Singlet-energy transfer in quadruple hydrogen-bonded oligo(p-phenylenevinylene)perylene-diimide dyads
Received
10th September 2002
, Accepted 12th November 2002
First published on 29th November 2002
Abstract
The photophysical properties of a supramolecular donor–acceptor dyad consisting of an oligo(p-phenylenevinylene) unit and a perylene-diimide unit are described. The dyad is created by functionalising the two chromophores with quadruple hydrogen bonding 2-ureido-4[1H]-pyrimidinone units, which provide a high association constant (K
≈ 108 M−1 in toluene). This feature enabled us to study the time-resolved photoinduced singlet-energy transfer reaction between the two chromophores in dilute solution with transient pump-probe spectroscopy. This energy transfer occurs with a time constant of 5.1 ps.
Introduction
Non-covalent interactions between organic molecules are ubiquitous in nature and serve to assemble, position and organize extended architectures that fulfil complex functions. The photosynthetic reaction centre is an intriguing example of such a structural design. Here, photoinduced energy and electron transfer reactions between carefully aligned molecular arrays of photo- and redox-active components work in concert to convert and store solar energy.1 Because hydrogen bonding is often a key element in structuring the natural systems, the study of photoinduced energy and electron transfer reactions between donors and acceptors assembled via hydrogen bonding interactions has attracted considerable interest in recent years.2,3 Hydrogen bonds, however, represent a fairly weak interaction and generally result in low association constants. Consequently, only a small fraction of the donors and acceptors remain associated, while the remaining molecules are free to diffuse in solution, especially at low concentrations. This problem can be alleviated by using multiple hydrogen-bonded arrays designed to gain strength and directionality.4 The 2-ureido-4[1H]-pyrimidinone (UP) quadruple hydrogen-bonding unit, for example, dimerises in a self-complementary array of four cooperative hydrogen bonds and provides association constants in excess of 107 M−1 in organic solvents.5 Utilizing these high association constants it is possible to construct supramolecular polymers.6 Recently the UP unit has been utilized to create hydrogen-bonded donor–acceptor dyads7 based on photo- and redox-active oligo(p-phenylenevinylene)
(OPV)8 and [60]fullerene (C60)9 derivatives. Photovoltaic cells based on supramolecular OPV polymers have demonstrated that the hydrogen bonds of the UP unit can be incorporated into working opto-electronic devices.10
Perylene-diimides have been extensively studied as organic semiconductors in electronic and optical applications such as field-effect transistors,11 fluorescent solar collectors,12 electrophotographic devices,13 photovoltaic devices,14 dye lasers,15 and molecular switches.16 These perylene-diimides have outstanding chemical, thermal and photochemical stability.17 We recently reported on photoinduced electron transfer in liquid crystalline oligo(p-phenylenevinylene)–perylene-diimide–oligo(p-phenylenevinylene)
(OPV-PERY-OPV) arrays and in π-stacks of triple hydrogen-bonded OPV–PERY–OPV trimers.18,19 Here we extend these studies and describe the synthesis of a perylene-diimide with the donor–donor–acceptor–acceptor (DDAA) four-point hydrogen bonding motif UP-unit (PERY-UP) and study its photophysical properties in hetero-assemblies with OPV-UP (Fig. 1) using fluorescence spectroscopy and femtosecond pump-probe spectroscopy. The high association constant of the heterodimer allowed us to time resolve the singlet-energy transfer reaction in this supramolecular system by femtosecond pump-probe spectroscopy.
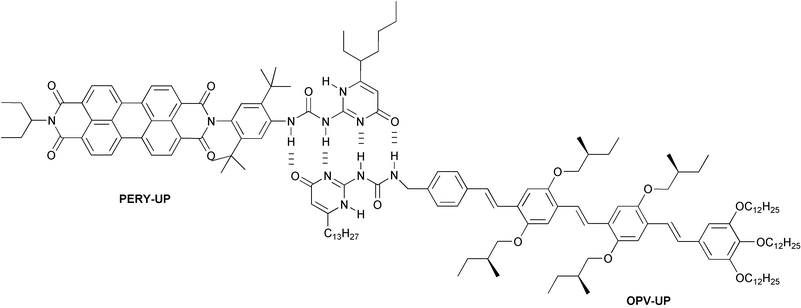 |
| | Fig. 1 Heterodimer of PERY-UP and OPV-UP bound by quadruple hydrogen bonds. | |
Results and discussion
Synthesis
The preparation of OPV-UP has been described previously.8 The synthetic route to PERY-UP is shown in Scheme 1. N,N′-bis(1-ethylpropyl)perylene-3,4:9,10-tetracarboxylic diimide 120 was partially hydrolysed with potassium hydroxide in tert-butanol to the anhydride imide 2.21,22 Anhydride imide 2 was subsequently reacted with 2,5-di-tert-butylbenzene-1,4-diamine23 in the presence of zinc acetate in molten imidazole to afford the asymmetric perylene-diimide 3. PERY-UP was obtained by reaction of 3 with carboxamide 424 in chloroform.
Keto–enol equilibrium
The UP-unit of OPV-UP and PERY-UP can exist in two different tautomeric forms.25 Apart from the pyrimidinone tautomer (keto) with a DDAA motif, the pyrimidinol tautomer (enol) exists. Both keto and enol forms are self-complementary with DDAA and DADA motifs, respectively, but cannot associate with each other. The keto–enol equilibrium depends on the polarity of the solvent and the nature of the substituent on the isocytosine moiety. The keto tautomer is known to have a higher dimerisation constant (K
= 6 × 107 M−1 in water-saturated chloroform; 6 × 108 M−1 in toluene) than the enol tautomer (K
=
∼105 M−1 in chloroform),25,26 as a result of more favourable secondary interactions.27,28
1H NMR spectroscopy has been used to determine the relative amounts of the keto and enol tautomers of PERY-UP in different solvents by comparing the integrated signals of the N–H protons. For PERY-UP the amount of keto is 80% in CDCl3, while it decreases to 50% in toluene-d8. These ratios are >99%
(CDCl3) and 90%
(toluene-d8) for OPV-UP.7 As a consequence, not all PERY-UP molecules can bind to OPV-UP molecules and we assume that in a 1 : 1 mixture of PERY-UP and OPV-UP, both homodimers and the heterodimer are simultaneously present.
Optical properties
The absorption and photoluminescence spectra of PERY-UP, OPV-UP and a 1 : 1 mixture of the two compounds in toluene are shown in Fig. 2. The characteristic absorption peaks of the perylene-diimide are at 459 nm, 490 nm and 527 nm for PERY-UP. The absorption spectrum of OPV-UP is not structured and maximizes at 432 nm. Both compounds fluoresce in toluene solution with very high quantum yields.7,20 The fluorescence spectra reveal that the singlet-excited state (S1) of OPV-UP (E(S1)
= 2.52 eV) is higher in energy than the singlet-excited state of PERY-UP (E(S1)
= 2.32 eV). Fluorescence lifetimes (τ) for OPV-UP and PERY-UP are 1.23 and 3.61 ns, respectively, as determined from time-correlated single photon counting (TCSPC) with excitation at 400 nm. The absorption spectrum of the mixture closely resembles the superposition of the spectra of the two compounds and gives no evidence of a significant electronic interaction. In dichloromethane, the oxidation potential of OPV-UP is Eox
= 0.71 V vs. SCE and the first reduction potential of PERY-UP is Ered
=
−0.61 V vs. SCE.
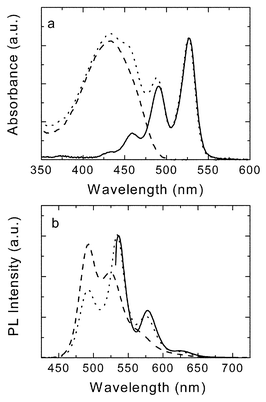 |
| | Fig. 2 Absorption (a) and emission (b) spectra of PERY-UP (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), OPV-UP ( ), OPV-UP (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and a 1 : 1 mixture of PERY-UP and OPV-UP (⋯) in toluene solution. The excitation wavelength is 527 nm for PERY-UP and 410 nm for OPV-UP and the mixture. ) and a 1 : 1 mixture of PERY-UP and OPV-UP (⋯) in toluene solution. The excitation wavelength is 527 nm for PERY-UP and 410 nm for OPV-UP and the mixture. | |
Fluorescence quenching
Fluorescence quenching has been used to study the steady-state photophysics of OPV-UP–PERY-UP heterodimers. As a result of the ordering of the singlet states and the difference between oxidation and reduction potentials, the singlet-excited state of OPV-UP can act as a donor towards PERY-UP in both photoinduced energy and photoinduced electron transfer reactions. Both processes are expected to quench the OPV-UP fluorescence, when their rate is competitive with the intrinsic decay of OPV-UP.
The OPV-UP fluorescence quenching was studied by adding a mixture of 2 × 10−5 M PERY-UP and 10−6 M OPV-UP in small aliquots to a solution of 10−6 M OPV-UP in toluene. Almost selective excitation of OPV-UP in the mixtures was achieved by irradiation at 410 nm and the fluorescence signal at 493 nm (OPV-UP fluorescence only) was followed as a function of the PERY-UP concentration in the mixture. A significant quenching of the OPV-UP fluorescence was observed. The quenching factor Q, i.e. the ratio of initial OPV-UP fluorescence (I0) and that of the mixture (I) is plotted in Fig. 3 after correction for absorption by PERY-UP.29 The Stern–Volmer constant, defined as KSV
=
(Q-1)/[PERY-UP], amounts to KSV
= 5.9 × 105 M−1 for concentrations of PERY-UP up to 1.13 × 10−6 M. Two control experiments were performed, in each experiment one of the two chromophores lacks the hydrogen bonding UP-group. The first control used 1 instead of PERY-UP together with OPV-UP and in the second experiment PERY-UP was mixed with a methyl end-capped oligo(p-phenylenevinylene) with four phenyl rings (OPV4)30 instead of OPV-UP. In both cases no fluorescence quenching was observed (Fig. 3). This indicates that dynamic or collisional quenching is not important at these low concentrations and that the quenching can unambiguously be ascribed to the formation of OPV-UP–PERY-UP heterodimers.
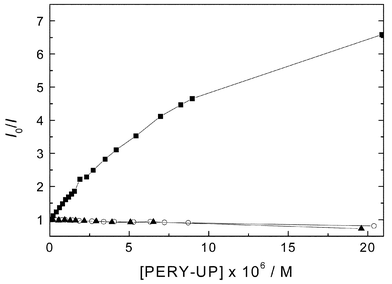 |
| | Fig. 3 Stern–Volmer plot of OPV fluorescence quenching by PERY derivatives in toluene solution. Curves are shown for OPV-UP + PERY-UP (■); OPV-UP +
1
(○); and OPV4 + PERY-UP (▲). The OPV chromophore concentration is 10−6 M in each experiment and the excitation wavelength 410 nm. The detection wavelength is 493 nm for OPV-UP and 500 nm for OPV4. | |
The origin of the OPV fluorescence quenching in the OPV-UP–PERY-UP heterodimers can be inferred from the photoluminescence spectrum (Fig. 2b) of the mixture. When the 1 : 1 mixture is excited at 410 nm, the fluorescence of the PERY-UP unit dominates the photoluminescence spectrum even though the absorption at 410 nm of this chromophore is negligible. Hence the OPV-UP unit acts as a sensitiser for the PERY-UP fluorescence. This indicates that a singlet-energy transfer, rather than electron transfer, occurs in the heterodimer in toluene in which the photoexcited OPV-UP S1 state evolves to PERY-UP S1.
Transient photoinduced absorption
Sub-picosecond transient pump-probe spectroscopy (150 fs pulses) was performed at room temperature on OPV-UP–PERY-UP solutions to assess the temporal evolution of the singlet-energy transfer on short timescales. Upon preferential photoexcitation of the OPV-UP moiety at 450 nm in a mixture of 4 × 10−5 M OPV-UP and 2 × 10−4 M PERY-UP in toluene, a negative differential transmission was observed by probing at 900 nm (Fig. 4). At 900 nm the Sn
← S1 photoinduced absorption of the OPV chromophore is present.31 The dynamics in the low picosecond time range (Fig. 4, ○) show a rapid decay which can be fitted to a mono-exponential decay with a time constant of 5.1 ps, i.e. a rate of k
= 1.9 × 1011 s−1. On longer time scales (Fig. 4, ●) a much slower decay is visible with a mono-exponential time constant of 1.1 ns. The two different decay times account for the fact that under these conditions both OPV-UP–PERY-UP heterodimers and OPV-UP–OPV-UP homodimers are present. It is important to note that a control experiment confirmed that the Sn
← S1 photoinduced absorption of the PERY chromophore has only a small (∼10%) contribution to the photoinduced absorption at 900 nm. The fast (5.1 ps) decay is attributed to the singlet-energy transfer reaction from OPV-UP(S1) to PERY-UP in the heterodimer, while the slow (1.1 ns) decay represents the intrinsic relaxation of OPV-UP(S1) in the homodimer. The lifetime of the latter process is close to the photoluminescence lifetime of 1.23 ns of OPV-UP homodimers, determined by TCSPC.7
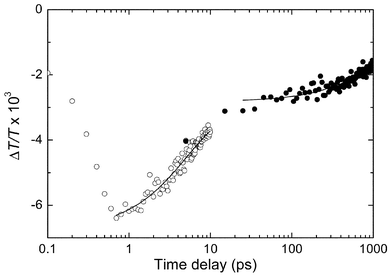 |
| | Fig. 4 Differential transmission dynamics of the OPV (Sn
← S1) absorption at 900 nm as a function of the pump-probe time delay after photoexcitation at 450 nm. The concentration PERY-UP is 2 × 10−4 M and the concentration OPV-UP is 4 × 10−5 M in toluene. Solid lines are fits to decays with time constants of 5 ps and 1100 ps. | |
Förster energy transfer
The fluorescence quenching studies provide a lower limit for the rate for singlet-energy transfer via the Stern–Volmer constant by: | | 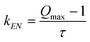 | (1) |
using the highest quenching observed in the titration experiment, Qmax
= 6.6, and τ
= 1.23 ns, the lower limit for kEN is 4.5 × 109 s−1. Of course, Qmax is limited by the presence of OPV-UP homodimers and the highest ratio of PERY-UP : OPV-UP (21.6 : 1) used in the experiment. The actual value, kEN
= 1.9 × 1011 s−1, determined from the transient spectroscopy, is over 40 times faster.
The Förster equation32 can be used to estimate the distance d between the centres of the donor and acceptor chromophores in the energy transfer process.
| | 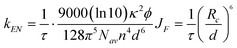 | (2) |
In
eqn. (2), the parameters
Nav and
n represent Avogadro's number and the refractive index of the medium,
ϕ is the fluorescence quantum yield of the donor in the absence of transfer,
Rc the Förster radius and
d is the actual centre-to-centre distance of the two
chromophores involved in the energy transfer.
JF represents the overlap between the absorption (
ε(
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/char_e0ce.gif)
)) of the acceptor (PERY-UP) and the fluorescence (
F(
)) of the donor (OPV-UP) on an energy scale (cm
−1) defined as:
| | 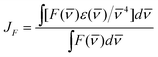 | (3) |
The factor κ is the so-called orientation factor, which incorporates the dependence of the energy transfer rate on the mutual orientation of the transition dipole moments of donor and acceptor chromophores. It can be expressed as:
| | | κ
= cos(ΘDA)
− cos(ϕDR) cos(ϕAR) | (4) |
where
ΘDA is the angle between the transition dipole moments of donor and acceptor and
ϕDR
(
ϕAR) is the angle between the transition dipole moment of the donor (acceptor)
chromophore and the line joining the centres of the two chromophoric units. In calculating values for
κ2, we have assumed that rotation around the bond joining the UP unit and the
methylene group connecting the UP and the OPV moieties is possible. For various conformations we find values for
κ2 ranging from 2.5 to 3.5. Using the following numbers for the OPV-UP–PERY-UP combination,
JF
= 1.55 × 10
−13 cm
6 mol
−1,
kEN
= 1.9 × 10
11 s
−1,
τ
= 1.23 ns,
ϕ
= 0.84,
7κ2
= 3, we obtain using
eqns. (2) and (3):
Rc
= 64 Å and
d
= 26 Å. The latter value is slightly less than the centre-to-centre distance estimated from molecular modelling (∼33 Å). The difference can be rationalized by considering that the singlet-excited states delocalise and effectively reduce the centre-to-centre distance. A similar result has been found for energy transfer reactions in covalent OPV4-C
60 donor–acceptor systems.
31
Electron transfer
Why does singlet-energy transfer occur from the OPV-UP singlet state rather than electron transfer and is electron transfer possibly subsequent to energy transfer from the singlet-excited state of PERY-UP? To address these questions it is instructive to consider the change in free energy for charge separation using the Weller equation:33,34| | 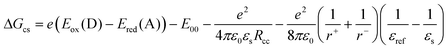 | (5) |
Table 1 shows that according to eqn. (5) photoinduced electron transfer is energetically possible for the OPV-UP–PERY-UP heterodimer in solvents of varying polarity from either the OPV-UP or the PERY-UP singlet-excited states.
Table 1 Change in free energy for charge separation ΔGCSa/eV from OPV(S1) and PERY(S1), and solvent reorganization energy λs/eV for OPV-UP–PERY-UP heterodimers in different solvents from eqns (5) and (7)
| |
ΔGCSOPV |
ΔGCSPERY |
λ
s
|
|
ΔGCSOPV uses E00
= 2.52 eV; ΔGCSPERY uses E00
= 2.32 eV.
|
| Toluene |
−0.47 |
−0.27 |
0.07 |
| odcb |
−1.28 |
−1.08 |
0.79 |
| Benzonitrile |
−1.43 |
−1.23 |
0.98 |
To investigate the possibility for electron transfer in the heterodimer in more detail, the fluorescence signal of PERY-UP at 578 nm was monitored after selective photoexcitation of PERY-UP at 527 nm as a function of an increasing amount of OPV-UP. For this purpose, a solution of 2 × 10−5 M OPV-UP and 10−6 M PERY-UP was added in steps to a 10−6 M solution of PERY-UP. Irrespective of the polarity of the solvent (toluene, ε
= 2.38; o-dichlorobenzene (odcb), ε
= 9.93; benzonitrile, ε
= 25.18) the PERY-UP fluorescence signal remained constant. From this experiment it can be concluded that photoinduced electron transfer does not occur in the heterodimer from the PERY-UP S1 state.
If electron transfer is absent but thermodynamically feasible, it is likely to be hampered kinetically. The expression for the rate constant for non-adiabatic charge separation:
| | 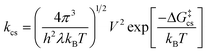 | (6) |
shows that
kcs is determined by the coupling (
V) between donor and acceptor in the excited state, the reorganization energy
λ and the barrier for charge separation (Δ
G‡cs
=
(Δ
Gcs
+
λ)
2/4
λ). The reorganization energy is the sum of internal (
λi) and
solvent (
λs) contributions. The latter can be estimated from:
35| | 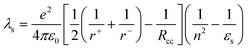 | (7) |
and the results for OPV-UP–PERY-UP are collected in
Table 1. The internal reorganization energy
λi is probably not very high for these extended conjugated systems and is estimated to be in the range of 0.2 to 0.5 eV. Within these limits the barrier Δ
G‡cs for charge separation from PERY-UP(S
1) remains less than 0.01 eV in odcb and
benzonitrile, and is only slightly higher in
toluene (<0.04 eV). The absence of
electron transfer in each of these
solvents is therefore ascribed to a very weak electronic coupling
V of donor and acceptor in the excited state, rather than a high barrier.
V is known to be exponentially dependent on the distance between donor and acceptor. Molecular modelling and the value derived from the Förster equation, indicate an appreciable distance between OPV-UP and PERY-UP units in the heterodimer, causing a low
V. In agreement with this proposition, a photoinduced electron transfer reaction does occur between OPV and PERY
chromophores in systems were the two redox-active
chromophores are at a much shorter distance.
18,19
Conclusion
Photoluminescence studies reveal that a singlet-energy transfer reaction occurs in quadruple hydrogen-bonded OPV-UP–PERY-UP heterodimers (Fig. 1) after excitation of the OPV chromophore. As a result of the high association constant of the quadruple hydrogen bond, significant amounts of heterodimers and homodimers are present, even in dilute solutions. This feature enabled us to follow the temporal evolution of the singlet-energy transfer reaction using sub-picosecond transient absorption spectroscopy in the heterodimers on short timescales. The time constant of 5.1 ps was obtained for this reaction is in fair agreement with Förster theory. The singlet-excited in OPV-UP homodimers, is much longer lived and decays with a time constant of 1.1 ns. Although exergonic, electron transfer does not occur after photoexcitation as a result of a too weak electronic coupling between OPV and PERY chromophores in the excited state. To achieve photoinduced electron transfer in quadruple hydrogen-bonded systems based on the UP unit a judicious design of the relative positioning of donor and acceptor will be required.
Experimental
General methods
1H NMR and 13C NMR spectra were recorded at room temperature on a Varian Gemini 300 MHz or a Varian Mercury 400 MHz spectrometer. Chemical shifts are given in ppm (δ) relative to tetramethylsilane. Matrix assisted laser desorption ionisation time of flight (MALDI-TOF) mass spectrometry was conducted on a Perseptive Biosystems Voyager DE-Pro MALDI-TOF mass spectrometer. UV/Vis absorption spectra were recorded on a Perkin Elmer Lambda 900 spectrophotometer. Fluorescence spectra were recorded on an Edinburgh Instruments FS920 double-monochromator spectrometer and a Peltier-cooled red-sensitive photomultiplier. Fluorescence lifetimes were determined using an Edinburgh Instruments LifeSpec-PS spectrometer, equipped with a 400 nm picosecond laser (PicoQuant PDL 800B) operated at 2.5 MHz and a Peltier-cooled Hamamatsu micro-channel plate photomultiplier (R3809U-50). Cyclic voltammograms were measured in 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte in dichloromethane using a Potentioscan Wenking POS73 potentiostat. The working electrode was a Pt disk (0.2 cm2), the counter electrode was a Pt plate (0.5 cm2), and a saturated calomel electrode (SCE) was used as the reference electrode, calibrated against Fc/Fc+
(+0.43 V).
N-(1-ethylpropyl)perylene-3,4:9,10-tetracarboxylic 3,4-anhydride 9,10-imide (2)
A mixture of N,N′-bis(1-ethylpropyl)perylene-3,4:9,10-tetracarboxylic diimide 1
(2.87 g, 5.4 mmol), potassium hydroxide (0.91 g, 0.016 mol) in tert-butanol (100 ml) was heated at 100 °C for 30 min. The reaction mixture was then poured into 400 ml of 10% HCl and the precipitate was filtered. The residue was stirred in 200 ml of a warm aqueous solution containing potassium hydroxide (20 g, 0.36 mol) and potassium chloride (16 g, 0.21 mol). The solid was filtered and subsequently washed with the aqueous solution until the solution was no longer coloured yellow–green. The solid was then stirred in water and subsequently filtered. The dark red coloured filtrate was precipitated by addition of hydrochloric acid to a final total percentage of 10% HCl concentration. The precipitate was filtered, washed with water and dried at 90 °C under vacuum to yield 0.50 g (20%) of a black solid. 1H NMR (CDCl3)
δ 8.80–8.60 (m, 8H), 5.07 (m, 1H), 2.35–2.20 (m, 2H), 2.00–1.90 (m, 2H), 0.93 (t, J
= 7.5 Hz). 13C NMR (CDCl3)
δ 160.15, 136.61, 133.86, 133.76, 131.81, 124.10, 123.35, 123.24, 119.23, 58.03, 57.86, 25.16, 11.50, Electron impact MS: (MW = 461)
m/z
= 461.
N-(1-ethylpropyl)-N′-(4-amino-2,5-di-tert-butylphenyl)perylene-3,4:9,10-tetracarboxylic diimide (3)
A mixture of N-(1-ethylpropyl)perylene-3,4:9,10-tetracarboxylic 3,4-anhydride 9,10-imide 2
(0.048 g, 0.10 mmol), 2,5-di-tert-butylbenzene-1,4-diamine (0.14 g, 0.64 mmol), imidazole (2.5 g) and a few grains of zinc acetate were stirred at 160 °C for 3.5 h under argon. After cooling to room temperature the reaction mixture was purified extensively by column chromatography (SiO2, CH2Cl2–ethanol 95 : 5, CH2Cl2–methanol 99 : 1–97.5 : 2.5, ethyl acetate–n-hexane 4 : 1 and ethyl acetate–n-hexane 3 : 2). After washing the solid with n-hexane 0.046 g (67%) of 3 was obtained pure as a red solid. 1H NMR (CDCl3)
δ 8.80–8.65 (m, 8H), 6.87 (s, 1H), 6.82 (s, 1H), 5.08 (m, 1H), 3.93 (s, 2H), 2.40–2.20 (m, 2H), 2.10–1.90 (m, 2H), 1.54 (s, 18H), 0.94 (t, 6H, J
= 7.3 Hz). 13C NMR (CDCl3)
δ 164.88, 145.24, 145.07, 135.08, 134.71, 133.07, 131.99, 130.05, 129.79, 129.40, 126.96, 126.66, 124.02, 123.73, 123.31, 123.24, 118.18, 100.34, 57.92, 34.04, 31.76, 29.73, 25.21, 11.51. MALDI-TOF MS (MW = 663.31)
m/z
= 663.02 [M−].
N-(1-ethylpropyl)-N′-{4-[2-ureido-6-(1-ethylpentyl)-4[1H]-pyrimidinone]-2′,5′-di-tert-butylphenyl}perylene-3,4:9,10-tetracarboxylic diimide (PERY-UP)
N-(1-ethylpropyl)-N′-(4-amino-2,5-di-tert-butylphenyl)perylene-3,4:9,10-tetracarboxylic diimide (3): (0.0262 g, 0.04 mmol) and N-[6-(1-ethylpentyl)-4-oxo-1,4-dihydropyrimidin-2-yl]imidazolyl-1-carboxamide 4
(about 0.02 g, 0.07 mmol) were refluxed in dry chloroform (dried over molecular sieves) for 18 h under argon. After cooling to room temperature the reaction mixture was washed with 1M HCl (5 ml), saturated aqueous NaHCO3, water, brine and was dried over Na2SO4. The product was triturated with acetone to yield 0.020 g (54%) of a red solid. 1H NMR (CDCl3)δ two tautomers: 13.27 (s, 1H), 12.59 (s, 1H), 11.67 (s, 1H), minor 11.49 (s), 8.77 (d, 2H, J
= 8.06 Hz), 8.69 (d, 2H, J
= 8.06 Hz), 8.67 (d, 2H, J
= 8.06 Hz), 8.66 (d, 2H, J
= 8.06 Hz), minor 7.63 (s), 7.47 (s, 1H), 7.10 (s, 1H), minor 6.27 (s), 5.89 (s, 1H), 5.08 (m, 1H), minor 2.53 (br s), 2.45–2.10 (m, 3H), 1.98 (m, 2H), 1.80–1.50 (m, 4H), 1.44 (s, 9H), 1.30 (s, 13H), 1.00–0.80 (m, 12H). 13C NMR (CDCl3)
δ two tautomers: 164.50, 156.06, 155.13, 145.53, 135.22, 134.61, 132.12, 130.07, 129.73, 126.94, 126.60, 123.69, 123.43, 123.27, 57.88, 45.43, 32.88, 31.68, 31.02, 29.41, 26.73, 25.15, 22.65, 14.03, 11.83, 11.57. MALDI-TOF MS (MW = 898.44). Found: m/z
= 898.43 [M−].
Pump-probe spectroscopy
The femtosecond laser system used for pump-probe experiments consists of an amplified Ti-sapphire laser (Spectra Physics Hurricane), providing 150 fs pulses at 800 nm with an energy of 750 µJ at 1 kHz. Pump (450 nm, fluence 0.5 µJ/mm2) and probe (900 nm) pulses were created by optical parametric amplification followed by frequency quadrupling or doubling using two OPA's (Spectra Physics OPA-C). The pump beam was linearly polarized at the magic angle (54.7°) with respect to the probe beam. The temporal evolution was recorded using a Si detector and standard lock-in detection at 500 Hz.
Acknowledgements
We thank A. Rizzuti, H. M. Keizer and M. van Gerwen for generous gifts of reactants, and A. El-ghayoury for a sample of OPV-UP. This work was financially supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (CW-NWO) and the Eindhoven University of Technology in the PIONIER program (98400). The research of SCJM has also been made possible by a fellowship of the Royal Dutch Academy of Arts and Sciences.
References
-
J. Deisenhofer and J. R. Norris, The Photosynthetic Reaction Center, Academic Press, New York, 1993 Search PubMed.
- For reviews see:
(a)
V. Balzani and F. Scandola, Supramolecular Photochemistry, Ellis Horwood, Chichester, 1991 Search PubMed;
(b)
J. L. Sessler, B. Wang, S. L. Springs and C. T. Brown, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle and Y. Murakami, Pergamon, Oxford, 1996, vol. 4, p. 311 Search PubMed;
(c) M. D. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC;
(d) P. Piotrowiak, Chem. Soc. Rev., 1999, 28, 143 RSC;
(e)
C. J. Chang, J. D. Brown, M. C. Y. Chang, E. A. Baker and D. G. Nocera, in Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, vol. 3, 409 Search PubMed;
(f) M. D. Ward, C. M. White, F. Barigelletti, N. Amaroli, G. Calogero and L. Flamigni, Coord. Chem. Rev., 1998, 171, 481 CrossRef CAS.
-
(a) P. Tecilla, R. P. Dixon, G. Slobodkin, D. S. Alavi, D. H. Waldeck and A. D. Hamilton, J. Am. Chem. Soc., 1990, 112, 9408 CrossRef CAS;
(b) A. Harriman, D. J. Magda and J. L. Sessler, J. Phys. Chem., 1991, 95, 1530 CrossRef CAS;
(c) A. Harriman, Y. Kubo and J. L. Sessler, J. Am. Chem. Soc., 1992, 114, 388 CrossRef CAS;
(d) C. Turro, C. K. Chang, G. E. Leroi, R. I. Cukier and D. G. Nocera, J. Am. Chem. Soc., 1992, 114, 4013 CrossRef CAS;
(e) J. L. Sessler, B. Wang and A. Harriman, J. Am. Chem. Soc., 1993, 115, 10418 CrossRef CAS;
(f) J. L. Sessler, B. Wang and A. Harriman, J. Am. Chem. Soc., 1995, 117, 704 CrossRef CAS;
(g) P. J. F. de Rege, S. A. Williams and M. J. Therien, Science, 1995, 269, 1409 CAS;
(h) J. A. Roberts, J. P. Kirby and D. G. Nocera, J. Am. Chem. Soc., 1995, 117, 8051 CrossRef CAS;
(i) J. P. Kirby, J. A. Roberts and D. G. Nocera, J. Am. Chem. Soc., 1997, 119, 9230 CrossRef CAS;
(j) E. Prasad and K. R. Gopidas, J. Am. Chem. Soc., 2000, 122, 3191 CrossRef CAS;
(k) A. J. Myles and N. R. Branda, J. Am. Chem. Soc., 2001, 123, 177 CrossRef CAS;
(l) M. A. Smitha, E. Prasad and K. R. Gopidas, J. Am. Chem. Soc., 2001, 123, 1159 CrossRef CAS;
(m) J. L. Sessler, M. Sathiosatham, C. T. Brown, T. A. Rhodes and G. Wiederrecht, J. Am. Chem. Soc., 2001, 123, 3655 CrossRef CAS;
(n) M. A. Smitha and K. R. Gopidas, Chem. Phys. Lett., 2001, 350, 86 CrossRef CAS.
- For a recent review see: D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83 Search PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS.
- E. H. A. Beckers, P. A. van Hal, A. P. H. J. Schenning, A. El-ghayoury, E. Peeters, M. T. Rispens, J. C. Hummelen, E. W. Meijer and R. A. J. Janssen, J. Mater. Chem., 2002, 12, 2054 RSC.
- A. El-ghayoury, E. Peeters, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2000, 1969 RSC.
-
(a) M. T. Rispens, L. Sánchez, J. Knol and J. C. Hummelen, Chem. Commun., 2001, 161 RSC;
(b) J. J. González, S. González, E. María Priego, C. Luo, D. M. Guldi, J. de Mendoza and N. Martín, Chem. Commun., 2001, 163 RSC;
(c) L. Sánchez, M. T. Rispens and J. C. Hummelen, Angew. Chem., Int. Ed., 2002, 41, 838 CrossRef CAS.
- A. El-ghayoury, A. P. H. J. Schenning, P. A. van Hal, J. K. J. van Duren, R. A. J. Janssen and E. W. Meijer, Angew. Chem., Int. Ed., 2001, 40, 3660 CrossRef CAS.
- P. R. L. Malenfant, C. D. Dimitrakopoulos, J. D. Gelorme, L. L. Kosbar, T. O. Graham, A. Curioni and W. Andreoni, Appl. Phys. Lett., 2002, 80, 2517 CrossRef CAS.
- G. Seybold and G. Wagenblast, Dyes Pigm., 1989, 11, 303 CrossRef.
- K. Y. Law, Chem. Rev., 1993, 93, 449 CrossRef CAS.
-
(a) D. Wohrle and D. Meissner, Adv. Mater., 1991, 3, 129 CrossRef CAS;
(b) D. Schlettwein, D. Wohrle, E. Karmann and U. Melville, Chem. Mater., 1994, 6, 3 CrossRef CAS;
(c) S. Ferrere, A. Zaban and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 4490 CrossRef CAS;
(d) L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS.
- M. Sadari, L. Hadel, R. R. Sauers, S. Husain, K. Krogh-Jespersen, J. D. Westbrook and G. R. Bird, J. Phys. Chem., 1992, 96, 7988 CrossRef.
- M. P. O'Neil, M. P. Niemczyk, W. A. Svec, D. Gosztola, G. L. Gaines and M. R. Wasielewski, Science, 1992, 257, 63 CAS.
-
W. A. Fisher, in Pigment Handbook Volume I: Properties and Economics; ed. T. C. Patton, John-Wiley & Sons, New York, 1973, p 667 Search PubMed.
- E. Peeters, P. A. van Hal, S. C. J. Meskers, R. A. J. Janssen and E. W. Meijer, Chem. Eur. J., 2002, 8, 4470 CrossRef CAS.
- A. P. H. J. Schenning, J. van Herrikhuyzen, P. Jonkheijm, Z. Chen, F. Würthner and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 10252 CrossRef CAS.
- S. Demmig and H. Langhals, Chem. Ber., 1988, 121, 225 Search PubMed.
- H. Kaiser, J. Lindner and H. Langhals, Chem. Ber., 1991, 124, 529 CrossRef CAS.
- Y. Nagao, T. Naito, Y. Abe and T. Misono, Dyes Pigm., 1996, 32, 71 CrossRef CAS.
- F. Bell, J. Chem. Soc., 1958, 120 RSC.
-
H. M. Keizer, to be published.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487 CrossRef.
- W. L. Jorgensen and J. Pranata, J. Am. Chem. Soc., 1990, 112, 2008 CrossRef.
- J. Pranata, S. G. Wierschke and W. L. Jorgensen, J. Am. Chem. Soc., 1991, 113, 2810 CrossRef CAS.
- The data points are corrected for the simultaneous excitation of the PERY-UP at 410 nm and for the re-absorption of the OPV-UP emission by PERY-UP at 493 nm (especially important at high excess of PERY-UP).
- E. Peeters, A. Marcos, S. C. J. Meskers and R. A. J. Janssen, J. Chem. Phys., 2000, 112, 9445 CrossRef CAS.
- P. A. van Hal, R. A. J. Janssen, G. Lanzani, G. Cerullo, M. Zavelani-Rossi and S. De Silvestri, Phys. Rev. B, 2001, 64, 075206 CrossRef.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7 RSC.
- A. Weller, Z. Phys. Chem. Neue Folge, 1982, 133, 93 Search PubMed.
- In eqn. (5), −e is the electron charge, ε0 the vacuum permittivity, εs the polarity of the solvent, εref the polarity of the solvent used to determine the redox potentials Eox(D) and Ered(A), Rcc the centre-to-centre distance of positive and negative charges and r+ and r− the radii of the positive and negative ions. E00 is the energy of the excited state from which electron transfer occurs. For OPV-UP, r+
= 5.05 Å
(Ref. 31). For the perylene diimide segment of PERY-UP, r−
= 4.71 Å was taken from the density (ρ
= 1.59 g cm−1) of N,N′-dimethylperylene-3,4:9,10-tertacarboxylic diimide derived from the X-ray crystallographic data (E. Hädicke and F. Graser, Acta Cryst. C, 1986, 42, 189)
viar−
=
[3M/(4πρNA)]⅓. Rcc was set to 33 Å.
- H. Oevering, M. N. Paddon-Row, M. Heppener, A. M. Oliver, E. Cotsaris, J. W. Verhoeven and N. S. Hush, J. Am. Chem. Soc., 1987, 109, 3258 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2003 |
Click here to see how this site uses Cookies. View our privacy policy here.