Catalytic asymmetric Henry reactions of silyl nitronates with aldehydes
Tine Risgaard, Kurt V. Gothelf and Karl Anker Jørgensen*
Danish National Research Foundation: Center for Catalysis, Department of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: kaj@chem.au.dk; Fax: 45 8619 6199; Tel: 45 8942 3910
First published on 3rd December 2002
Abstract
A catalytic enantioselective Henry reaction of silyl nitronates with aldehydes has been developed. Different chiral Lewis acids have been tested for the reaction and it has been found that a variety of chiral copper–ligand complexes can catalyze the Henry reaction. The best yield, diastereo- and enantioselectivity of the nitroalcohols formed are obtained by the application of a copper(II)–diphenyl–bisoxazoline complex as the catalyst in the presence of tetrabutylammonium triphenylsilyldifluorosilicate (TBAT). In order to minimize the epimerization of the nitroaldol products they were converted into the corresponding Mosher esters. The reaction proceeds well for different aromatic aldehydes reacting with alkyl nitronates.
Introduction
β-Hydroxynitroalkanes, prepared by the Henry (nitroaldol) reaction, are valuable synthetic intermediates in organic synthesis.1The development of catalytic enantioselective versions of the nitroaldol reaction has recently been in focus. Sibasaki et al. have shown that the use of heterobimetallic complexes with lanthanide BINOL systems provide an efficient catalyst for the Henry reaction of nitromethane with different types of aldehydes.2 Catalytic enantioselective nitroaldol reactions of nitromethane with aldehydes have also been developed by Trost et al. by the use of a chiral dinuclear catalyst to obtain impressive enantioselectivities.3 Our approach to catalytic enantioselective nitroaldol reactions has been the development of the highly enantioselective addition of nitromethane to α-ketoesters catalyzed by a chiral tert-butyl bisoxazoline copper(II) complex.4 This turned out to be a quite general reaction for many different α-ketoesters and provided an easy entry to optically active β-nitro-α-hydroxy esters and β-amino-α-hydroxy esters.
One of the drawbacks for the catalytic enantioselective nitroaldol reaction developed is that it is mainly restricted to nitromethane acting as the nucleophile, with the exception of the lanthanide–lithium–BINOL complex which can also catalyse the reaction of alkyl aldehydes with other nitroalkanes.2c
An alternative approach to the Henry reaction is the application of silyl nitronates 1 instead of nitro compounds for the addition to aldehydes 2 (Scheme 1). Seebach et al. have described the diastereoselective fluoride catalyzed addition of silyl nitronates to aldehydes.5 Recently, we described the asymmetric addition of silyl nitronates to imines in the presence of catalytic amounts of chiral Lewis acids.6
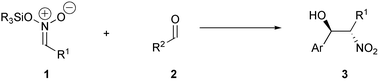 | ||
| Scheme 1 The silyl nitronate Henry reaction. | ||
In this paper we present the development of a catalytic diastereo- and enantioselective nitroaldol reaction of nitronates 1 with aldehydes 2 in the presence of chiral Lewis complexes.
Results and discussion
The nitroaldol reaction of the nitronate 1a with benzaldehyde 2a proceeds well in the presence of different chiral Lewis acid complexes as the catalyst and the nitroalcohol 3a can be obtained (Scheme 2). However, the isolation and characterization of the nitroalcohol 3a obtained in the present type of reaction turned out to be tedious, since this compound is unstable and tends to epimerize and undergo a retro-Henry reaction.7 Thus compound 3a was subsequently converted into the Mosher ester 5a by reaction with (S)-α-methoxy-α-trifluoromethylphenylacetyl chloride ((S)-MTPA-Cl), 4. The Mosher esters 5 are stable compounds and were isolated. The enantiomeric excess7 of the nitroaldol reaction was determined from the diastereomeric excess of the Mosher esters.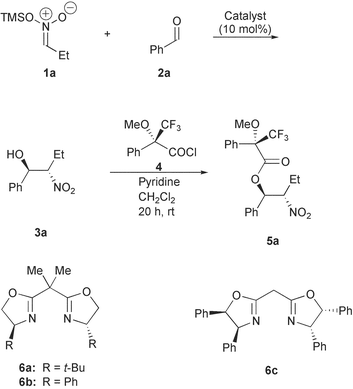 | ||
| Scheme 2 Catalytic diastereo- and enantioselective nitroaldol reaction of nitronates 1a with aldehydes 2a, and subsequent conversion into the Mosher ester 5a. | ||
Among a series of different chiral ligands BINAP and 1,2-bis((2S,5S)-2,5-dimethylphospholano)benzene (DUPHOS) (P,P-chelating), bisoxazolines (BOX) and Py–BOX (N,N-chelating), and phosphinoxazoline (N,P-chelating) and dibenzofuradiylphenyloxazoline (DBFOX) the BOX ligands 6a–c gave the most promising results.
In Table 1 is presented some of the screening results for the nitroaldol reaction of nitronate 1a with benzaldehyde 2a catalyzed by different copper salts and applying the BOX ligands 6a–c under various reaction conditions.
| Entry | Catalyst | Solvent | Conv. (%) | Dr threo : erythro | Eeathreo(%) |
|---|---|---|---|---|---|
| a The ee of the nitroaldol reaction is derived from the de of the Mosher ester.b 20 mol% of the catalyst was applied in the presence of 20 mol% tetrabutylammonium triphenylsilyldifluorosilicate (TBAT) and the reaction was performed at room temperature. | |||||
| 1 | 6a-Cu(OTf)2 | THF | 51 | 3 : 1 | 19 |
| 2 | 6b-Cu(OTf)2 | THF | 66 | 3.5 : 1 | 0 |
| 3 | 6c-Cu(OTf)2 | THF | 70 | 1.2 : 1 | 66 |
| 4 | 6c-Cu(OTf)2 | CH2Cl2 | 73 | 1.1 : 1 | 35 |
| 5 | 6c-Cu(OTf)2 | Et2O | 68 | 1.2 : 1 | 56 |
| 6 | 6c-Cu(SbF6)2 | CH2Cl2 | 73 | 1.1 : 1 | 35 |
| 7 | 6c-CuPF6 | THF | 70 | 1.5 : 1 | 20 |
| 8 | 6c-CuClO4 | THF | 69 | 1 : 1 | 30 |
| 9 | 6c-Cu(OTf)2b | THF | 94 | 5 : 1 | 65 |
Among the Lewis acids tested for the reaction both copper(I) and copper(II) salts showed the most promising properties, as a good conversion was found with these salts. The results in Table 1 for the reaction in the presence of the chiral BOX ligands 6a–c show that the corresponding Mosher ester is obtained with a low threo : erythro ratio, but as it appears from the results this selectivity is very dependent of both the ligand and the solvent. The highest enantiomeric excess is obtained when applying the bis[(4R,5S)-diphenyloxazoline] ligand 6c in combination with Cu(II) salts where up to 66% ee is found (entry 3). The reaction presented in entry 3, Table 1 has also been performed at 0 °C and room temperature, and under these conditions the conversion was 85% and 94%, the diastereomeric ratio 2 : 1 and 5 : 1, respectively, and the enantioselectivity was in both cases 65% ee of threo-5a. The results obtained for the reaction in the presence of 6c–CuClO4 were not promising (entry 8). When the reaction was performed in the presence of 20 mol% of 6c–Cu(TfO)2 and the fluoride donor tetrabutylammonium triphenylsilyldifluorosilicate (TBAT),8 the reaction proceeded at room temperature and the results were improved with respect to both conversion, threo-selectivity and enantioselectivity (entry 9). The enantiomeric excess of the erythro-isomer was in all cases lower than that of the obtained threo-isomer.
To develop this reaction for other silyl nitronates and aldehydes the nitroaldol reaction was performed using nitronates 1a,b and aldehydes 2a–g in the presence of 6c–Cu(TfO)2 as the catalyst and TBAT in THF (Scheme 3, Table 2). The crude nitroaldol compounds obtained from the first step of the reaction were subsequently converted to the Mosher esters after filtration and evaporation of the solvent.
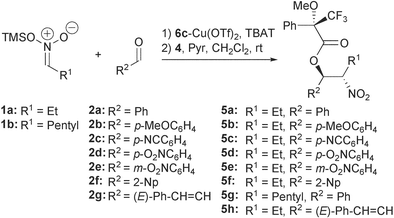 | ||
| Scheme 3 Catalytic diastereo- and enantioselective nitroaldol reaction of nitronates 1 with aldehydes 2. | ||
| Entry | Nitronate | Aldehyde | Yield (%) | Dr threo : erythro | Eeathreo (%) |
|---|---|---|---|---|---|
| a The ee of the nitroaldol reaction is derived from the de of the Mosher ester. | |||||
| 1 | 1a | 2a | 5a – 76 | 5 : 1 | 65 |
| 2 | 1a | 2b | 5b – 34 | 3 : 1 | 59 |
| 3 | 1a | 2c | 5c – 47 | >10 : 1 | 40 |
| 4 | 1a | 2d | 5d – 63 | >10 : 1 | 50 |
| 5 | 1a | 2e | 5e – 67 | >10 : 1 | 45 |
| 6 | 1a | 2f | 5f – 34 | 2.5 : 1 | 60 |
| 7 | 1b | 2a | 5g – 81 | 3 : 1 | 43 |
| 8 | 1a | 2g | 5h – 44 | >10 : 1 | 45 |
The nitronate 1a, derived from nitropropane, reacts well with the different aldehydes and the nitroalcohols are obtained with moderate to very high diastereoselectivity. For the aromatic aldehydes, substituted with electron-withdrawing substituents (2c–e) only the threo-isomer could be detected (Table 2, entries 3-6). However, for these substrates the enantioselectivity of the reactions is moderate. The reaction of the nitronates 1a,b with benzaldehyde 2a and p-methoxybenzaldehyde 2b proceeds with lower diastereoselectivity, however, the enantioselectivity of the nitroalcohols 3a,b,g are improved (entries 1,2,7) compared to those substituted with electron-withdrawing substituents.
The enantiomeric excess of the nitroaldol reactions listed in Table 2 are derived from the de of the obtained Mosher ester and the highest ee obtained so far is 65%. In all of the entries where the erythro-isomers were detected the ee's of the erythro-isomers were lower than those obtained for the corresponding threo-isomer. 1H NMR analysis of the crude nitroaldols obtained in the reactions show higher ratios of the erythro-isomer than observed after conversion into the Mosher esters. This strongly indicates that the unstable nitroaldols undergo partial epimerization before being converted into the stable Mosher esters 5a–h. We thus have reason to believe that the ee's of the Henry reaction, derived from the de's of the Mosher esters, represent the minimal ee's actually induced in the reaction.
In summary we have developed the first catalytic asymmetric Henry reaction of silyl nitronates with aldehydes. This is made possible by trapping the unstable nitroaldols using (S)-MTPA-Cl to form the corresponding Mosher esters 5a–h. For several of the entries good yields, high threo-selectivities and moderate enantioselectivities were obtained. In future work we will try to completely avoid epimerization of the crude nitroaldols, which will reveal if higher enantioselectivities can be obtained by this approach.
Experimental
General methods
Commercially available starting materials were used without further purification. Solvents were dried according to standard procedures. Purification of the products was carried out by flash chromatography (FC) using Merck silica gel 60 (230–400 mesh). 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, using CDCl3 as the solvent and were reported in ppm downfield from TMS (δ = 0) for 1H NMR and relative to the central CDCl3 resonance (δ = 77.00) for 13C NMR. Mass spectra and high resolution mass spectra were obtained on an LC-TOF spectrometer (Micromass). All glassware was dried at 120 °C for 24 h and flame dried before use. Cu(SbF6)2 was made by mixing CuBr2 with AgSbF6 in CH2Cl2. The solution was filtered through Celite before use. The silyl nitronates 1a and 1b were synthesized from the corresponding nitro compounds and trimethylsilyl chloride according to literature procedures.9 All other reagents and materials were purchased from Aldrich or Lancaster.Preparation of catalyst 6c–Cu(OTf)2
2,2′-Methylenebis[(4R,5S)-4,5-diphenyl-2-oxazoline] (20.18 mg, 0.044 mmol, 22 mol%) and Cu(OTf)2 (14.4 mg, 0.04 mmol, 20 mol%) were mixed and stirred under vacuum for 30 min. After adding THF (2 mL) and stirring for 10 min, TBAT (21.6 mg, 0.040 mmol, 20 mol%) in THF (0.5 mL) was added and stirring was continued for 10 min.General procedure for the asymmetric Henry reaction of nitronates 1a–b and aldehydes 2a–g to obtain the Mosher esters 5a–h
To catalyst 6c–Cu(OTf)2 prepared as described above was added the aldehyde (0.2 mmol) and after adjusting the reaction mixture to the appropriate temperature the TMS–nitronate (0.3 mmol) was added and the reaction was stirred at room temperature for 16 h. Then the solvent was removed by evaporation and to the residue was added CH2Cl2 (0.6 mL), pyridine (0.6 mL, 8 mmol) and (S)-MTPA-Cl (45 µL, 0.24 mmol) and the mixture was stirred for 16 h. The reaction was quenched by adding water and then extracted twice with Et2O. The collected organic phases were washed with dilute HCl and brine and finally dried using MgSO4. The crude product was purified by flash chromatography (FC) on silica using mixtures of Et2O and pentane as the eluent.2-Nitro-1-phenylbutyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5a)
Prepared according to the general procedure at room temperature using nitronate 1a (42 µL, 0.3 mmol) and benzaldehyde 2a (22 µL, 0.2 mmol) as the starting material. As determined by 1H NMR the threo : erythro ratio was 5 : 1. The diastereomeric excess of threo-5a was 65% de as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 10 : 90 as the eluent which gave threo-5a in 76% yield (62.2 mg). 1H NMR (CDCl3) δ 7.39–7.08 (m, 10H), 6.24 (d, J = 10.4 Hz, 1H, 1 diastereomer), 6.14 (d, J = 10.4 Hz, 1H, 2. diastereomer), 4.78–4.67 (m, 1H), 3.39 (s, 3H, 1 diastereomer), 3.28 (s, 3H, 2. diastereomer), 1.76–1.61 (m, 2H), 0.82–0.78 (m, 3H); 13C NMR (CDCl3) δ 164.95, 134.96, 133.31, 130.11, 129.97, 129.90, 129.82, 129.62, 129.51, 128.99, 128.92, 128.46, 128.36, 127.95, 127.91, 127.47, 127.24, 127.16, 126.91, 125.51, 124.45, 91.39, 91.26, 78.40, 77.87, 55.70, 55.43, 23.79, 23.76, 9.80, 9.77; MS (TOF ES+): m/z 434 (M + Na)+; HRMS calc. for C20H20F3NO5Na 434.1191; found 434.1188.1-(4-Methoxyphenyl)-2-nitrobutyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5b)
Prepared according to the general procedure using nitronate 1a (42 µL, 0.3 mmol) and p-methoxybenzaldehyde 2b (31.2 mg, 0.2 mmol) as the starting material. The reaction was initiated at −78 °C and allowed to warm to room temperature over 16 h. As determined by 1H NMR the threo : erythro ratio was 3 : 1. The diastereomeric excess of threo-5c was 59% de as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 10 : 90 as the eluent which gave threo-5b in 34% yield (31.2 mg). 1H NMR (CDCl3) δ 7.38–7.25 (m, 5H), 7.12 (d, J = 9.3 Hz, 2H, 1. diastereomer), 7.08 (d, J = 9.3 Hz, 2H, 2 diastereomers), 6.86 (d, J = 9.3 Hz, 2H, 1. diastereomer), 6.78 (d, J = 9.3 Hz, 2H, 2 diastereomers), 6.22 (d, J = 13.9 Hz, 1H, 1. diastereomer), 6.08 (d, J = 13.9 Hz, 1H, 2 diastereomers), 4.78–4.64 (m, 1H), 3.96 (s, 3H, 2 diastereomers), 3.59 (s, 3H, 1. diastereomer), 3.28 (s, 3H, 2 diastereomers), 2.96 (s, 3H, 1. diastereomer), 1.77–1.64 (m, 2H, 2 diastereomers), 1.43–1.34 (m, 2H, 1. diastereomer), 0.95–0.86 (m, 3H); 13C NMR (CDCl3) δ 160.86, 160.72, 160.60, 160.01, 135.98, 135.10, 130.89, 130.11, 129.78, 129.47, 129.40, 129.37, 129.27, 128.92, 128.21, 128.04, 114.95, 114.54, 114.29, 113.87, 91.97, 91.43, 91.29, 90.76, 78.54, 78.17, 56.12, 55.58, 55.34, 55.01, 23.83, 23.43, 10.21, 9.80; MS (TOF ES+): m/z 464 (M + Na)+; HRMS calc. for C21H22F3NO6Na 464.1297; found 464.1298.1-(4-Cyanophenyl)-2-nitrobutyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5c)
Prepared according to the general procedure using nitronate 1a (42 µL, 0.3 mmol) and p-cyanobenzaldehyde 2c (26.2 µL, 0.2 mmol) as the starting material. The reaction was initiated at −78 °C and allowed to warm to room temperature over 16 h. As determined by 1H NMR the threo : erythro ratio was >10 : 1. The diastereomeric excess of threo-5c was 40% as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 30 : 70 as the eluent which gave threo-5c in 47% yield (41.3 mg). 1H NMR (CDCl3) δ 7.49–7.25 (m, 9H), 6.30 (d, J = 10.6 Hz, 1H, 1 diastereomer), 6.21 (d, J = 10.6 Hz, 1H, 2 diastereomers), 4.78–4.70 (m, 1H), 3.45 (s, 3H, 1 diastereomer), 3.34 (s, 3H, 2 diastereomers), 1.83–1.77 (m, 2H, 1 diastereomer), 1.42–1.37 (m, 2H, 2 diastereomers), 0.93–0.85 (m, 3H); 13C NMR (CDCl3) δ 165.74, 165.13, 138.57, 138.29, 133.21, 132.64, 131.43, 130.15, 129.81, 129.34, 128.55, 128.13, 127.96,127.21, 126.75, 126.31, 117.70, 117.13, 114.92, 114.20, 114.03, 113.74, 110.00, 109.83, 90.74, 90.20, 78.94, 78.19, 56.39, 56,09, 23.60, 23.41, 10.06, 9.78; MS (TOF ES+): m/z 459 (M+Na)+; HRMS calc. for C21H19F3N2O5Na 459.1144, found 459.1130.2-Nitro-1-(4-nitrophenyl)butyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5d)
Prepared according to the general procedure at room temperature using nitronate 1a (42 µL, 0.3 mmol) and p-nitrobenzaldehyde 2d (30.2 mg, 0.2 mmol) as the starting material. As determined by 1H NMR the threo : erythro ratio was >10 : 1. The diastereomeric excess of threo-5d was 50% as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 20 : 80 as the eluent which gave threo-5d in 63% yield (57.6 mg). 1H NMR (CDCl3) δ 7.58–7.51 (m, 4H), 7.37–7.24 (m, 5H), 6.29 (d, J = 11.1 Hz, 1H, 2 diastereomers), 6.21 (d, J = 11.1 Hz, 1H, 1. diastereomer), 4.76–4.62 (m, 1H), 3.38 (s, 3H, 1 diastereomer), 3.26 (s, 3H, 2 diastereomers), 1.81–1.72 (m, 2H, 1 diastereomer), 1.36–1.29 (m, 2H, 2 diastereomers), 0.89–0.82 (m, 3H); 13C NMR (CDCl3) δ 164.94, 164.32, 151.07, 150.78, 148.70, 148.23, 134.94, 134.21, 130.92, 130.47, 130.09, 129.83, 129.19, 128.72, 128.41, 128.15, 126.72, 126.05, 124.29, 123.86, 121.24, 121.03, 90.77, 90.23, 75.91, 75.25, 55.77, 55.21, 24.08, 23.60, 10.02, 9.76; MS (TOF ES+): m/z 479 (M+Na)+; HRMS calc. for C20H19F3N2O7Na 479.1042, found 479.1050.2-Nitro-1-(3-nitrophenyl)butyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5e)
Prepared according to the general procedure at room temperature using nitronate 1a (42 µL, 0.3 mmol) and m-nitrobenzaldehyde 2e (30.2 mg, 0.2 mmol) as the starting material. As determined by 1H NMR the threo : erythro ratio was >10 : 1. The diastereomeric excess of threo-5e was 45% as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 15 : 85 as the eluent which gave threo-5e in 67% yield (61.1 mg). 1H NMR (CDCl3) δ 7.51–7.04 (m, 9H), 6.38 (d, J = 11.1 Hz, 1H, 1 diastereomer), 6.32 (d, J = 11.1 Hz, 1H, 2 diastereomers), 4.78–4.64 (m, 1H), 3.41 (s, 3H, 1 diastereomer), 3.29 (s, 3H, 2 diastereomers), 2.19–2.09 (m, 2H, 1 diastereomer), 1.88–1.75 (m, 2H, 2 diastereomers), 0.86–0.81 (m, 3H); 13C NMR (CDCl3) δ 163.46, 149.02, 148.72, 148.43, 158.13, 137.33, 136.84, 134.62, 134.28, 130.74, 130.37, 130.04, 129.78, 128.59, 128.03, 128.33, 127.93, 126.57, 166.21, 124.78, 124.49, 124.38, 124.01, 122.50, 122.12, 90.78, 90.41, 74.91, 74.03, 55.85, 55.32, 24.03, 23.63, 10.24, 9.75; MS (TOF ES+): m/z 479 (M + Na)+; HRMS calc. for C20H19F3N2O7Na 479.1042, found 479.1044.1-(2-Naphthyl)-2-nitrobutyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5f)
Prepared according to the general procedure using nitronate 1a (42 µL, 0.3 mmol) and 2-naphthaldehyde 2d (42 µL, 0.2 mmol) as the starting material. The reaction was initiated at −78 °C and allowed to warm to room temperature over 16 h. As determined by 1H NMR the threo : erythro ratio was 2.5 : 1. The diastereomeric excess of threo-5c was 60% as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 10 : 90 as the eluent which gave threo-5f in 34% yield (31.2 mg). 1H NMR (CDCl3) δ 7.82–7.61 (m, 4H), 7.47–7.20 (m, 8H), 6.41 (d, J = 12.4 Hz, 1H, 1 diastereomer), 6.30 (d, J = 12.4 Hz, 1H, 2 diastereomers), 4.88–4.74 (m, 1H), 3.41 (s, 3H, 1 diastereomer), 3.28 (s, 3H, 2 diastereomers), 1.83–1.72 (m, 2H, 1 diastereomer), 1.21–1.04 (m, 2H, 2 diastereomers), 0.84–0.79 (m, 3H); 13C NMR (CDCl3) δ 173.72, 173.39, 136.42, 136.02, 134.62, 134.06, 132.60, 132.17, 129.51, 129.49, 129.12, 129.09, 128.85, 128.43, 128.38, 128.25, 128.21, 128.06, 127.98, 127.76, 127.67, 127.51, 127.46, 127.24, 127.21, 127.08, 126.85, 126.53, 124.17, 123.92, 122.71, 122.35, 102.31, 101.94, 91.49, 91.16, 78.88, 78.56, 49.65, 49.21, 24.06, 23.84, 10.13, 9.82; MS (TOF ES+): m/z 484 (M + Na)+; HRMS calc. for C24H22F3NO5Na 484.1348, found 484.1349.2-Nitro-1-phenylheptyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5g)
Prepared according to the general procedure using nitronate 1b (61 mg, 0.3 mmol) and benzaldehyde 2a (22 µL, 0.2 mmol) as the starting material. The reaction was initiated at −78 °C and allowed to warm to room temperature over 16 h. As determined by 1H NMR the threo : erythro ratio was 3 : 1. The diastereomeric excess of threo-5g was 43% as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 30 : 70 as the eluent which gave threo-5g in 81% yield (74.6 mg). 1H NMR (CDCl3) δ 7.59–7.19 (m, 10H), 6.22 (d, J = 12.2 Hz, 1H, 1 diastereomer), 6.12 (d, J = 12.2 Hz, 1H, 2 diastereomers), 4.81–4.72 (m, 1H), 3.39 (s, 3H, 1diastereomer), 3.36 (s, 3H, 2 diastereomers), 1.78–1.71 (m, 2H, 1 diastereomer), 1.61–1.53 (m, 2H, 2 diastereomers), 1.32–1.21 (m, 6H), 0.77–0.69 (m, 3H); 13C NMR (CDCl3) δ 164.94, 133.32, 133.23, 130.09, 130.05, 129.89, 29.83, 129.60, 129.49, 128.98, 128.90, 128.34, 128.21, 127.48, 127.31, 126.90, 126.81, 124.45, 121.57, 104.73, 90.01, 89.88, 78.59, 78.05, 75.72, 75.37, 31.52, 31.00, 25.84, 25.44, 24.93, 24.86, 22.28, 22.25, 13.82, 13.79; MS (TOF ES+): m/z 476 (M+Na)+; HRMS calc. for C23H26F3NO5Na 476.1661, found 476.1656.2-Nitro-1-(2-phenylethenyl)butyl 2-trifluoromethyl-2-methoxy-2-phenylethanoate (5h)
Prepared according to the general procedure using nitronate 1a (42 mg, 0.3 mmol) and (E)-cinnamaldehyde 2g (22 µL, 0.2 mmol) as the starting material. The reaction was initiated at −78 °C and allowed to warm to room temperature over 16 h. As determined by 1H NMR the threo : erythro ratio was >10 : 1. The diastereomeric excess of threo-5h was 45% as determined by 19F NMR of the crude product. FC was performed using Et2O–pentane 10 : 90 as the eluent which gave threo-5h in 44% yield (38.6 mg). 1H NMR (CDCl3) δ 7.36–7.23 (m, 10H), 6.75 (d, J = 13.8 Hz, 1H, 2 diastereomers), 6.68 (d, J = 13.8 Hz, 1H, 1 diastereomer), 6.02–5.94 (m, 1H), 5.83–78 (m, 1H), 4.43–4.54 (m, 1H), 2.03–1.78 (m, 2H), 3.42 (s, 3H, 1 diastereomer), 3.31 (s, 3H, 2 diastereomers), 0.98–0.91 (m, 3H); 13C NMR (CDCl3) δ 139.15, 138.83, 129.87, 129.62, 129.52, 129.24, 128.97, 128.84, 128.80, 128.43, 128.36, 127.89, 127.65, 127.54, 127.35, 127.04, 126.99, 126.94, 126.78, 121.60, 121.29, 119.21, 118.93, 90.46, 90.10, 76.34, 76.01, 53.92, 53.28, 23.94, 23.14, 10.23, 9.83; MS (TOF ES+): m/z 460 (M+Na)+; HRMS calc. for C22H22F3NO5Na 460.1348, found 460.1356.Acknowledgements
The authors are indebted to The Danish National Research Foundation for financial support.References
- For recent reviews dealing with the Henry reaction and its use see e.g.: (a) F. A. Luzzio, Tetrahedron, 2001, 57, 915 CrossRef CAS; (b) G. Rosini, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1999, vol. 1, 321. Search PubMed; (c) G. Rosini and R. Ballini, Synthesis, 1988, 833 CrossRef; (d) H. W. Pinnick, in Organic Reactions, ed. L. A. Paquette, Wiley, New York, 1990, vol. 38, chapter 3. Search PubMed; (e) M. Shibasaki and H. Gröger, in Comprehensive Asymmetric Catalysis I-IIII, eds. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, Berlin-Heidelberg, 1999, chapter 29, 3 Search PubMed.
- (a) H. Sasai, T. Suzuki, S. Arai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1992, 114, 4418 CrossRef CAS; (b) H. Sasai, T. Suzuki, N. Itoh and M. Shibasaki, Tetrahedron Lett., 1993, 34, 851 CrossRef CAS; (c) H. Sasai, T. Tokunaga, S. Watanabe, T. Suzuki, N. Itoh and M. Shibasaki, J. Org. Chem., 1995, 60, 7388 CrossRef CAS; (d) K. Iseki, S. Oishi and S. M. Shibasaki, Tetrahedron Lett., 1996, 37, 9081 CrossRef CAS; (e) T. Arai, Y. M. A. Yamada, H. Sasai and M. Shibasaki, Chem. Eur. J., 1996, 2, 1368 CrossRef CAS.
- (a) B. M. Trost and V. S. C. Yeh, Angew. Chem., Int. Ed., 2002, 41, 861 CrossRef CAS; (b) B. M. Trost, V. S. C. Yeh, H. Ito and N. Bremeyer, Org. Lett., 2002, 4, 2621 CrossRef CAS.
- (a) C. Christensen, K. Juhl and K. A. Jørgensen, Chem. Commun., 2001, 2222 RSC; (b) C. Christensen, K. Juhl, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 2002, 67, 4875 CrossRef CAS.
- Examples of diastereoselective reactions: (a) E. W. Colvin and D. Seebach, J. Chem. Soc., Chem. Commun., 1978, 689 RSC; (b) D. Seebach, A. K. Beck, T. Mukhopadhyay and E. Thomas, Helv. Chim. Acta, 1982, 65, 1101 CrossRef CAS; (c) S. Hanessian and J. Kloss, Tetrahedron Lett., 1985, 26, 1261 CrossRef; (d) A. G. M. Barrett, C. Roby and C. D. Spilling, J. Org. Chem., 1989, 54, 1233 CrossRef CAS.
- (a) K. R. Knudsen, T. Risgaard, N. Nishiwaki, K. V. Gothelf and K. A. Jørgensen, J. Am. Chem. Soc., 2001, 123, 5843 CrossRef CAS . See also; (b) N. Nishiwaki, K. R. Knudsen, K. V. Gothelf and K. A. Jørgensen, Angew. Chem. Int. Ed., 2001, 40, 2992 CrossRef CAS.
- See also ref 2c for a discussion of this problem..
- (a) A. S. Pilcher, H. L. Ammon and Deshong, J. Am. Chem. Soc., 1995, 117, 5166 CrossRef CAS; (b) J. Krüger and E. M. Carreira, J. Am. Chem. Soc., 1998, 120, 827 CrossRef CAS.
- K. Torssell and O. Zeuthen, Acta Chem. Scand., 1978, B 32, 188 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |