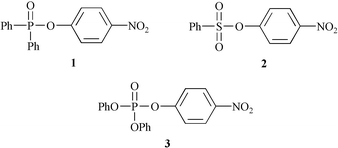
Alkali-metal ion catalysis and inhibition in nucleophilic displacement reactions at carbon, phosphorus and sulfur centres. IX.†p-Nitrophenyl diphenyl phosphate‡
Received
29th August 2002
, Accepted 10th October 2002
First published on 3rd December 2002
Abstract
We report on the catalytic effects by alkali-metal ions in the ethanolysis of p-nitrophenyl diphenyl phosphate, in continuation of our studies on alkali-metal ion catalysis and inhibition in nucleophilic displacement reactions at carbon, phosphorus and sulfur centres. The following selectivity order of catalytic reactivity was observed for nucleophilic displacement at the phosphorus center with p-nitrophenoxide as leaving group: Li+ > Na+ > K+ > Cs+. A minor reaction pathway with phenoxide leaving was also found. The kobs data have been dissected into reaction pathways by free ions (kEtO−) and by ion pairs (kMOEt), with the latter being dominant, in a 4-membered transition-state. Further analysis is given in terms of initial-state and transition-state stabilization by the alkali-metal ions in terms of the Eisenman model (electrostatic interaction vs. desolvation). Results of ab-initio MO calculations are presented based on interaction between M+ and a model bipyramidal phosphorane intermediate and compared with the sulfurane analogue.
Introduction
Metal ions are known to play an important role in biological processes, and especially on the mechanism of phosphoryl transfer reactions.2 The majority of studies have been concerned with the catalytic effects of divalent metal ions, e.g. Mg2+, Ca2+, Mn2+, Fe2+, Co2+, Cu2+ and Zn2+ in the hydrolysis of phosphate esters.3
The ubiquitous presence of alkali metals in biological systems4 calls for an examination of their effects on phosphoryl transfer and such studies were initiated in our laboratory. Unexpectedly, we found that ethanolysis of the phosphinate ester, p-nitrophenyl diphenyl phosphinate, 1, exhibited catalysis by alkali metals, with the selectivity order Li+ > Na+ > K+.5
In contrast, in the ethanolysis of p-nitrophenyl benzenesulfonate, 2, Li+ brought about inhibition of rate and the catalytic order of the other alkali-metals was K+ > Cs+ > Na+.6
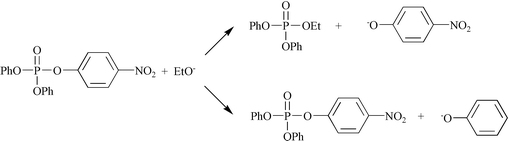
The present work is concerned with study of catalytic effects on the ethanolysis of the phosphate ester, p-nitrophenyl diphenyl phosphate, 3. In this case, in addition to nucleophilic displacement at the phosphorus center with p-nitrophenoxide as leaving group, a minor reaction pathway with phenoxide leaving was also identified. The structural change in the phosphorus(V) center, could be expected to have a marked effect on reactivity. Kinetic analysis has enabled dissection of metal ion interactions in the ground-state and transition-state, with comparison among the substrates 1–3.
Previous studies with p-nitrophenyl diphenyl phosphate have included micellar catalyzed hydrolysis by Bunton and coworkers,7 by Moss et al.,8 and by Menger and Whitesell,9 which included metallomicelles, while Breslow and coworkers have reported on the catalytic hydrolysis by tetracoordinated zinc complexes.10
Results
Reaction pathways for alkali-metal ethoxides
The rates of ethanolysis reactions of 3 with LiOEt, NaOEt, KOEt and CsOEt were determined spectrophotometrically in anhydrous ethanol at 25 °C under pseudo-first-order conditions with the base in excess. Rate constants, kobs, were calculated from linear plots of log(Ainf
−
At)
vs. time where the absorbances (A) were taken at 400 nm corresponding to formation of p-nitrophenoxide ion. Results are given in Supplementary Tables S1–S4‡ and shown graphically in Fig. 1.
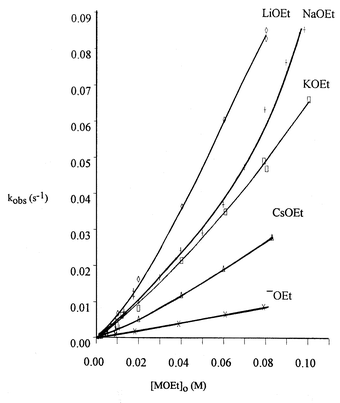 |
| | Fig. 1 Kinetic data for the reaction of 3 with LiOEt, KOEt, CsOEt and KOEt in EtOH and in the presence of excess 18-Crown-6 at 25 °C (data in Tables S1–S5‡). | |
Repetitive scanning of the LiOEt reaction solution over 210–450 nm revealed also absorbance increases at 230 nm and 290 nm corresponding to the concurrent release of phenoxide ion, albeit at a much slower rate. The generation of PhO− similarly followed first-order kinetics with a rate constant ca. 1/250, slower than for formation of p-nitrophenoxide ion. This secondary process (Scheme 1) was also observed by Breslow10 in the OH− hydrolysis of 3 and was not investigated further in this work.
 |
| | Scheme 1 | |
Effect of added complexing agents
The effect of added 18-Crown-6 ether to the reaction of 3 with KOEt is shown in Fig. 2
(see also Table S5‡). The rate decreases until one equivalent is added, at which point further addition of complexing agent has no effect. A similar result had been found in the phosphinate system.5b However these results contrast with typical SN2 type reactions where metal complexing agents produce rate acceleration. A rate enhancing effect was also observed4,6 for the addition of complexing agents to the reaction of LiOEt with the benzenesulfonate 2
(vide infra), in contrast with the present work.
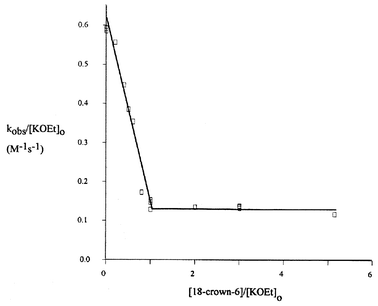 |
| | Fig. 2 Kinetic data for the reaction of 3 with KOEt in the presence of 18-Crown-6, in EtOH at 25 °C. (Data in Table S5‡). | |
Effect of added metal ions
Added potassium iodide and acetate to the reaction of 3 with KOEt increased the rate significantly; as was similarly the case on addition of LiNO3 and LiCl to the LiOEt reaction (Fig. 3, Table S6‡). However, addition of (n-Bu)4NCl as an inert salt to the reaction of 3 with KOEt in the presence of 18C6 gave only a small (ca. 4%) increase in rate (Table S7‡), indicating a small salt effect. These results confirm the catalytic effect of alkali metal cations on the ethanolysis of 3 in the order Li+ > K+.
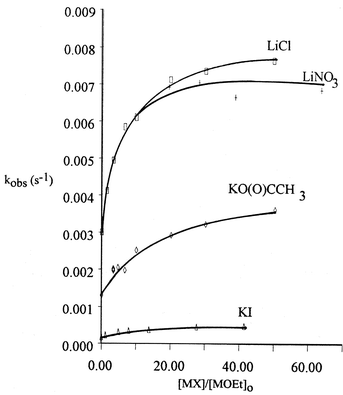 |
| | Fig. 3 Kinetic data for the reaction of 3 with LiOEt in the presence of LiCl and LiNO3 and with KOEt in EtOH at 25 °C in the presence of KO(O)CCH3 and KI. (Data shown in Tables S6(a)–(d)).‡ | |
Discussion
Reactivities of alkali-metal ethoxide ion pairs vs. free ions
Notable immediately on comparing the reactivities of the phosphate ester 3 with the phosphinate ester 1, is the reactivity order 1 > 3. Direct comparison of the rate constants, kEtO−, for the two substrates can be had from the reactions of MOEt in presence of excess 18C6 which yields kEtO−(1)/kEtO−(3)
= 10. This reactivity difference finds explanation in O → P π-back-bonding in 3, but its absence in 1, which thus results in 1 having the more electrophilic P center.
The upward curvature in the plots of kobsvs. [MOEt]
(Fig. 1), which was also observed in the reaction of 1,5b contrasts with expected behavior if free ethoxide were more reactive than the ion-paired species. In fact, a number of SN2 processes at saturated carbon centers have been found to be characterized by downward curvature of the kvs. [Nu] plots,11a–11e which was interpreted as arising from decreased reactivity of the ion-paired species as compared to the dissociated anions. The contrasting behavior in the present system finds explanation in terms of reactivities of free ions vs. ion pairs toward the phosphorus ester E, which can be expressed in terms of Scheme 2 and equations 1 and 2:
 |
| | Scheme 2 | |
| | | kobs
=
kEtO−[EtO−]
+
kMOEt[MOEt] | (1) |
| | | kobs/[EtO−]
=
kEtO−
+
kMOEtKa[EtO−] | (2) |
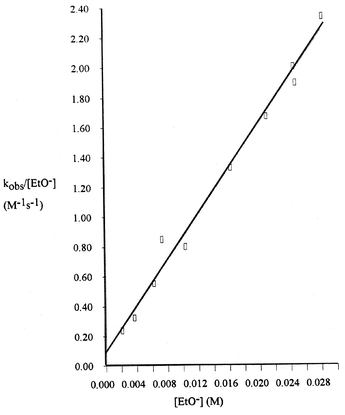
The plots of kobs/[EtO−]
vs. [EtO−] are generally linear (R2: Li, 0.998; Na, 0.942; K, 0.987; Cs, 0.999) but the small intercept values have rather large associated errors, e.g. Fig. 4
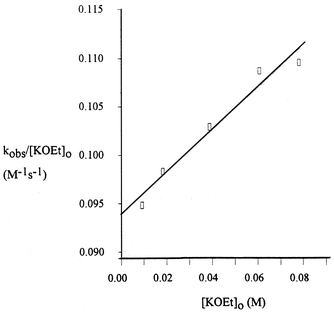
(Table S8‡), for KOEt. A more reliable value of kEtO− could be obtained from plots of kobs/[KOEt]
vs. [KOEt] in the presence of excess 18C6, Fig. 5
(Table S9‡) for KOEt, which yields kEtO−
= 0.0939 ± 0.001 M−1s−1. Values of kMOEt obtained from the various plots are collected in Table 1. The overall reactivity order is LiOEt > NaOEt > KOEt > CsOEt > EtO−.
 |
| | Fig. 4 Ion pairing treatment of the data for the reaction of 3 with KOEt in EtOH at 25 °C. (Data in Table S8‡). | |
 |
| | Fig. 5 Ion pairing treatment of the data for the reaction of 3 with KOEt in the presence of excess complexing agent. (Data in Table S9‡). | |
Table 1 Calculated rate constants for free ions and ion pairs in ethanolysis of 3 at 25 °C
| MOEt/M |
k
MOEt/s−1M−1 |
| LiOEt |
1.36 ± 0.026 |
| NaOEt |
1.03 ± 0.057 |
| KOEt |
0.85 ± 0.035 |
| CsOEt |
0.42 ± 0.0008 |
| KOEt/18C6 (Table S72) |
0.094 ± 0.001 |
The mechanism we suggest for catalysis by MOEt ion pairs is one where the metal cation coordinates to the phosphoryl oxygen concertedly with nucleophilic attack by ethoxide in a 4-membered transition-state. Alternatively, one could consider pre-equilibrium attachment of M+ followed by nucleophilic attack. However these two alternatives can be shown to be kinetically indistinguishable. There is evidence for participation by ion pairs in similar systems12 and the following discussion is based on catalysis occurring by MOEt ion pairs.
Metal ion interactions in ground- and transition-states: dissection of stabilization energies
The catalytic role of metal ions in the reactions of alkali metal ethoxides with the compounds under investigation can be analyzed in terms of differing metal–ion interactions in the ground- and transition-states. Catalysis by ion pairs will be assumed, as opposed to the pre-association mechanism, as stated above.
To investigate the catalytic role of metal ions in the reaction mechanism, the relative stabilization of the ground-states (GS) to that of the transition-states (TS) should be considered. We use a method devised by Kurz14a and in different forms by Mandolini14b and Tee.14c
Association constants for interactions of alkali metals and ethoxides (Ka, in Scheme 1) are known (LiOEt, 212 M−1; NaOEt, 102 M−1; KOEt, 90 M−1; CsOEt, 121 M−1),13 and it is assumed here (for lack of other evidence) that interaction between M+ and E in the ground-state can be neglected. Using the thermodynamic cycle in Scheme 3 one can then calculate a corresponding virtual association constant, K′a, for the TS.
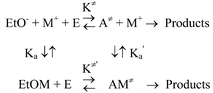 |
| | Scheme 3 | |
From transition-state theory, one can then derive eqn. (3) which combines the GS association constants with reaction rates to obtain association constants Ka′ for the TS. The latter in turn can be expressed in terms of free energies of stabilization, δGts and δGip, with the net catalytic effect δGcat being given by the difference in free energies of activation between the catalyzed and uncatalyzed reactions in eqn. 4. This relationship between GS and TS stabilization is illustrated in Fig. 6.
| | | ΔGcat
=
ΔGc≠
−
ΔGu≠
=
δGts
−
δGip | (4) |
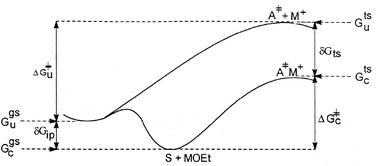 |
| | Fig. 6 Free energies involved in the uncatalyzed and metal ion catalyzed reactions of substrate S with alkali metal ethoxides, MOEt. | |
The δGcat, δGip and δGts data for 1, 2 and 3 are presented in Table 2. It is seen that for the phosphorus based esters 1 and 3, all the alkali metal ions stabilize the TS to a greater extent than the GS and thus act as catalysts (ΔGcat < 0). While both esters exhibit the same trends in stabilization, the phosphinate 1 shows a greater range of values from Li+, the most effective catalyst, to Cs+, the least effective catalyst (ΔΔGcat
= 4.9 kJ mol−1) than the phosphate ester 3
(ΔΔGcat
= 2.9 kJ mol−1). This is interpreted to signify that the phosphinate ester is more sensitive to the nature of the alkali-metal ion than the phosphate ester. For the sulfonate ester 2, Li+ acts as an inhibitor with ΔGcat > 0, i.e. association in the GS is stronger than with the TS. Also, in the case of 2, K+ showed the greatest value for ΔGcat, having the strongest association with the TS, i.e. a selectivity order very different from that observed for the two phosphorus esters.
Table 2 Free energies of metal ion stabilization of ethoxide (δGip), transition states (δGts), and the net catalytic effect (ΔGcat) for the reaction of ethoxide with 1, 2 and 3 at 25 °C in ethanol. All values are in kJ mol−1
| Ion |
δG
ip
|
δG
ts
|
ΔGcat |
| EtO− |
3
|
1
|
2
|
3
|
1
|
2
|
| Li+ |
−13.3 |
−19.7 |
−21.2 |
−12.1 |
−6.4 |
−7.9 |
+1.2 |
| Na+ |
−11.5 |
−17.2 |
−17.6 |
−14.0 |
−5.7 |
−6.1 |
−2.5 |
| K+ |
−11.2 |
−16.4 |
−15.1 |
−15.0 |
−5.2 |
−3.9 |
−3.8 |
| Cs+ |
−11.9 |
−15.4 |
−13.5 |
−14.9 |
−3.5 |
−3.0 |
−3.0 |
In examining the above contrasting selectivities, an interesting relationship becomes apparent with selectivity patterns of alkali-metal cations for ion-exchange resins and glass electrodes which has been discussed elegantly by Eisenman.15 Two opposing factors were considered to govern the strength of the interaction between a cation and a fixed ionic group: electrostatic interactions engendered in approach of ions of opposite charge, and the energy expended in rearranging solvent molecules around the ions to enable close contact, i.e. the difference in solvation energies of the ions. The overall free energy change for interchange of cations 1 and 2 (ΔG°½) at a fixed anionic site will be given by the differences in electrostatic interactions and solvent rearrangement terms as shown in eqn. 5 where e is the electronic charge, r1, r2 and rA are the ratii of cations 1 and 2 and of the anion, respectively, and ΔG1, ΔG2 solvation energies of cations 1 and 2.
| | | ΔG°½
=
[e2/(rA
+
r2)]
−
[e2/(rA
+
r1)]
−
(ΔG2
−
ΔG1) | (5) |
One would predict that for an anionic group with localized charge, or high field strength, the electrostatic term would be dominant over the solvation energy term, allowing the ions to come in direct contact. Thus smaller cations would be bound most strongly, leading to the selectivity order Li+ > Na+ > K+ > Cs+. One also predicts a linear relationship between ΔGts(M+) and the inverse of the crystal radius (Fig. 7) as was also reported for the phosphinate system 1.
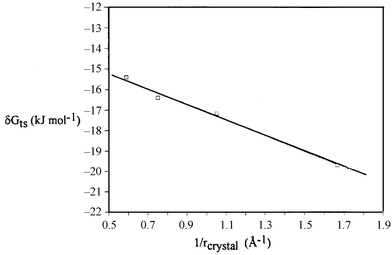 |
| | Fig. 7 Plot of δGtsversus the reciprocal of the crystal radius of the metal ion for the reactions of 3 with alkali metal ethoxides in ethanol at 25 °C. | |
On the other hand, the sulfonate system, with charge delocalized over two oxygens in the TS, would correspond to a low electric-field strength situation and here the solvation term would be dominant over the electrostatic term. A linear relationship between ΔGts
(M+) and the inverse of the solvated (Stokes) radius of M+ is followed.
In parallel theoretical studies transition-state models were optimized at the ab initio HF/3-21 + G* level as penta-coordinate intermediates resulting from attack of OH− on phosphorane H3PO2 and sulfurane H2SO3.16 The optimized trigonal bipyramidal structures along with charge distributions are shown in Fig. 8. Relative energies for stabilization of the sulfurane and phosphorane species by Li+ and Na+ ions and the geometries of the metal ion complexes, at the B3LYP/6-31+G*//HF/3-21+G* level, are presented in Fig. 9. It is apparent that, in the gas phase, there is weaker stabilization in the sulfurane species compared to the phosphorane on interaction with the cations. In the Eisenman scheme, this could lead to interaction with solvated cations in the sulfonate TS compared to bare cations for the phosphorane TS. The calculational results are hence condordant with our observations of selectivities of alkali-metals in the reactions under investigation as discussed herein.
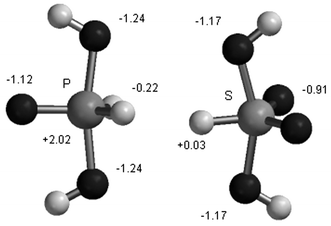 |
| | Fig. 8 Structures and charge distributions of sulfurane and phosphorane intermediates from ab initio calculations. | |
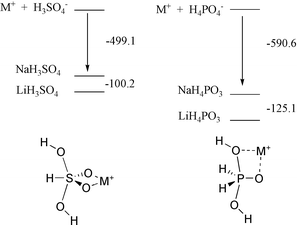 |
| | Fig. 9 Relative energies (kJ mol−1) for stabilization of sulfurane and phosphorane intermediates by Li+ and Na+ and geometries of metal ion complexes: sulfurane (diequatorial); phosphorane (apical-equatorial). The structures presented are the lowest energy stationary points from exploration of the conformational space and different coordination geometries. Relative energies include ZPE and were calculated at B3LYP/6-31+G*//HF/3-21+G(*). | |
Experimental
p-Nitrophenyl diphenyl phosphate was prepared according to literature procedures (10) and fully characterized. Alkali-metal ethoxide solutions were prepared as previously and were stored under N2 in the refrigerator. Base concentrations were determined by titration with standard HCl. Salts used were of reagent quality. 18-Crown-6 ether (Aldrich) was recrystallized from acetonitrile and dried over P2O5in vacuo. Bu4NCl was recrystallized from acetone/ether and dried in vacuo at 60 °C.
Kinetics were performed using a Varian Cary 3 or a Hewlett Packard 8452A diode array spectrophotometer equipped with a HP 89075C Multicell transport and thermostatted cell holders. Pseudo-first-order conditions with base at least 20 times greater than substrate concentration were used. Typically reactions were followed for 4–10 half-lives and kobs calculated using a linear regression of the equation
| | | ln(Ainf
−
At)
=
kobst
+ ln(Ainf
−
Ao). |
(6)
|
Kinetic runs were performed generally in replicate;
kobs values are presented as Supplementary Material.
‡
Acknowledgements
Support of this research by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) is gratefully acknowledged. R. N. is the recipient of a Postgraduate Scholarship from NSERC.
Footnotes and References
- R. Nagelkerke, M. J. Pregel, E. J. Dunn, G. R. J. Thatcher and E. Buncel, Org. React. (Tartu), 1995, 102, 11 Search PubMed.
-
(a)
A. Fersht, Enzyme Structure and Mechanism, ed. W. H. Freeman and company, New York, 2nd edn., 1985 Search PubMed;
(b) G. R. J. Thatcher and R. Kluger, Adv. Phys. Org. Chem., 1989, 25, 99 Search PubMed;
(c) J. Chin, Acc. Chem. Res., 1991, 24, 145 CrossRef CAS.
-
(a) R. Breslow, Adv. Enzymol., 1986, 58, 1 Search PubMed;
(b) D. Herschlag and W.P. Jencks, J. Am. Chem. Soc., 1987, 109, 4665 CrossRef CAS.
-
(a)
L. Stryer, Biochemistry, ed. W. H. Freeman and company, New York, 3rd edn., 1988 Search PubMed;
(b) T. M. Fyles, T. D. James and K. C. Kaye, J. Am. Chem. Soc., 1993, 115, 12315 CrossRef CAS;
(c) M. J. Pregel, L. Jullien, J. Canceill, L. Lacombe and J.-M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1995, 417 RSC;
(d)
J. J. R. Fraústo da Silva and R. J. P. Williams, The Biological Chemistry of the Elements, Clarendon Press, Oxford, 1991 Search PubMed.
-
(a) E. Buncel, E. J. Dunn, R. A. B. Bannard and J. P. Purdon, J. Chem. Soc., Chem. Commun., 1984, 162 RSC;
(b) E. J. Dunn and E. Buncel, Can. J. Chem., 1989, 67, 1440 CAS;
(c) M. J. Pregel, E. J. Dunn and E. Buncel, Actualidades de Fisico-Quimica Organica, 1991, 110 Search PubMed.
-
(a) E. Buncel and M. J. Pregel, J. Chem. Soc., Chem. Commun., 1989, 1566 RSC;
(b) M. J. Pregel, E. J. Dunn and E. Buncel, Can. J. Chem., 1990, 68, 1846 CAS;
(c) M.J. Pregel and E. Buncel, J. Org. Chem., 1991, 56, 5583 CrossRef CAS;
(d) M. J. Pregel and E. Buncel, J. Chem. Soc., Perkin Trans. 2, 1991, 307 RSC;
(e) M. J. Pregel, E. J. Dunn and E. Buncel, J. Am. Chem. Soc., 1991, 113, 3545 CrossRef CAS.
-
(a) C. A. Bunton and L. Robinson, J. Org. Chem., 1969, 34, 773 CrossRef CAS;
(b) C. A. Bunton and L. G. Ionescu, J. Am. Chem. Soc., 1973, 95, 2912 CrossRef CAS.
- R. A. Moss, K. W. Alevis and J.-S. Shin, J. Am. Chem. Soc., 1984, 106, 2651 CrossRef CAS.
-
(a) F. M. Menger and L. G. Whitesell, J. Am. Chem. Soc., 1985, 107, 707 CrossRef CAS;
(b) F. M. Menger, L. H. Gan, E. Johnson and D. H. Durst, J. Am. Chem. Soc., 1987, 109, 2800 CrossRef CAS.
- S. H. Gellman, R. Petter and R. Breslow, J. Am. Chem. Soc., 1986, 108, 2388 CrossRef CAS.
-
(a) C. C. Evans and S. Sugden, J. Chem. Soc., 1949, 270 RSC;
(b) J. R. Bevan and C.B. Monk, J. Chem. Soc., 1956, 1396 RSC;
(c) P. Jones, P. Harrison and L. Wynne-Jones, J. Chem. Soc., Perkin Trans. 2, 1979, 1679 RSC;
(d) P. Beronius and L. Pataki, Acta. Chem. Scand., Ser. A, 1979, A33, 675 Search PubMed;
(e) N. N. Lichtin and K. N. Rao, J. Am. Chem. Soc., 1961, 83, 2417 CrossRef CAS.
-
(a) A. Papoutsis, G. Papanastasiou, D. Jannakoudakis and C. Georgulis, J. Chim. Phys., 1985, 82, 913 Search PubMed;
(b) J. R. Jones, Prog. React. Kinet., 1973, 7, 1 Search PubMed.
- J. Barthel, G. Bader and M. Raach-Lenz, Z. Phys. Chem., 1973, 84, 100 Search PubMed.
-
(a) J. L. Kurz, J. Am. Chem. Soc., 1963, 85, 987 CrossRef CAS;
(b) R. Cacciapaglia and L. Mandolini, Chem. Soc. Rev., 1993, 22, 221 RSC;
(c) O. S. Tee, Adv. Phys. Org. Chem., 1994, 29, 1 Search PubMed.
- D. Eisenman, Biophys. J. Suppl., 1962, 2, 259 Search PubMed.
-
(a) M. J. Pregel, E. J. Dunn, R. Nagelkerke, G. R. J. Thatcher and E. Buncel, Chem. Soc. Rev., 1995, 24, 445 RSC;
(b)
R. Nagelkerke, Ph.D. Thesis, Queen's University, 1993;
(c) A. M. P. Borrajo, J. F. Gal, P. C. Maria, M. Decouzon, D. C. Ripley, E. Buncel and G. R. J. Thatcher, J. Org. Chem., 1997, 62, 9203 CrossRef CAS.
- W. M. Gulick and D. H. Geske, J. Am. Chem. Soc., 1966, 88, 2928 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2003 |
Click here to see how this site uses Cookies. View our privacy policy here.