moderate conversion into products (10–25%). In order to determine the kH/kD ratios, determination of the relative amount of the Ar-CHO and Ar-CDO oxidation products (Scheme 4) was done by GC-MS analyses after a 5 h reaction time.Chemical oxidations
Oxidations with K5CoIIIW12O40 (viz. Co(III)W) (60 mmol) of a substrate (30 mmol) were conducted in 2 mL citrate buffer, at room temperature for 1 or 3 days; conventional work-up with CHCl3 or ethyl acetate followed. In the oxidations with ‘preformed ABTS++’, 20 µmol of ABTS were dissolved in 1.5 mL of 2 M H2SO4; 40 µmol of (NH4)2CeIV(NO3)6 (viz. CAN) dissolved in 1.5 mL of 2 M H2SO4 were added, and the red colour of the dication developed immediately; 20 µmol of substrate were added very quickly at this point, and the resulting solution stirred at room temperature for 3 min, or until the red colour had turned blue (i.e., ABTS˙+). Conventional work-up followed.Abbreviations
| HBT | 1-Hydroxybenzotriazole |
| HPI | N-Hydroxyphthalimide |
| VLA | Violuric acid |
| ABTS | 2,2′-Azinobis(3-ethylbenzothiazoline-6-sulfonic acid) |
Acknowledgements
Thanks are due to Novo Nordisk Biotech (Denmark) for a generous gift of laccase. We also thank the EU for financial support (grant QLK5-CT-1999-01277) to the OXYDELIGN project.References
- M. Tien and T. K. Kirk, Science, 1983, 221, 661–663 CAS.
- M. Kuwahara, J. K. Glenn, A. Morgan and M. H. Gold, FEBS Lett., 1984, 169, 247–250 CrossRef CAS.
- A. Messerschmidt, Multi-Copper Oxidases, World Scientific, Singapore, 1997 Search PubMed.
- (a) F. Xu, J. J. Kulys, K. Duke, K. L. K. Krikstopaitis, H.-J. W. Deussen, E. Abbate, V. Galinyte and P. Schneider, Appl. Environ. Microbiol., 2000, 66, 2052–2056 CrossRef CAS; (b) F. Xu, W. Shin, S. H. Brown, J. A. Wahleithner, U. M. Sundaram and E. I. Solomon, Biochim. Biophys. Acta, 1996, 1292, 303–311 CAS; (c) K. Li, F. Xu and K.-E. L. Eriksson, Appl. Environ. Microbiol., 1999, 65, 2654–2660 CAS.
- (a) R. Bourbonnais and M. G. Paice, FEBS Lett., 1990, 267, 99–102 CrossRef CAS; (b) R. Bourbonnais and M. G. Paice, Appl. Microbiol. Biot., 1992, 36, 823–827 Search PubMed; (c) R. Bourbonnais, M. G. Paice, B. Freiermuth, E. Bodie and S. Borneman, Appl. Environ. Microb., 1997, 63, 4627–4632 Search PubMed.
- C. Crestini and D. S. Argyropoulos, Bioorgan. Med. Chem., 1998, 6, 2161–2169 Search PubMed.
- M. Fabbrini, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., 2002, 16, 231–240 CrossRef CAS.
- L. Banci, S. Ciofi-Baffoni and M. Tien, Biochemistry, 1999, 38, 3205–3210 CrossRef CAS.
- F. d'Acunzo, C. Galli and B. Masci, Eur. J. Biochem., 2002, 269, 5330–5335 CrossRef CAS.
- M. Fabbrini, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., 2002, 18, 169–171 CrossRef CAS.
- C. Eggert, U. Temp, J. F. D. Dean and K.-E. L. Eriksson, FEBS Lett., 1996, 391, 144–148 CrossRef CAS.
- H.-P. Call and I. Mücke, J. Biotechnol., 1997, 53, 163–202 CrossRef CAS.
- M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551–7553 CrossRef CAS.
- F. d'Acunzo, P. Baiocco, M. Fabbrini, C. Galli and P. Gentili, Eur. J. Org. Chem., 2003 , in the press.
- E. Baciocchi, F. d'Acunzo, C. Galli and O. Lanzalunga, J. Chem. Soc., Perkin Trans. 2, 1996, 133–140 RSC.
- G. Cantarella, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., submitted Search PubMed.
- R. ten Have and P. J. M. Teunissen, Chem. Rev., 2001, 101, 3397–3413 CrossRef CAS.
- (a) A. Majcherczyk, C. Johannes and A. Hüttermann, Appl. Microbiol. Biotechnol., 1999, 51, 267–276 CrossRef CAS; (b) A. Muheim, A. Fiechter, P. J. Harvey and H. E. Schoemaker, Holzforschung, 1992, 46, 121–126 Search PubMed.
- E. Baciocchi, M. Bietti and O. Lanzalunga, Acc. Chem. Res., 2000, 33, 243–251 CrossRef CAS.
- R. Bourbonnais, D. Leech and M. G. Paice, Biochim. Biophys. Acta, 1998, 1379, 381–390 CrossRef CAS.
- A. Potthast, T. Rosenau and K. Fischer, Holzforschung, 2001, 55, 47–56 Search PubMed.
- (a) M. Bietti, E. Baciocchi and S. Steenken, J. Phys. Chem. A, 1998, 102, 7337–7342 CrossRef CAS; (b) J. Howell, J. M. Goncalves, C. Amatore, L. Klasinc, R. M. Wightman and J. K. Kochi, J. Am. Chem. Soc., 1984, 106, 3968–3976 CrossRef CAS; (c) P. J. Kersten, B. Kalyanaraman, K. E. Hammel, B. Reinhammar and T. K. Kirk, Biochem. J., 1990, 268, 475–480 CAS.
- T. Iwahama, Y. Yoshino, T. Keitoku, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2000, 65, 6502–6507 CrossRef CAS.
- F. Minisci, C. Punta, F. Recupero, F. Fontana and G. F. Pedulli, J. Org. Chem., 2002, 67, 2671–2676 CrossRef CAS.
- F. d'Acunzo, P. Baiocco, M. Fabbrini, C. Galli and P. Gentili, New J. Chem., 2002, 26, 1791–1794 RSC.
- F. Minisci, C. Punta, F. Recupero, F. Fontana and G. F. Pedulli, Chem. Commun., 2002, 688–689 RSC.
- D. F. McMillen and D. M. Golden, Ann. Rev. Phys Chem., 1982, 33, 493–532 Search PubMed.
- C. Walling, A. L. Rieger and D. D. Tanner, J. Am. Chem. Soc., 1963, 85, 3129–3134 CrossRef CAS.
- E. Baciocchi and O. Lanzalunga, Tetrahedron, 1993, 49, 7267–7276 CrossRef CAS.
- T. H. Lowry and K. S. Richardson, Mechanism and Theory in Organic Chemistry, 2nd ed., Harper & Row, New York, 1981 Search PubMed.
- G. J. Gleicher, J. Org. Chem., 1968, 33, 332–336 CrossRef CAS.
- E. Baciocchi, T. Del Giacco, C. Rol and G. V. Sebastiani, Tetrahedron Lett., 1989, 30, 3573–3576 CrossRef CAS.
- E. Baciocchi, S. Belvedere, M. Bietti and O. Lanzalunga, Eur. J. Org. Chem., 1998, 299–302 CrossRef CAS.
- K. Li, R. F. Helm and K.-E. L. Eriksson, Biotechnol. Appl. Bioc., 1998, 27, 239–243 Search PubMed.
- E. Baciocchi, M. Mattioli, R. Romano and R. Ruzziconi, J. Org. Chem., 1991, 56, 7154–7160 CrossRef CAS.
- E. Baciocchi, M. Bietti, L. Putignani and S. Steenken, J. Am. Chem. Soc., 1996, 118, 5952–5960 CrossRef CAS.
- E. Baciocchi, S. Belvedere and M. Bietti, Tetrahedron Lett., 1998, 39, 4711–4714 CrossRef CAS.
- This inference is made on the basis of the redox potentials of the structurally comparable 3 [E° 1.8 V] and 5 [E° 1.4 V]: see Table 1.
- E. Baciocchi, M. Crescenzi, E. Fasella and M. Mattioli, J. Org. Chem., 1992, 57, 4684–4689 CrossRef CAS.
- (a) G. Labat and B. Meunier, J. Org. Chem., 1989, 54, 5008–5011 CrossRef CAS; (b) B. Meunier, La Chimica e l'Industria (Milan), 1990, 72, 433–439 Search PubMed.
- D. Rochefort, R. Bourbonnais, D. Leech and M. G. Paice, Chem. Commun., 2002, 1182–1183 RSC.
- C. Galli et al., work in progress. To be presented to the 12th International Symposium on Wood and Pulping Chemistry, (june 2003), Madison, WI, USA.
- C. Ueda, M. Noyama, H. Ohmori and M. Masui, Chem. Pharm. Bull., 1987, 35, 1372–1377 CAS.
- E. Baciocchi, M. F. Gerini, O. Lanzalunga, A. Lapi, M. G. Lo Piparo and S. Mancinelli, Eur. J. Org. Chem., 2001, 2305–2310 CrossRef CAS.
- E. Baciocchi, M. Fabbrini, O. Lanzalunga, L. Manduchi and G. Pochetti, Eur. J. Biochem., 2001, 268, 665–672 CrossRef CAS.
- F. Xu, Biochemistry, 1996, 35, 7608–7614 CrossRef CAS.
- B. S. Wolfenden and R. L. Willson, J. Chem. Soc., Perkin Trans. 2, 1982, 805–812 RSC.
| This journal is © The Royal Society of Chemistry 2003 |