Through-space interactions between face-to-face, center-to-edge oriented arenes: importance of polar–π effects
Franco
Cozzi
*a,
Rita
Annunziata
a,
Maurizio
Benaglia
a,
Mauro
Cinquini
a,
Laura
Raimondi
a,
Kim K.
Baldridge
b and
Jay S.
Siegel
*b
aCNR-ISTM and Dipartimento di Chimica Organica e Industriale, Universita' di Milano, via Golgi, 19-20133 Milano, Italy
bDepartment of Chemistry and Biochemistry -0358 University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0358, USA
First published on 6th December 2002
Abstract
Two series of conformationally restricted polycyclic compounds (1–3 and 4–7) have been synthesized as model systems for studying the through-space interactions between face-to-face, center-to-edge (parallel-offset) oriented arenes. These compounds feature different X substituents on one of the interacting rings. By monitoring the variation of the ΔG≠ for the rotation around the aryl–aryl bond in 1–7 as a function of X by 2D [1H,1H] EXSY NMR spectroscopy, it was found that the barriers increase on passing from electron-donating to electron-withdrawing substituted derivatives. Quantum mechanical calculations [MP2/DVZ (2d,p)//B3LYP/DVZ(2d,p)] gave barrier values and variations in agreement with the experimental data. The results are consistent with a repulsive arene–arene interaction dominated by electrostatic effects.
Introduction
Through-space interactions between π-systems1 are important because they play a major role in controlling disparate chemical phenomena including molecular recognition,2 stereoselectivity of organic reactions,3 nucleic acid and protein macroscopic structures,4 aggregation of extended π-systems,5 and crystal packing.6 Several theoretical studies have been aimed at elucidating the nature of the arene–arene interactions in the simple case of the benzene dimer (Fig. 1).1,7 From these works emerged the idea that the parallel-stacked orientation A is unfavorable, the edge-to-face one B is favorable,8 and the energy of the face-to-face, center-to-edge (parallel-offset) arrangement C depends on the contact surface area. A simple picture of the electronic distribution of benzene possessing a negatively charged core surrounded by a positively charged periphery,7b,7i rationalizes these results on an electrostatic basis.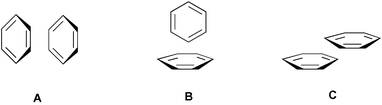 | ||
| Fig. 1 Relative orientations of rings in the benzene dimer. | ||
The electrostatic nature of the repulsive interaction in the parallel-stacked orientation A has experimentally been demonstrated by us by studying a number of variously substituted 1,8-diarylnaphthalenes,9 and confirmed by work carried out on other systems.10 These findings can be invoked to rationalize a variety of experimental results in different fields,2–5 and nicely explain the tendency of the mixed arene–perfluoroarene systems to adopt the parallel-stacked orientation.11
Experimental work has also been devoted to elucidate the type B interaction between edge-to-face oriented arenes.12,13 The results of a quantitative study led Hunter and coworkers to conclude that “the interactions between the CH groups of the edge ring and the π electron density of the face ring are sensitive to changes in the local charge distribution on the two rings”.12a,b In contrast, Wilcox and coworkers12c designed a model system that was unresponsive to changes in arene substitution, a result that “casts substantial doubt on the importance of electrostatic interactions in edge-to-face binding events.”12c
Face-to-face, center-to-edge interactions (type C), also called parallel-offset interactions, have received considerably less attention, despite the fact that this structural motif can be found in many structures.14 Recently,15,16 some conformationally flexible derivatives of N-benzyl-2-phenylpyridinium bromide adopting, among others, a parallel-offset conformation in water, have been reported. Herein, we investigate type C interactions in two conformationally restricted polycyclic systems.
Results and discussion
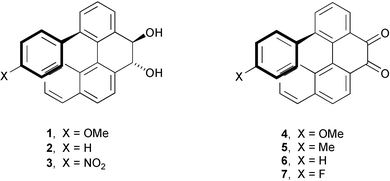 | ||
| Chart 1 Structures of compounds 1–7. | ||
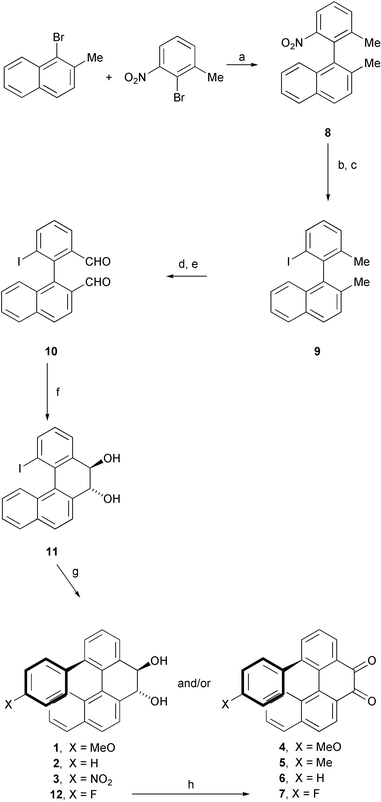 | ||
| Scheme 1 Synthesis of compounds 1–7.Reagents and conditions: a, Cu bronze, neat, 200 °C. b, SnCl2·H2O, AcOEt, 80 °C, 15 h. c, H2SO4, NaNO2, H2O, 0–5 °C, 30 min; then KI 5 to 20 °C, 15 h. d, NBS cat. PhCO2O, hν, CCl4, 80 °C, 48 h. e, AgNO3, dioxane, H2O, 100 °C, 15 h. f, SmI2, THF, −78 °C, 1 h. g, Pd2(dba)3, AsPh3, DMF, 150 °C, 40 h. h, PySO3, DMSO, RT, 2 h. | ||
Ullmann reaction between commercially available 2-bromo-3-nitrotoluene and 1-bromo-2-methylnaphthalene (3 mol equiv.)17 gave the cross-coupling product 8 in 43% yield. Reduction of the nitro group (87% yield) followed by Sandmeyer reaction of the resulting amine afforded the iodo compound 9 in 57% yield. Perbromination with excess NBS (5.5 mol equiv.) gave the bis(dibromomethyl) derivative (87% yield) that was then hydrolyzed to the corresponding bis(aldehyde) 10 by silver nitrate promoted reaction in dioxane–water (70% yield). Reductive intramolecular coupling carried out under McMurry conditions18 to generate 1-iodobenzo[c]phenanthrene led to concomitant cleavage of the carbon–iodine bond. An attempt to prepare the same compound by reaction of 10 with hydrazine in acetic acid19 failed, as did the synthesis of 1-iodo-5,6-epoxybenzo[c]phenanthrene by reaction of 10 with tris(dimethylaminophosphine) in toluene.20 Ultimately, SmI221 promoted pinacol reaction of 10 in THF at −78 °C gave the racemic diequatorial diol 11 (J = 10.5 Hz) in 75% yield.22
The reaction of 11 with 4X-substituted phenyl trimethylstannanes (4 mol equiv.)23 in the presence of 0.2 mol equiv. of Pd2(dba)3 and 0.8 mol equiv. of AsPh324 in degassed DMF (150 °C, 40 h) gave diols 1–3, albeit in low yields (1, 29%; 2, 12%; 3, 11%). Surprisingly, 1 and 2 were accompanied by comparable amounts of the corresponding diketones 4 (24% yield) and 6 (11% yield). Starting from 4-methylphenyl trimethylstannane, diketone 5 was the only isolated product (15% yield). Finally, fluorodiketone 7 was obtained from the corresponding crude diol 12 (that could not be obtained in pure form) by Py·SO3 oxidation25 in 12% overall yield from 11.
The ΔG≠ for the rotation around the aryl–aryl bond in the series of compounds 1–3 and 4–7 was determined by 2D [1H,1H] EXSY NMR experiments (Table 1).27,28 The data for the diol and, independently, the diketone series show a trend for the barrier to rotation to increase from ED to EW X substituents on the phenyl ring. The plots of ΔG≠ values vs the σpara for 1–3 and 4–7 showed excellent linear relationship (Fig. 2).
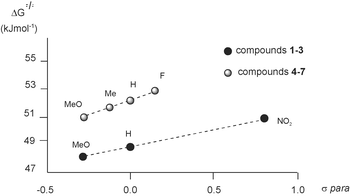 | ||
| Fig. 2 Plots of ΔG≠ (kJ mol−1) vs.σpara for compounds 1–3 (R = 0.9999) and 4–7 (R = 0.9998). | ||
Also in type A interactions, the barrier to rotation around aryl–aryl bonds increased when the interacting rings were made less electron rich by the introduction of EW groups. This effect was apparently due to a decreased repulsion between the π electrons of the arenes at the ground state (GS) level.9 In compounds 1–7, the distance between the interacting rings is longer and the overlap between the π clouds is less extended than in the type A analogs previously studied.9 Specifically, a typical angle between planes in 1–7 is ca. 55° with a center-to-center distance of 4.1 Å (closest contact 3.2 Å; furthest contact 5.2 Å). Thus, the repulsion between the π electrons is lower and the substituent effects should be less dramatic than previously seen. Indeed, the difference in the barriers between 1 and 3 is 3.0 kJ mol−1, while in the 1,8-diarylnaphthalene case a similar change in substitution led to ΔΔG≠ values of up to 14 kJ mol−1.9a At the GS level compounds 1–7 should experience an attractive electrostatic interaction between the hydrogens of one ring and the π electrons of the other. However, the effect of different substituents on this interaction should be small,12a and the contribution of the CH–π factor to the overall interaction likely negligible.
Quantum mechanical calculations [MP2/DVZ (2d,p)//B3LYP/DVZ(2d,p)] on diketone 6 showed a centroid to centroid distance of 4.3 Å and a type C conformation between the reference and rotating rings in the GS. At the transition state (TS, 1 negative eigenvalue) calculations showed that one of the ortho hydrogens of the rotating ring points towards the carbon atom rim of the reference ring with a distance of ca. 2.2 Å; the same hydrogen is 2.8 Å from the center of the ring, significantly further than the rim-atom interaction. The computational barrier (TS–GS) is 48.2 kJ mol−1, in reasonable agreement with experiment (52.4 kJ mol−1). Thus the computation model supports the supposition that we are studying an interaction loosely analogous to type C in the series 1–7 and that this series behaves similarly to the type A analogs studied previously.9
Two additional derivatives of 6 were calculated: 6-NH2 and 6-NO2. They provide some perspective on the trends across a Hammett series. The predicted barriers to rotation for 6-NH2, 6, and 6–NO2 are 47.5, 48.2, and 49.0 kJ mol−1, respectively. Albeit a small effect, the barrier increases with decreasing donor character of the para substituent. This trend parallels the one experimentally observed for both the diketone and diol series, and the magnitude of the barriers is in accord with those measured by NMR, showing that the type C interactions can be modeled well by this level of theory.
Conclusions
In conclusion, both theory and experiments agree in indicating that aryl rotation is responsible for the spectral changes observed by the variable temperature NMR spectroscopy, and that the through-space (type C) interactions between parallel-offset oriented arenes is strongly influenced by electrostatic effects. Since these effects were also found to be important in the type A and type B interactions between parallel-stacked and center-to-edge oriented arenes, respectively, the results herein described lend additional support to the notion that polar–π interactions are decisive factors for understanding the arene–arene interactions.Experimental
2-Methyl-1-(2-methyl-6-nitrophenyl)naphthalene 8
To a stirred mixture of 1-bromo-2-methylnaphthalene (2.16 mL, 13.88 mmol) and copper bronze (2.93 g, 46.2 mmol) heated up to 200 °C, 2-bromo-3-nitrotoluene (1.0 g, 4.62 mmol) was added portionwise over a period of 6 h. After the end of the addition the mixture was stirred at 200 °C for 20 h, whereupon it was cooled to RT and acetone (20 mL) was added. The suspension was stirred for 1 h at RT and then filtered. The filtrate was concentrated under vacuum and the resulting crude product was purified by flash chromatography with a 95 : 5 hexanes : Et2O mixture as eluant. The product, an orange thick oil, was isolated in 43% yield (0.556 g). IR (thin film): 3048, 2940, 1640, 1380, 1265 cm−1. 1H NMR: δ 7.87 (d, J = 7.9 Hz, 1H, hydrogen ortho to NO2), 7.84 (dd, J = 8.9, 1.3 Hz, 1H, H–C5 of naphthalene), 7.81 (d, J = 8.9 Hz, 1H, H–C4 of naphthalene), 7.61 (d, J = 7.9 Hz, 1H, hydrogen para to NO2), 7.50 (t, J = 7.9 Hz, 1H, hydrogen meta to NO2), 7.47 (d, J = 8.9 Hz, 1H, H–C3 of naphthalene), 7.40 (dt, J = 8.9, 1.3 Hz, 1H, H–C6 of naphthalene), 7.31 (dt, 8.9, 1.3 Hz, 1H, H–C7 of naphthalene), 7.09 (dd, J = 8.9, 1.3 Hz, 1H, H–C8 of naphthalene), 2.15 (s, 3H, Me of naphthalene), 1.90 (s, 3H, Me of phenyl group); 13C NMR: δ 148.0, 140.1, 134.3, 133.5, 133.2, 132.0, 131.9, 131.7, 128.5, 128.3, 128.25, 128.2, 126.5, 125.1, 124.3, 121.7, 20.1, 19.8. C18H15NO2 requires: C, 77.96; H, 5.45; N, 5.05%; found: C, 77.77; H, 5.58; N, 4.97%.3-Methyl-2-(2-methyl-1-naphthyl)aniline
A mixture of compound 8 (0.979 g, 3.53 mmol) and SnCl2 dihydrate (4.0 g, 17.65 mmol) in refluxing ethyl acetate (50 mL) was stirred for ca 15 h. When TLC analysis showed the disappearance of the starting material, the mixture was cooled to RT and made slightly alkaline by the careful addition of a saturated aqueous solution of NaHCO3. The mixture was then filtered on a Celite cake, the organic phase was separated, and the aqueous phase was extracted with Et2O (2 × 20 mL). The combined organic phases were dried over NaSO4, filtered and concentrated under vacuum. The crude product was purified by flash chromatography with a 80 : 20 hexanes : Et2O mixture as eluant. The product, a yellow thick oil, was isolated in 87% yield (0.759 g). IR (thin film): 3469, 3388, 3053, 2950, 1612, 1265 cm−1. 1H NMR: δ 7.92 (d, J = 8.0 Hz, 1H, H–C5 of naphthalene), 7.87 (d, 8.4 Hz, 1H, H–C4 of naphthalene), 7.53 (d, J = 8.4 Hz, 1H, H–C3 of naphthalene), 7.50–7.40 (m, 3H, H–C6, H–C7 and H–C8 of naphthalene), 7.23 (t, J = 7.7 Hz, 1H, hydrogen meta to NH2), 6.86 (d J = 7.7 Hz, 1H, hydrogen para to NH2), 6.74 (d, J = 7.7 Hz, 1H, hydrogen ortho to NH2), 2.29 (s, 3H, Me of naphthalene), 1.88 (s, 3H, Me of phenyl group); 13C NMR: δ 144.2, 138.7, 134.8, 133.4, 132.6, 132.2, 129.0, 128.3, 128.1, 127.7, 126.4, 125.2, 125.0, 120.1, 112.8, 112.2, 24.0, 19.9. C18H17N requires: C, 87.41; H, 6.93; N, 5.66%; found: C, 87.67; H, 7.11; N, 5.49%.2-Methyl-1-(2-iodo-6-methylphenyl)naphthalene 9
To a stirred suspension of the above described amine (0.73 g, 2.95 mmol) in concentrated sulfuric acid (0.75 mL) and water (0.2 mL) cooled at −10 °C, a solution of sodium nitrite (0.305 g, 4.43 mmol) in water (0.2 mL) was added. After 15 min stirring at −10 °C, a solution of KI (1.45 g, 8.85 mmol) in water (0.6 mL) was slowly added. The deep red solution was stirred overnight while allowing the temperature to slowly rise to RT. The mixture was made neutral by the addition of a saturated aqueous solution of NaHCO3, and was then washed with a saturated aqueous solution of Na2SO3 (10 mL). The resulting mixture was extracted with Et2O (2 × 20 mL) and CH2Cl2 (2 × 20 mL). The combined organic phases were dried over NaSO4, filtered, and concentrated under vacuum. The residue was purified by flash chromatography with hexanes as eluant. The product, a colorless oil, was obtained in 57% yield (0.602 g). IR (thin film): 3053, 2955, 1615, 1260 cm−1. 1H NMR: δ 7.91–7.86 (m, 2H, hydrogen ortho to I and H–C5 of naphthalene), 7.84 (d, J = 8.6, 1H, H–C4 of naphthalene), 7.44 (d, J = 8.6, 1H, H–C3 of naphthalene), 7.42 (dt, J = 8.0, 1.2 Hz, 1H, H–C6), 7.35 (dt, J = 8.0, 1.2 Hz, 1H, H–C7 of naphthalene), 7.33 (d, J = 8.0 Hz, 1H, hydrogen para to I), 7.15 (d, J = 8.0 Hz, 1H, H–C8 of naphthalene), 7.04 (t, J = 8.0 Hz, 1H, hydrogen meta to I), 2.13 (s, 3H, Me of naphthalene), 1.91 (s, 3H, Me of phenyl group); 13C NMR: δ 144.2, 139.2, 138.7, 136.8, 132.9, 132.1, 131.3, 129.9, 129.1, 128.7, 128.0, 127.7, 126.3, 125.0, 124.6, 101.8, 20.3, 19.5. C18H15I requires: C, 60.35; H, 4.22%; found: C, 60.58; H, 4.29%.1-(2-Formyl-6-iodophenyl)-2-naphthaldehyde 10
This compound was prepared in two steps as follows. First step: A solution of compound 9 (0.147 g, 0.41 mmol), NBS (0.404 g, 2.25 mmol), and benzoylperoxide (0.003 g) in CCl4 (4 mL) was refluxed overnight under a 150 W lamp. An additional portion of NBS (0.073 g, 0.41 mmol) was then added, and stirring was continued for 4 h. The suspension was filtered when still hot, and the filtrate was concentrated under vacuum. The residue was filtered through silica gel with a 80 : 20 hexanes : Et2O mixture as eluant to give the product (0.245 g, ) that was used without further purification in the following step. Second step: To a stirred, refluxing solution of the tetrabromide (0.387 g, 0.57 mmol) in dioxane (15 mL), a solution of silver nitrate (0.585 g, 3.44 mmol) in water (3.0 mL) was added dropwise. The resulting mixture was stirred at reflux overnight, and the precipitated silver bromide was then removed by filtration of the hot suspension. The filtrate was concentrated under vacuum, the residue was taken up into THF (7 mL), and a 10% aqueous solution of HCl (5 mL) was then added. The mixture was refluxed for 1 h, cooled down to RT, and extracted with Et2O (3 × 20 mL). The combined organic phases were dried over NaSO4, filtered, and concentrated under vacuum. The residue was dissolved in CH2Cl2 (20 mL) and anhydrous magnesium sulfate (1 g) was added. The suspension was then refluxed for 2 h under a Dean–Stark apparatus containing molecular sieves. The cooled solution was then concentrated under vacuum and the residue was purified by flash chromatography with a 70 : 30 hexanes : Et2O mixture as eluant. The product, a pale yellow oil that becomes a waxeous solid when digested with pentane, was obtained in 70% yield (0.154 g). IR (thin film): 3050, 1692, 1620, cm−1. 1H NMR: δ 9.79 (s, 1H, CHO of phenyl group), 9.33 (s, 1H, CHO of naphthalene), 8.30 (dd, J = 8.0, 1.2 Hz, 1H, hydrogen ortho to I), 8.16 (dd, J = 8.0, 1.2 Hz, 1H, hydrogen para to I), 8.11 (AB system, J = 8.0 Hz, 2H, H–C3 and H–C4 of naphthalene), 8.00 (dd, J = 7.9, 1.2 Hz, 1H, H–C5), 7.67 (dt, J = 7.9, 1.2 Hz, 1H, H–C6 of naphthalene), 7.49 (dt, J = 7.9, 1.2 Hz, 1H, H–C7 of naphthalene), 7.45 (t, J = 8.0 Hz, 1H, hydrogen meta to I), 7.25 (dd, J = 7.9, 1.2 Hz, 1H, H–C8 of naphthalene); 13C NMR: δ 190.7, 190.4, 144.5, 143.7, 143.4, 137.5, 136.0, 132.0, 131.8, 130.7, 130.0, 129.4, 128.7, 128.0, 127.9, 126.4, 122.8, 103.5. C18H11I O2 requires: C, 55.98; H, 2.87%; found: C, 56.18; H, 2.78%.trans-5-Iodo-9,10-dihydroxy-9,10-dihydrobenzo[c]phenanthrene 11
To a solution of compound 10 (0.165 g, 0.43 mmol) in dry THF (7 mL) cooled at −78 °C and kept under a nitrogen atmosphere, a freshly opened 0.1 M solution of samarium diiodide in THF (9 mL) was added by syringe. After 30 min stirring at −78 °C, an additional 9 mL aliquot of samarium diiodide solution was added to the yellow solution, whereupon the color of the mixture became deep blue. Stirring was continued for 30 min at −78 °C and the reaction was quenched by the addition of a 10% aqueous solution of HCl (5 mL). After addition of Et2O (20 mL) the organic phase was separated, washed with a saturated aqueous solution of Na2SO3 (10 mL), dried over NaSO4, filtered, and concentrated under vacuum. The residue was purified by flash chromatography first with 40 : 60 and then with 20 : 80 hexanes : Et2O mixtures as eluants. The product, mp 155–157 °C, was obtained in 75% yield (0.125 g). IR (CH2Cl2): 3650, 3055, 2960, 1610, 1425, 1265 cm−1. 1H NMR: δ 7.84 (dd, J = 7.8, 1.2 Hz, 1H, H–C8 of naphthalene), 7.80 (d, J = 8.1 Hz, 1H, hydrogen ortho to I), 7.74 (br s, 2H, H–C4 and H–C3 of naphthalene), 7.71 (dd, J = 7.8, 1.2 Hz, 1H, H–C5 of naphthalene), 7.64 (d, J = 8.1 Hz, 1H, hydrogen para to I), 7.33–7.25 (m, 2H, H–C6 and H–C7 of naphthalene), 6.95 (t, J = 8.1 Hz, 1H, hydrogen meta to I), 4.44 (B part of AB system, J = 10.6 Hz, 1H, CH–OH bound to naphthalene), 4.23 (A part of AB system, J = 10.6 Hz, 1H, CH–OH bound to phenyl); 13C NMR: δ 142.2, 139.9, 137.6, 136.3, 133.5, 130.2, 129.2, 129.2, 128.5, 128.4, 128.0, 125.5, 125.3, 124.3, 121.9, 96.8, 74.1, 74.0. C18H13I O2 requires: C, 55.69; H, 3.37%; found: C, 55.78; H, 3.41%.Synthesis of diols 1–3, 12 and diketones 4–6
trans-5-(4-Methoxyphenyl)-9,10-dihydroxy-9,10-dihydrobenzo[c]phenanthrene 1 and 5-(4-methoxyphenyl)benzo[c]phenanthrene-9,10-dione 4
To a solution of iodo compound 11 (0.065 g, 0.167 mmol) and 4-methoxyphenyltrimethylstannane (0.204 g, 0.713 mmol) in dry, degassed DMF (5 mL) kept under nitrogen, Pd2(dba)3 (0.0345 g, 0.0334 mmol) and AsPh3 (0.041 g, 0.1336 mmol), both dissolved in degassed DMF (1 mL), were added in this order. The resulting mixture was stirred at reflux for 40 h. Most of the solvent was then evaporated under vacuum and the residue was dissolved in Et2O (15 mL). The solution was washed with water (4 × 5 mL) to remove the remaining DMF, and the organic phase was dried over NaSO4, filtered, and concentrated under vacuum. The residue was purified by flash chromatography with pentane : Et2O mixtures of increasing polarity as eluants (first eluant: 90 : 10; for diketone 4 isolation: 70 : 30; for diol 1 isolation: 30 : 70). Analytically pure samples were obtained by gravity chromatography of the diol and diketone compounds using flash chromatography silica gel as the stationary phase and the same eluants. Diol 1, a white solid with mp 178–180 °C (decomp.) was obtained in 29% yield (0.0178 g). IR (CH2Cl2): 3650, 3054, 2987, 1422, 1264 cm−1. 1H NMR: δ 7.90–7.83 (m, 2H, H–C1 and H–C2), 7.73 (dd, J = 8.5, 1.3 Hz, 1H, H–C8), 7.64 (d, J = 8.4 Hz, 1H, H–C3′), 7.56–7.52 (m, 3H, H–C4′, H–C6, and H–C7), 7.16 (t, J = 8.4 Hz, 1H, H–C3″), 6.93 (dt, J = 8.4, 1.3 Hz, 1H, H–C4″), 6.91 (d, J = 8.9 Hz, 2H, hydrogen meta to methoxy in phenyl ring), 6.51 (d, J = 8.9 Hz, 2H, hydrogen ortho to methoxy in phenyl ring), 4.83 (B part of AB system, J = 10.5 Hz, 1H, H–C10), 4.58 (A part of AB system, J = 10.5 Hz, 1H, H–C9), 3.64 (s, 3H, MeO); 13C NMR: δ 158.5, 140.5, 139.0, 135.7, 135.0, 133.2, 130.5, 130.0, 129.9, 128.7, 128.6, 128.5, 128.2, 127.3, 126.5, 124.9, 124.8, 121.9, 120.7, 113.5, 74.8, 74.6, 55.1. C25H20O3 requires: C, 81.50; H, 5.47%; found: C, 81.37; H, 5.38%.Diketone 4, a red solid with mp 169–171 °C was obtained in 24% yield (0.0146 g). IR (CH2Cl2): 3040, 2964, 1682, 1514, 1252, 1031 cm−1. 1H NMR: δ 8.13 (dd, J = 8.5, 1.3 Hz, 1H, H–C8), 8.05 (B part of an AB system, J = 8.4 Hz, 1H, H–C1), 7.83 (dd, J = 8.5, 1.3 Hz, 1H, H–C6), 7.81 (B part of an AB system, J = 8.4 Hz, 1H, H–C2), 7.68 (dd, J = 8.4, 1.2 Hz, 1H, H–C4′), 7.62 (d, J = 8.4 Hz, 1H, H–C3′), 7.60 (t, J = 8.4 Hz, 1H, H–C7), 7.32 (dt, J = 8.4, 1.0 Hz, H–C3″), 7.00 (dt, J = 8.4, 1.0 Hz, H–C4″), 6.92 (broad multiplet, 2H, hydrogens meta to MeO in phenyl ring), 6.55 (d, J = 8.9 Hz, 2H, hydrogens ortho to methoxy in phenyl ring), 3.66 (s, 3H, MeO); 13C NMR: δ 184.0, 159.8, 142.7, 139.2, 138.2, 137.2, 135.0, 133.5, 131.9, 130.2, 130.0, 129.8, 129.4, 128.9, 128.8, 128.3, 127.7, 127.5, 126.1, 122.6, 113.2, 55.2. C25H16O3 requires: C, 82.40; H, 4.43%; found: C, 82.37; H, 4.58%.
trans-5-Phenyl--9,10-dihydroxy-9,10-dihydrobenzo[c]phenanthrene 2
A white solid with mp 180–183 °C, was obtained in 12% yield. IR (CH2Cl2): 3655, 3055, 2986, 1422, 1264 cm−1. 1H NMR: δ 7.92–7.82 (m, 2H, H–C1 and H–C2), 7.77 (d, J = 8.5 Hz, 1H, H–C8), 7.66 (d, J = 8.4 Hz, 1H, H–C3′), 7.60–7.50 (m, 3H, H–C4″, H–C6 and H–C7), 7.15 (t, J = 8.4 Hz, 1H, H–C3″), 7.00–6.93 (broad multiplet, 5H of phenyl ring), 6.90 (t, J = 8.4 Hz, 1H, H–C4″), 4.87 (B part of AB system, J = 10.5 Hz, 1H, H–C10), 4.60 (A part of AB system, J = 10.5 Hz, 1H, H–C9); 13C NMR: δ 142.5, 140.5, 139.2, 135.5, 133.0, 130.7, 130.0, 128.9, 128.8, 128.7, 128.6, 128.2, 128.0, 127.9, 127.8, 126.3, 124.9, 124.8, 122.4, 120.8, 74.6, 74.5. C24H18O2 requires: C, 85.18; H, 5.36%; found: C, 85.41; H, 5.29%.5-Phenylbenzo[c]phenanthrene-9,10-dione 6
An orange solid with mp 138–140 °C (decomp.) was obtained in 11% yield. IR (CH2Cl2): 3050, 2960, 1682, 1510, 1250, 1025 cm−1. 1H NMR: δ 8.15 (dd, J = 8.0, 1.4 Hz, 1H, H–C8), 8.07 (B part of an AB system, J = 8.5 Hz, 1H, H–C1), 7.86 (dd, J = 8.0, 1.4 Hz, 1H, H–C6), 7.81 (A part of an AB system, J = 8.5 Hz, 1H, H–C2), 7.68 (dd, J = 8.4, 1.0 Hz, 1H, H–C4′), 7.63 (t, J = 8.0 Hz, 1H, H–C7), 7.62 (d, J = 8.0 Hz, 1H, H–C3′), 7.29 (dt, J = 8.0, 1.0 Hz, 1H, H–C3″), 7.07–7.00 (broad multiplet, 5H of phenyl ring), 6.97 (dt, J = 8.0, 1.0 Hz, 1H, H–C4″); 13C NMR: δ 184.0, 143.5, 140.0, 139.2, 138.4, 137.5, 135.9, 132.5, 129.9, 129.8, 129.4, 129.1, 129.0, 128.9, 128.7, 128.4, 127.7, 127.4, 127.2, 126.2, 122.6. C24H14O2 requires: C, 86.21; H, 4.22%; found: C, 85.99; H, 4.39%.trans-5-(4-Nitrophenyl)--9,10-dihydroxy-9,10-dihydrobenzo[c]phenanthrene 3
A red solid with mp >200 °C was obtained in 11% yield. IR (CH2Cl2): 3650, 3053, 2986, 1423, 1266, 1045 cm−1. 1H NMR: δ 7.93–7.89 (m, 2H, H–C1 and H–C2), 7.88 (d, J = 8.5 Hz, H–C8), 7.83 (d, J = 8.8 Hz, 2H, hydrogens ortho to NO2 in phenyl ring), 7.67 (d, J = 8.1 Hz, H–C3′), 7.60 (t, J = 8.5 Hz, 1H, H–C7), 7.53 (d, 8.5 Hz, 1H, H–C6), 7.45 (d, J = 8.1 Hz, 1H, H–C4′), 7.17–7.12 (m, 3H, H–C3″ and hydrogens meta to NO2 in phenyl ring), 6.92 (t, J = 8.1 Hz, 1H, H–C4″), 4.86 (B part of AB system, J = 10.5 Hz, 1H, H–C10), 4.62 (A part of AB system, J = 10.5 Hz, 1H, H–C9); 13C NMR: δ 149.5, 146.2, 140.5, 140.0, 136.2, 133.4, 130.5, 130.3, 129.5, 129.3, 128.5, 128.1, 128.0, 125.7, 125.3, 125.2, 124.2, 124.1, 123.2, 121.1, 74.6, 74.5. C24H17NO4 requires: C, 75.20; H, 4.47; N, 3.65%; found: C, 75.49; H, 4.56; N, 3.59%.5-(4-Methylphenyl)benzo[c]phenanthrene-9,10-dione 5
An orange solid with mp 115–117 °C was obtained in 15% yield. IR (CH2Cl2): 3055, 2988, 1684, 1488, 1266 cm−1. 1H NMR: δ 8.12 (dd, J = 8.5, 1.3 Hz, 1H, H–C8), 8.05 (B part of an AB system, J = 8.4 Hz, 1H, H–C1), 7.82 (dd, J = 8.5, 1.3 Hz, 1H, H–C6), 7.80 (A part of an AB system, J = 8.5 Hz, 1H, H–C2), 7.68 (d, J = 8.4 Hz, 1H, H–C4′), 7.63 (d, J = 8.4 Hz, 1H, H–C3′), 7.60 (t, J = 8.4 Hz, 1H, H–C7), 7.28 (dt, J = 8.4, 1.0 Hz, H–C3″), 6.97 (dt, J = 8.4, 1.0 Hz, H–C4″), 6.88 (broad multiplet, 2H, hydrogens meta to Me in phenyl ring), 6.80 (A part of an AB system, J = 8.8 Hz, 2H, hydrogens ortho to Me in phenyl ring), 2.13 (s, 3H, Me in phenyl ring); 13C NMR: δ 184.0, 143.0, 139.2, 138.5, 138.3, 137.5, 137.1, 135.2, 131.9, 129.9, 129.8, 129.4, 129.1, 129.0, 128.9, 128.8, 128.5, 127.7, 127.5, 126.1, 122.6, 22.2. C25H16O2 requires: C, 86.19; H, 4.63%; found: C, 86.28; H, 4.66%.5-(4-Fluorophenyl)benzo[c]phenanthrene-9,10-dione 7
The inseparable mixture of crude diketone 7 and diol 12, obtained as described above, was subjected to the following procedure: To a stirred mixture of 7 and 12 (0.016 g, approximately 0.045 mmol) in dry DMSO (0.5 mL) kept at RT under nitrogen, triethylamine (0.022 mL, 0.16 mmol) was added, followed by Py·SO3 (0.0178 g, 0.112 mmol) in 0.5 mL of dry DMSO. After 5 h stirring at RT, the reaction was quenched by the addition of water (5 mL), and the solution was extracted with Et2O (5 × 5 mL). The diethyl ether solution was washed with water (5 mL) to remove the remaining DMSO, and the organic phase was dried over NaSO4, filtered, and concentrated under vacuum. The residue was purified by flash chromatography with pentane : Et2O mixtures of increasing polarity as eluants (first eluant: 90 : 10; for diketone isolation: 80 : 20). Compound 7, a red solid with mp 93–95 °C was obtained in 12% overall yield from 11. IR (CH2Cl2): 3050, 2986, 1683, 1485, 1266 cm−1. 1H NMR: δ 8.15 (dd, J = 7.8, 1.3 Hz, 1H, H–C8), 8.07 (B part of an AB system, J = 8.5 Hz, 1H, H–C1), 7.83 (A part of an AB system, J = 8.5 Hz, 1H, H–C2), 7.81 (dd, J = 7.8, 1.3 Hz, 1H, H–C6), 7.68–7.64 (m, 2H, H–C3′ and H–C4′), 7.63 (t, J = 7.8 Hz, 1H, H–C7), 7.34 (t, J = 8.9 Hz, H–C3″), 7.01 (t, J = 8.9 Hz, H–C4″), 6.97 (broad multiplet, 2H, hydrogen meta to F in phenyl ring), 6.72 (t, J = 8.0 Hz, 2H, hydrogens ortho to F in phenyl ring); 13C NMR: δ 184.0, 142.3, 142.2, 139.2, 138.1, 137.8, 137.5, 135.7, 132.1, 130.8, 130.1, 130.0, 129.5, 129.4, 129.0, 128.8, 127.9, 127.3, 126.3, 122.7, 115.4; 19F NMR: δ 117.0. C24H13FO2 requires: C, 81.81; H, 3.72%; found: C, 81.59; H, 3.86%.2D EXSY Experiments
For all the 2D EXSY experiments, the samples were dissolved in THF-d8 and were degassed by several freeze–thaw cycles on a high vacuum line to remove dissolved oxygen. The spectra were recorded at ≤210 K temperature. Pure absorption 2D EXSY spectra were recorded employing the following parameters: spectral width 300 Hz; 256 time increments of 64 transients each; mixing times of 0.8 and 1 s for compounds, 1–3 and 4–7, respectively; relaxation delays of 2.0 s (ca. 3 T1 values of the relevant protons at the EXSY temperature).Acknowledgements
National Science Foundation (CHE-9904275) and CNR-Progetto Strategico provided support for this work.References and Notes
- (a) C. A. Hunter, Angew. Chem. Int. Ed. Engl., 1993, 32, 1584–1586 CrossRef; (b) C. A. Hunter, Chem. Soc. Rev., 1994, 101–109 RSC.
- (a) F. Diederich, Angew. Chem. Int. Ed. Engl., 1988, 27, 362–386 CrossRef; (b) D. B. Smithrud, T. B. Wyman and F. Diederich, J. Am. Chem. Soc., 1991, 113, 5420–5426 CrossRef CAS and references therein.; (c) M. M. Conn, G. Deslongchamps, J. de Mendoza and J. Rebek, J. Am. Chem. Soc., 1993, 115, 3458–3557 CrossRef; (d) D. Philip and J. F. Stoddardt, Angew. Chem. Int. Ed. Engl., 1996, 35, 1154–1196 CrossRef; (e) A. V. Muehldorf, D. Van Egen, J. C. Warner and A. D. Hamilton, J. Am. Chem. Soc., 1988, 110, 6561–6562 CrossRef CAS; (f) S. C. Zimmermann, Z. Zeng, W. Wu and D. E. Reichert, J. Am. Chem. Soc., 1991, 113, 183–196 CrossRef; (g) L. T. Newcomb and S. H. Gellman, J. Am. Chem. Soc., 1994, 116, 4993–4994 CrossRef CAS; (h) J. E. Cochran, T. J. Parrott, B. J. Whitlock and H. W. Whitlock, J. Am. Chem. Soc., 1992, 114, 2269–2270 CrossRef CAS; (i) E. C. Breinlinger and V. M. Rotello, J. Am. Chem. Soc., 1997, 119, 1165–1166 CrossRef CAS.
- Review: G. B. Jones, Tetrahedron, 2001, 57, 7999–8016 Search PubMed.
- (a) W. Saenger, Principles of Nucleic Acid Structures, Springer Verlag, New York, 1984 Search PubMed; (b) S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CAS; (c) C. A. Hunter, J. Singh and J. M. Thornton, J. Mol. Biol., 1991, 218, 837–846 CrossRef CAS; (d) C. A. Hunter, J. Mol. Biol., 1993, 230, 1025–1054 CrossRef CAS; (e) O. F. Schall and G. W. Gokel, J. Org. Chem., 1996, 61, 1149–1458 CrossRef CAS; (f) K. M. Guckian, B. A. Schweitzer, R. X. F. Ren, C. J. Sheils, P. L. Paris, D. C. Tanmasseri and E. T. Kool, J. Am. Chem. Soc., 1996, 118, 8182–8183 CrossRef CAS; (g) T. L. Ho, P. Y. Liao and K. T. Wang, J. Chem. Soc., Chem. Commun., 1995, 2437–2438 RSC; (h) D. Ranganathan, V. Haridas, R. Gilardi and J. L. Karle, J. Am. Chem. Soc., 1998, 120, 10793–10800 CrossRef CAS; (i) F. Chelli, F. L. Gervasio, P. Procacci and V. Schettino, J. Am. Chem. Soc., 2002, 124, 6133–6143 CrossRef and references therein.; (j) F. Chelli, F. L. Gervasio, P. Procacci and V. Schettino, Proteins, 2002, 48, 117–125 CrossRef.
- (a) C. A. Hunter, M. N. Meah and J.K.M. Sanders, J. Am. Chem. Soc., 1990, 112, 5773–5780 CrossRef; (b) A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019–1027 CrossRef CAS and references therein.; (c) G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem, Soc., 1998, 120, 3641–3649 CrossRef CAS.
- (a) T. Dahl, Acta Chem. Scand., 1994, 48, 95–106 Search PubMed; (b) J. H. Williams, Acc. Chem. Res., 1993, 26, 593–598 CrossRef CAS; (c) H. L. Anderson, A. Bashall, K. Henrick, M. McPartlin and J. K. M. Sanders, Angew. Chem., Int. Ed. Engl., 1994, 33, 429–431 CrossRef; (d) I. Dance and M. Scuddar, Chem. Eur. J., 1996, 2, 481–486 CrossRef CAS; (e) Z. H. Wang, T. Hirose, K. Hiratani, Y. Yang and K. Kasuga, Chem. Lett., 1996, 603–604 CAS; (f) C. B. Martin, B. O. Patrick and A. Cammers-Goodwin, J. Org. Chem., 1999, 64, 7568–7578 CrossRef CAS; (g) Y. Nakamura, H. Suzuki, Y. Hayashida, T. Kudo and J. Nishimura, Liebigs Ann./Recueil, 1997, 1769–1776 Search PubMed.
- (a) W. L. Jorgensen and D. L. Severance, J. Am. Chem. Soc., 1990, 112, 4768–4774 CrossRef CAS; (b) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS; (c) P. Linse, J. Am. Chem. Soc., 1992, 114, 4366–4373 CrossRef CAS; (d) P. Hobza, H. L. Selzle and E. W. Schlag, J. Am. Chem. Soc., 1994, 116, 3500–3506 CrossRef CAS; (e) V. E. Williams, R. P. Lemieux and G. R. Thatcher, J. Org. Chem., 1996, 61, 1927–1933 CrossRef CAS; (f) C. Chipot, R. Jaffe, B. Maigret, D. A. Pearlman and P. A. Kollman, J. Am. Chem. Soc., 1996, 118, 11217–11224 CrossRef CAS; (g) J. M. Seminario, A. G. Zacarias and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 3970–3974 CrossRef CAS; (h) S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc, 2002, 124, 104–112 CrossRef CAS; (i) M. Claessens, L. Palombini, M. L. Stein and J. Reisse, Nouv. J. Chem., 1982, 6, 595 Search PubMed; (j) M. O. Sinnokrot, E. F. Valeev, C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, in press Search PubMed.
- The preference for the edge-to-face orientation has been demonstrated for gaseous, liquid, and crystalline benzene: (a) X. Shi and L. S. Bartell, J. Chem. Phys., 1988, 92, 5667–5673 CAS; (b) L. J. Lowden and D. J. Chandler, J. Chem. Phys., 1974, 61, 5228–5241 CrossRef CAS; (c) D. Hall and D. E. Williams, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1975, 31, 56–58 CrossRef and references therein.
- (a) F. Cozzi, M. Cinquini, R. Annunziata, T. Dwyer and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 5729–5733 CrossRef CAS; (b) F. Cozzi, M. Cinquini, R. Annunziata and J. S. Siegel, J. Am. Chem. Soc., 1993, 115, 5330–5331 CrossRef CAS; (c) F. Cozzi, F. Ponzini, R. Annunziata, M. Cinquini and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1995, 34, 1019–1020 CrossRef CAS; (d) F. Cozzi and J. S. Siegel, Pure Appl. Chem., 1995, 67, 683–689 CAS.
- (a) J. A. Zoltewicz, N. A. Maier and W. M. F. Fablan, J. Org. Chem., 1998, 63, 4985–4990 CrossRef CAS and references therein; (b) N. Kaneta, F. Mitamura, M. Uemura, Y. Murata and K. Komatsu, Tetrahedron Lett., 1996, 37, 5835–5838 CrossRef CAS.
- (a) C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CAS; (b) J. H. Williams, J. K. Cockroft and A. N. Fitch, Angew. Chem., Int. Ed. Engl., 1992, 31, 1655–1657 CrossRef; (c) R. E. Gillard, J. F. Stoddardt, A. J. P. White, B. J. Williams and D. J. Williams, J. Org. Chem., 1996, 61, 4504–4505 CrossRef CAS; (d) G. W. Coates, A. B. Dunn, L. M. Henling, D. A. Doughety and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1997, 36, 248–251 CrossRef CAS; (e) F. Ponzini, R. Zagha, K. Hardcastle and J. S. Siegel, Angew. Chem., Int. Ed., 2000, 39, 2323–2325 CrossRef CAS; (f) M. T. Messina, P. Metrangolo, S. Pappalardo, M.F. Parisi, T. Pilati and G. Resnati, Chem. Eur. J., 2000, 6, 3495–3500 CrossRef CAS; (g) C. Y. Kim, P. P. Chandra, A. Jain and D. W. Christianson, J. Am. Chem. Soc., 2001, 123, 9620–9627 CrossRef CAS and references therein.
- (a) F. J. Carver, C. A. Hunter and E. M. Seward, J. Chem. Soc., Chem. Commun., 1998, 775–776 RSC; (b) F. J. Carver, C. A. Hunter, D. J. Livingstone, J. F. McCabe and E. M. Seward, Chem. Eur. J., 2002, 8, 2847–2859 CrossRef CAS and references therein; (c) E. Kim, S. Paliwal and C. S. Wilcox, J. Am. Chem. Soc., 1998, 120, 11192–11193 CrossRef CAS and references therein; (d) K. Nakamura and K. N. Houk, Org. Lett., 1999, 1, 2049–2051 CrossRef CAS; (e) Review: W. B. Jennings, B. M. Farrell and J. F. Malone, Acc. Chem. Res., 2001, 34, 885–894 Search PubMed.
- For recent examples of molecular recognition between arenes based on an edge-to-face interaction, see: (a) A. P. Poisson and C. A. Hunter, J. Chem. Soc., Chem. Commun., 1996, 1723–1724 RSC; (b) M.J. Cloninger and H.W. Whitlock, J. Org. Chem., 1998, 63, 6153–6159 CrossRef CAS; (c) S. Superchi, M.I. Donnoli, G. Proni, G.P. Spada and C. Rosini, J. Org. Chem., 1999, 64, 4762–4767. CrossRef CAS For the influence exerted by the edge-to-face interactions on the relative orientation of different arenes, see; (d) A. Garcia-Martinez, J. Osio Barcina, A. de Fresno Cerezo and R. Gutierrez Rivas, J. Am. Chem. Soc., 1998, 120, 673–679 CrossRef; (e) B. Uno, T. Iwamoto and N. Okumura, J. Org. Chem., 1998, 63, 9794–9800 CrossRef CAS; (f) S. V. Lindeman, D. Kosynkin and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 13268–13269 CrossRef CAS; (g) For a review on the CH–π interactions, see: M. Nishio, Y. Umezawa, M. Hirota and Y. Takeuchi, Tetrahedron, 1995, 51, 8665–8701 Search PubMed.
- See for instance: P.R. Ashton, D. Joachimi, N. Spencer, J. F. Stoddardt, C. Tschierske, A. J. P. White, D. J. Williams and K. Zab, Angew. Chem., Int. Ed. Engl., 1994, 33, 1503–1506 Search PubMed.
- C. B. Martin, H. R. Mulla, P. G. Willis and A. Cammers-Goodwin, J. Org. Chem., 1999, 64, 7802–7806 CrossRef CAS.
- M. J. Rashkin and M. L. Waters, J. Am. Chem. Soc., 2002, 124, 1860–1861 CrossRef CAS.
- 1-Bromo-2-methylnaphthalene can be used in excess because it produces little of the homocoupling product under these conditions..
- J. E. McMurry, Chem. Rev., 1989, 89, 1513–1524 CrossRef CAS.
- R. G. R. Bacon and W. S. Lindsay, J. Chem. Soc., 1958, 1375–1381 RSC.
- E. Abu-Shagra and J. Blum, J. Heterocycl. Chem., 1991, 28, 473–475 Search PubMed.
- G.A. Molander and C.R. Harris, Chem. Rev., 1996, 96, 307–338 CrossRef CAS.
- For similar results see: (a) K. Ohmori, M. Kitamura and K. Suzuki, Angew. Chem., Int. Ed., 1999, 38, 1226–1229 CrossRef CAS; (b) N. Taniguchi, T. Hata and M. Uemura, Angew. Chem., Int. Ed., 1999, 38, 1232–1235 CrossRef CAS; (c) R. Annunziata, M. Benaglia, M. Cinquini and L. Raimondi, Eur. J. Org. Chem., 1999, 3369–3374 CrossRef CAS.
- M. Benaglia, S. Toyota, C. R. Woods and J. S. Siegel, Tetrahedron Lett., 1997, 38, 4737–4740 CrossRef CAS.
- (a) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef; (b) V. Farina, Pure Appl. Chem., 1996, 68, 73–78 CAS and references therein.
- A modification of the published procedure was used: R. G. Harvey, S. H. Goh and C. Cortez, J. Am. Chem. Soc., 1975, 97, 3468–3479 Search PubMed.
- For the rotation around the aryl–aryl bond in 1-phenylbenzo[c]phenanthrene derivatives a ΔG≠ of ca. 54 kJ mol−1 was determined; the enantiomerization of the benzo[c]phenanthrene skeleton requires a ΔG≠ of ca. 67 kJ mol−1: W. H. Laarhoven, W. H. M. Peters and A. H. A. Tinnemans, Tetrahedron, 1978, 34, 769–777 Search PubMed . In molecules related to diols 1–3 this process requires even higher energy (see ref. 22a).
- For a review on the use of 2D EXSY NMR spectroscopy to measure chemical dynamics, see: C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 Search PubMed.
- Because of the logarithmic relationship between k and ΔG≠, the major contribution to the error in determining ΔG≠ is the error in the temperature. All spectra were measured with the same instrumentation at similar temperatures. A liberal estimate of the error is 1 kJ mol−1.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. J. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle, J. A. Pople, Gaussian 98, Revision A.6, Pittsburgh PA, 1998 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS . Structural computations were performed using the hybrid density functional method, B3LYP; Becke's 3 parameter functional plus the nonlocal correlation of Lee-Yang-Parr with both local and nonlocal terms. Dunning's DZV(2d,p) double-ζ valence basis set was employed. Single point energy computations were performed on optimized structures using MP2 dynamic correlation treatment for superior energetic analysis.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–792 CrossRef CAS; (b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–207 CrossRef CAS.
- T. H. Dunning, Jr., P. J. Hay, Modern Theoretical Chemistry; Plenum, New York, 1976, pp. 1–28 Search PubMed.
- J. M. L. Martin, Chem. Phys. Lett, 1996, 262, 97–104 CrossRef CAS.
- C. Moller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |