Synthesis of novel chiral phosphinocyrhetrenyloxazoline ligands and their application in asymmetric catalysis
Carsten
Bolm
*,
Li
Xiao
and
Martin
Kesselgruber
Institut für Organische Chemie der RWTH Aachen, Professor-Pirlet-Strasse 1, 52056 Aachen, Germany. E-mail: carsten.bolm@oc.rwth-aachen.de; Fax: +49 (0)241 80 92 391
First published on 26th November 2002
Abstract
Several novel planar chiral phosphinocyrhetrenyloxazolines have been synthesized, and their catalytic activities have been evaluated in a variety of asymmetric catalytic reactions. Preferable effects as compared to their ferrocenyl analogues have been observed in asymmetric allylic amination and asymmetric hydrosilylation, and up to 97% ee and 72% ee were reached, respectively. The Lewis basicity of the phosphorus on the ferrocene and the cyrhetrene, which contributes to their different behavior in catalysis, has been deduced by 31P NMR spectroscopy analysis, as indicated by 1J(77Se–31P) in the corresponding phosphine selenides.
Introduction
Transition metal-catalyzed reactions have found widespread application as powerful tools in modern organic synthesis. In particular, transition metal–chiral ligand complexes have demonstrated their potency in many enantioselective transformations even on an industrial scale.1 One of the most prominent classes of chiral ligands utilized in asymmetric synthesis is based on the planar chiral transition-metal π-complexes.Enantiopure ferrocenes are among the most useful and popular ligands for enantioselective homogeneous catalysis both on a laboratory scale and in industry.2 Recently, we reported the synthesis of a ferrocene-based hydroxyoxazoline ligand and its application in the asymmetric addition of diphenylzinc to aldehydes to form diarylmethanols.3 Further optimization of this process was achieved by variation of the metal–π-fragment, which was expected to influence the steric and electronic properties of the catalyst. η5-Cyclopentadienyl(tricarbonyl)rhenium(I) (cyrhetrene) based 1 (Fig. 1) was employed for this purpose, and indeed, an enantiomeric excess of up to 99% was obtained in the above mentioned reaction.4 As compared with that of its ferrocenyl analogue the enantioselectivity was higher and, furthermore, the required catalyst loading was lower. This effect could be attributed to the existence of electron-withdrawing and bulky carbonyl groups. Being amongst the best π-acceptor ligands, carbonyl groups stabilize low oxidation states and electron density on the metal. If these electronic changes are then efficiently transferred to the catalytically active site, and are not simultaneously counteracted any steric modifications that occur, beneficial effects like those observed in the catalysis could result.
 | ||
| Fig. 1 | ||
To the best of our knowledge, 1 was the first example of a planar chiral cyrhetrene complex to be utilized as a chiral ligand in asymmetric catalysis. Since such cyrhetrene complexes could offer a broad range of new potential ligands, it appeared worthwhile investigating if this approach could be generalized.
Enantiopure Phosferrox-type5 ligands such as 26 (Fig. 1) have been applied to a variety of asymmetric catalytic reactions, such as Pd-catalyzed allylic alkylations and aminations,7 Pd-catalyzed Heck reactions,8 Ru-catalyzed transfer-hydrogenations,9 and hydrosilylations.10 Therefore, we decided to prepare its structurally analogous cyrhetrenyloxazolines 3,114 and 5 (Schemes 1 and 2) and examine their efficiency in such asymmetric reactions.
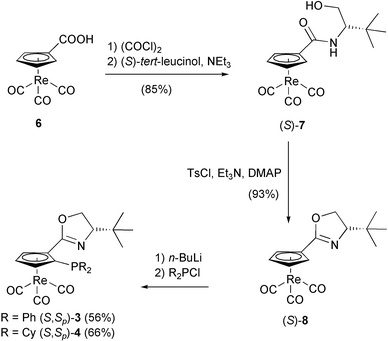 | ||
| Scheme 1 Synthesis of cyrhetrenyloxazolines 3 and 4. | ||
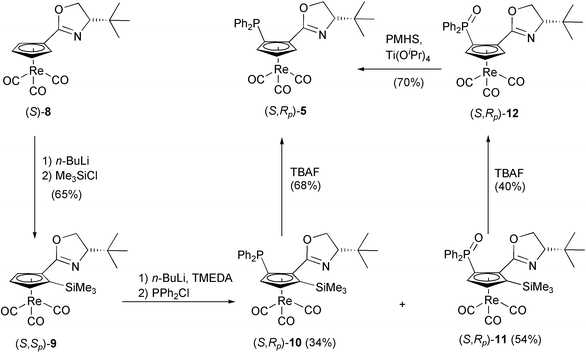 | ||
| Scheme 2 Synthesis of cyrhetrenyloxazoline 5. | ||
Results and discussion
The synthetic routes to 3–5 are depicted in Schemes 1 and 2. Cyrhetrenyloxazolines 3 and 5 differ in the sense of planar chirality, and 4 bears a more electron-rich and bulky phosphine substituent than 3.Treatment of cyrhetrenecarboxylic acid (6) with oxalyl chloride and subsequent reaction with (S)-tert-leucinol gave amide (S)-7 in 85% yield.11 The cyclization using Evans's protocol12 (TsCl, NEt3 and DMAP) went smoothly and afforded oxazoline (S)-8 in 93% yield. After a highly diastereoselective ortho-lithiation followed by quenching with R2PCl, the cyrhetrenyloxazoline phosphines (S,Sp)-3 (R = Ph, 56%) and (S,Sp)-4 (R = Cy, 66%) were obtained in a straightforward manner (Scheme 1).
To prepare (S,Rp)-5, the stereoisomer of (S,Sp)-3, the Sp position on the Cp ring was blocked by introducing a TMS group. Then, lithiation of (S,Sp)-9, which was obtained from (S)-8 in 65% yield, and trapping with Ph2PCl resulted in phosphine (S,Rp)-10 in 34% yield and unexpected phosphine oxide (S,Rp)-11 in 54% yield. Removal of the TMS-group by TBAF afforded phosphine (S,Rp)-5 in 68% yield starting from (S,R)-10, and phosphine oxide (S,Rp)-12 in 40% yield from (S,Rp)-11. Phosphine (S,Rp)-5 could also be obtained in 70% yield upon reduction of (S,Rp)-12 with poly(methylhydrosiloxane) (PMHS)–Ti(OiPr)4 (Scheme 2).
The catalytical properties of cyrhetrenyloxazolines (S,Sp)-3, (S,Sp)-4 and (S,Rp)-5 were examined in several catalytic model reactions such as allylic alkylation (Tables 1 and 2), allylic amination (Table 3), transfer hydrogenation (Table 4) and hydrosilylation (Table 5). A comparison of the new cyrhetrenyloxazolines with the corresponding ferrocenyl ligand (S,Sp)-2 under analogous conditions was also carried out.
As a starting point we chose to explore the applicability of the cyrhetrenyloxazolines in enantioselective Pd-catalyzed allylic substitutions, owing to its detailed mechanistic studies.13 Acyclic (reactions 1 and 2) and cyclic acetates (reactions 3–5) together with dimethyl malonate were used as substrates. The results are summarized in Tables 1 and 2.

| Entry | Reaction | Ligand | Reaction time/h | Yield (%)b | Ee (%)c | Abs. config.d |
|---|---|---|---|---|---|---|
| a See Experimental section. b After column chromatography. c For methyl (S,E)-2-methoxycarbonyl-3,5-diphenylpent-4-enoate (product of reaction 1) the ee was determined by HPLC using a chiral stationary phase (Chiracel AD); for methyl (S,E)-2-methoxycarbonyl-3-methylhex-4-enoate (product of reaction 2) the ee was calculated on the basis of the specific rotation. d The absolute configuration of the product was assigned by comparison of the sign of the specific rotation to the literature value. e 99% and 92.3% ees were reported in refs. 7a and 7b, respectively. f 35% ee was given in ref. 7a. | ||||||
| 1 | 1 | (S,Sp)-2 | 4 | 98 | 73e | (−)-(S) |
| 2 | 1 | (S,Sp)-3 | 2 | 99 | 65 | (−)-(S) |
| 3 | 1 | (S,Sp)-4 | 48 | 88 | 54 | (−)-(S) |
| 4 | 1 | (S,Rp)-5 | 72 | 80 | 21 | (−)-(S) |
| 5 | 2 | (S,Sp)-2 | 24 | 95 | 34f | (−)-(S) |
| 6 | 2 | (S,Sp)-3 | 24 | 93 | 34 | (−)-(S) |
| 7 | 2 | (S,Sp)-4 | 24 | 91 | 51 | (−)-(S) |
| 8 | 2 | (S,Rp)-5 | 24 | 92 | 53 | (−)-(S) |

| Entry | Reaction | Ligand | Yield (%)b | Ee (%)c | Abs. config.d |
|---|---|---|---|---|---|
| a See Experimental section, all the reactions were run for 24 h. b After column chromatography. No product was obtained in reactions reported in entries 6–8. c Calculated on the basis of specific rotation. d Absolute configuration of the product was assigned by comparison of the sign of specific rotation to literature data. | |||||
| 1 | 3 | (S,Sp)-2 | 95 | 64 | (+)-(R) |
| 2 | 3 | (S,Sp)-3 | 96 | 56 | (+)-(R) |
| 3 | 3 | (S,Sp)-4 | 94 | 51 | (+)-(R) |
| 4 | 3 | (S,Rp)-5 | 92 | 6 | (−)-(S) |
| 5 | 4 | (S,Sp)-2 | 94 | 65 | (+)-(R) |
| 6 | 4 | (S,Sp)-3 | — | — | — |
| 7 | 4 | (S,Sp)-4 | — | — | — |
| 8 | 4 | (S,Rp)-5 | — | — | — |
| 9 | 5 | (S,Sp)-2 | 97 | 83 | (+)-(R) |
| 10 | 5 | (S,Sp)-3 | 95 | 79 | (+)-(R) |
| 11 | 5 | (S,Sp)-4 | 60 | 42 | (−)-(S) |
| 12 | 5 | (S,Rp)-5 | 68 | 24 | (−)-(S) |
In the case of reactions with 1,3-diphenylallyl acetate (reaction 1), cyrhetrenyloxazoline (S,Sp)-3 was found to be the most active ligand. The reaction was complete after 2 h at room temperature giving the S-configurated product with 65% ee in 99% yield (Table 1, entry 2). The reaction with the sterically more bulky and electron-rich dicyclohexylphosphino ligand (S,Sp)-4 possessing the same absolute configuration required a longer reaction time and showed a slightly lower enantioselectivity (54% ee) (Table 1, entry 3). The diastereomeric ligand with opposite planar chirality (S,Rp)-5 was less active (80% yield after 3 days), and the ee of the product was significantly lower (21% ee) (Table 1, entry 4), indicating a mismatched combination of stereogenic elements in the ligand. Overall, these results are moderate as compared to the ferrocenyl analogue 2 (73% ee, Table 1, entry 1). As described for the latter type of ligand,7 the products resulting from the catalyses with the cyrhetrenyloxazolines had the same absolute configurations with both diastereomeric ligands.
The asymmetric alkylation of 1,3-dimethylallyl acetate (reaction 2) showed a very different trend. Here, (S,Rp)-5 gave the product with higher enantiomeric excess compared to (S,Sp)-3 (53% ee vs. 34% ee, Table 1, entries 6 and 8). Also, application of (S,Sp)-4 resulted in higher enantioselectivity (51% ee) than with (S,Sp)-3 (Table 1, entry 7). Furthermore it is noteworthy that with this substrate, two of the cyrhetrenyloxazolines [(S,Sp)-4 and (S,Rp)-5] performed better than their ferrocenyl counterpart (S,Sp)-2. The absolute configurations of the products were the same for all three cyrhetrenyloxazolines. This behavior is in agreement with the one determined for the ferrocenyl ligands, which showed that the chirality of the oxazoline subunit is the primary determinant of the stereochemical outcome of this allylation reaction.7b
The allylation reactions of cyclic substrates were also studied. With (S,Sp)-3 as ligand the best activity as well as enantioselectivity, 56% for 3-cyclopentyl acetate (Table 2, reaction 3) and 79% ee for 3-cycloheptyl acetate (reaction 5) (Table 2, entries 2 and 10) were achieved. Cyrhetrenyloxazoline (S,Rp)-5 with different planar chirality resulted in not only lower enantioselectivity but also opposite absolute configuration of the products (Table 2, entries 4 and 12). Interestingly, a strong substrate effect was observed with all ligands, especially with (S,Sp)-4, which gave an (R)-configurated product (51% ee) in the five (Table 2, entry 3), but an (S)-configurated one (42% ee) in the seven-membered ring system (Table 2, entry 11). No products have been obtained for the six-membered ring system (reaction 4) using cyrhetrenyl ligands.
The cyrhetrenyloxazolines have also been tested in asymmetric allylic amination reactions (Table 3). With (S,Sp)-3 as ligand the product was obtained with an excellent ee of 97% albeit in low yield (entry 2). Reactions with (S,Rp)-5 and (S,Sp)-4 yielded only traces of product (entries 3 and 4). Most interestingly, in terms of enantioselectivity cyrhetrenyl ligand (S,Sp)-3 proved to be superior here than its ferrocenyl analogue (S,Sp)-2, with which only 77% ee was reached (Table 3, entry 1).

| Entry | Ligand | Yield (%)b | Ee (%)c | Abs. config.d |
|---|---|---|---|---|
| a See Experimental section. b After column chromatography. c Determined by HPLC using a chiral stationary phase (Chiracel OJ). d The absolute configuration of the product was assigned by comparison of the sign of specific rotation to the literature value. e 47.7% ee was obtained at 40 °C in ref. 7b. | ||||
| 1 | (S,Sp)-2 | 40 | 77e | (−)-(R) |
| 2 | (S,Sp)-3 | 34 | 97 | (−)-(R) |
| 3 | (S,Sp)-4 | 2 | n.d. | n.d. |
| 4 | (S,Rp)-5 | 1 | n.d. | n.d. |
Transfer hydrogenation using propan-2-ol as a source of hydrogen is an attractive method for the reduction of ketones to alcohols, in view of the low cost of the reducing agent and operational simplicity.14 Phosferrox-type ligands have proved to be very successful for this type of reaction.9 Thus, it was of interest to test the activity of cyrhetrenyloxazolines in those ruthenium-catalyzed asymmetric transfer hydrogenations as well. The results with propiophenone as substrate are shown in Table 4. As compared to the ferrocenyl analogue 2, generally lower reactivities (only at reflux temperature for 2 h could a reasonable yield of the product be obtained) and reduced enantioselectivities were observed with cyrhetrenyl ligands 3–5. The highest ee of 64% was obtained with (S,Sp)-4, bearing an electron-rich phosphino group (Table 4, entry 3). Mismatched stereogenic elements led to a drop in the ee from 47% to 9% (Table 4, entries 2 and 4, respectively). Furthermore, the catalytic activity was reduced as indicated by the lower yield in the latter reaction.

| Entry | Ligand | Temp./°C | Reaction time/h | Yield (%)b | Ee (%)c |
|---|---|---|---|---|---|
| a See Experimental section. b After column chromatography. c Determined by HPLC using a chiral stationary phase (Chiracel OD–H). All products had R configuration. d Literature value.9b | |||||
| 1 | (S,Sp)-2 | 50 | 8 | 99 | 99.7d |
| 2 | (S,Sp)-3 | 82 | 2 | 86 | 47 |
| 3 | (S,Sp)-4 | 82 | 2 | 93 | 64 |
| 4 | (S,Rp)-5 | 82 | 2 | 13 | 9 |
The preparation of enantiomerically enriched alcohols by asymmetric hydrosilylation of ketones has proven to be very efficient, due to the exceedingly mild reaction conditions and technical simplicity.15 Herein, the Rh-catalyzed asymmetric hyrosilylation of acetophenone was used as a model reaction to test the cyrhetrenyl ligands. The results are listed in Table 5. The best result was obtained with (S,Sp)-3 at −20 °C using diethyl ether as the solvent (72% ee, entry 5). Use of cyrhetrenyloxazoline (S,Rp)-5 with the opposite planar chirality led to both lower reactivity and reduced enantioselectivity (17% ee, entry 8). The same effect was observed when (S,Sp)-4 bearing an electron-rich phosphino substituent was used as ligand (entry 7). Also in this case, cyrhetrenyloxazoline (S,Sp)-3 proved to be superior over its ferrocenyl analogue (S,Sp)-2 (72 vs. 58% ee, entries 5 and 6, respectively).

| Entry | Ligand | Solvent | Temp./°C | Reaction time/h | Yield (%)b | Ee (%)c |
|---|---|---|---|---|---|---|
| a See Experimental section. b After column chromatography. c Determined by GC using a chiral column. All products had R configuration. | ||||||
| 1 | (S,Sp)-3 | — | 0 | 24 | 87 | 28 |
| 2 | (S,Sp)-3 | THF | 0 | 24 | 91 | 34 |
| 3 | (S,Sp)-3 | Toluene | 0 | 24 | 73 | 46 |
| 4 | (S,Sp)-3 | Ether | 0 | 24 | 82 | 48 |
| 5 | (S,Sp)-3 | Ether | −20 | 60 | 55 | 72 |
| 6 | (S,Sp)-2 | Ether | −20 | 60 | 27 | 58 |
| 7 | (S,Sp)-4 | Ether | −20 | 60 | 41 | 33 |
| 8 | (S,Rp)-5 | Ether | 0 | 60 | 28 | 17 |
In conclusion, cyrhetrenyloxazolines 3–5 compared to the ferrocenyl analogue 2 showed preferable effects when applied as ligands in asymmetric allylic amination and hydrosilylation reactions. In allylic alkylations the results achieved with both ligand classes were comparable. Lower catalytic activities were found when the rhenium complexes were used as ligands in transfer hydrogenations. In most of the cases, the sense of asymmetric induction clearly depended on the configuration of the stereogenic center at the ligand. However, as in the ferrocene case,7b,16 the planar chirality also plays an important role in the catalyses.
The Re(CO)3 fragment differs from the FeCp one in both steric and electronic properties. A study to quantify the latter has thus been carried out in order to gain a more detailed view of the catalytic behaviors of the respective ligands. For this purpose, selenium derivatives of 2 and 3 were prepared and investigated by NMR spectroscopy.
Allen et al.17have reported that the electron-donating ability of phosphorus towards metal acceptors is indicated by the 1J(77Se–31P) coupling constants of the phosphine selenides in 31P NMR spectroscopy. The respective coupling constants increase as the groups attached to phosphorus become more electron withdrawing, indicating an increased s character for the phosphorus lone pair, forming an apparent weaker donor to metals. The phosphine selenides 13 and 14 (Fig. 2) were thus prepared by reacting (S,Sp)-2 and (S,Sp)-3, respectively, with elemental selenium in CHCl3.
 | ||
| Fig. 2 | ||
The higher coupling constant 1J(77Se–31P) = 394 Hz for complex 14 compared to 13 (325 Hz) and the stronger shielding (29.01 ppm vs. 35.26 ppm) revealed that the phosphorus on ferrocene was a better donor than the one on cyrhetrene. The Lewis basicity of ferrocene (S,Sp)-2 is thus higher than that of (S,Sp)-3.
Since both reactivity and selectivity are controlled by steric and electronic factors, it is not surprising that ligands with different phosphine basicity and different steric requirements lead to different results. To fully rationalize them and to draw general conclusions is difficult at the present stage of our investigations.
Conclusions
In summary, we have designed and synthesized a number of novel planar chiral phosphinocyrhetrenyloxazolines and applied them as ligands in a series of catalytic reactions. Although in most cases, the results cannot compete with the best ones reported in the literature, it became clear, that cyrhetrenes represent a new family of ligands with promising properties. Their modular construction allows an extensive and independent variation of the phosphine part (including the introduction of further stereogenic centers), the metal-containing backbone (for example, the CO could be substituted by CN, or phosphine), and the oxazoline (by introducing substituents other than the bulky tert-butyl group). The established understanding of the stereoelectronic properties of the cyrhetrene unit makes it possible to optimize a particular reaction by varying the ligand structure. Therefore, in view of this electronic and steric tunability, we are optimistic that cyrhetrene-based ligands such as the phosphinocyrhetrenyloxazolines reported here, will find further applications in asymmetric catalysis. Experiments towards modified ligands and investigations on additional catalytic reactions are currently ongoing in our laboratories.Experimental
General
Melting points were determined on a Büchi B-540 melting point apparatus and are uncorrected. NMR spectra were recorded on a Varian Gemini 300 spectrometer at 300 MHz (1H NMR) and 75 MHz (13C NMR), respectively, in CDCl3 unless otherwise noted. Chemical shifts δ are reported in ppm relative to the internal reference TMS. Mass spectra were measured on a Varian MAT 212 or a Finnigan SSQ 7000 spectrometer with EI ionization. IR spectra were measured on a Perkin Elmer 1760 S as KBr pellets. Elemental analyses were performed at the Institut für Organische Chemie der RWTH, Aachen, on a Heraeus CHNO-Rapid apparatus. Optical rotations were determined on a Perkin-Elmer polarimeter 241 in CHCl3 at ambient temperature. All manipulation except work up and purification were conducted under Ar atmosphere using standard Schlenk techniques. Et2O, toluene and THF were distilled from sodium–benzophenone ketyl radical, CH2Cl2, CHCl3 and propan-2-ol were dried over CaH2 prior to use. Flash chromatography was conducted using silica gel (MERCK, 40–63 µm) and light petroleum ether (30–60 °C). (S,Sp)-2-[2-(Diphenylphosphino)ferrocenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (2) was synthesized according to the literature procedure;6 (S)-[N-(2-hydroxy-1-tert-butylethyl)]cyrhetrenecarboxamide (7) was prepared according to the reported method;4 (S)-2-cyrhetrenyl-4-tert-butyl-4,5-dihydro-1,3-oxazole (8) was synthesized from 7 in 93% yield, with use of toluene-p-sulfonyl chloride (1 equiv.), Et3N (1.2 equiv.) and DMAP (0.1 equiv.) in CH2Cl2.12 The analytical data of compound 8 is in accordance with the reported data.4(S,Sp)-2-[2-(Diphenylphosphino)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (3)
To a degassed solution of (S)-2-cyrhetrenyl-4-tert-butyl-4,5-dihydro-1,3-oxazole (8, 1.68 g, 3.65 mmol) in Et2O (50 mL) was added dropwise n-BuLi (2.57 mL of a 1.7 M solution in hexane, 4.37 mmol) at −78 °C. The reaction mixture was stirred at this temperature for 15 min, then Ph2PCl (1.16 g, 5.25 mmol) was added. After 40 min at −78 °C the reaction was quenched with saturated NaHCO3 solution (10 mL), and the organic phase was separated, washed with brine, and dried over MgSO4. After removal of the solvent, the residue was purified by chromatography (silica gel). Elution with petroleum ether–diethyl ether (9 ∶ 1) afforded 3 as a white foam (1.32 g, 56%). [α]D = +2.8 (c = 0.5, CHCl3); anal. calcd for C27H25NO4PRe: C, 50.30; H, 3.91; N, 2.17; found: C, 50.58; H, 4.31; N, 1.92%; IR (KBr):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1
= 3419, 2954, 2026, 1928, 1667; 1H NMR: δ 0.88 (s, 9H, CH3), 3.73 (dd, 1H, J
= 8.5 and 8.2 Hz, CH2), 3.90 (dd, 1H, J
= 9.2 and 8.2 Hz, CH), 4.18 (dd, 1H, J
= 9.2 and 8.5 Hz, CH2), 4.70 (m, 1H, Cp–H), 5.20 (m, 1H, Cp–H), 6.09 (m, 1H, Cp–H), 7.30–7.43 (m, 10H, Ph); 13C NMR: δ 26.16 (CH3), 34.25 (C), 69.41 (CH2), 76.78 (CH), 82.65 (d, J
= 2.3 Hz, Cp–CH), 91.26 (Cp–CH), 92.57 (d, J
= 5.1 Hz, Cp–CH), 95.06 (d, J
= 16.0 Hz, Cp–C), 102.14 (d, J
= 28.6 Hz, Cp–C), 128.91 (d, J
= 6.8 Hz, Ph–CH), 129.05 (d, J
= 6.7 Hz, Ph–CH), 129.26 (Ph–CH), 129.88 (Ph–CH), 133.13 (d, J
= 20.0 Hz, Ph–CH), 134.97 (d, J
= 20.5 Hz, Ph–CH), 136.33 (d, J
= 14.2 Hz, Ph–C), 138.28 (d, J
= 11.4 Hz, Ph–C), 158.54 (C), 193.25 (CO); 31P NMR: δ
−16.02; MS m/z
(rel%)
= 645 (M+, 58), 617 (100), 559 (15), 483 (30).
/cm−1
= 3419, 2954, 2026, 1928, 1667; 1H NMR: δ 0.88 (s, 9H, CH3), 3.73 (dd, 1H, J
= 8.5 and 8.2 Hz, CH2), 3.90 (dd, 1H, J
= 9.2 and 8.2 Hz, CH), 4.18 (dd, 1H, J
= 9.2 and 8.5 Hz, CH2), 4.70 (m, 1H, Cp–H), 5.20 (m, 1H, Cp–H), 6.09 (m, 1H, Cp–H), 7.30–7.43 (m, 10H, Ph); 13C NMR: δ 26.16 (CH3), 34.25 (C), 69.41 (CH2), 76.78 (CH), 82.65 (d, J
= 2.3 Hz, Cp–CH), 91.26 (Cp–CH), 92.57 (d, J
= 5.1 Hz, Cp–CH), 95.06 (d, J
= 16.0 Hz, Cp–C), 102.14 (d, J
= 28.6 Hz, Cp–C), 128.91 (d, J
= 6.8 Hz, Ph–CH), 129.05 (d, J
= 6.7 Hz, Ph–CH), 129.26 (Ph–CH), 129.88 (Ph–CH), 133.13 (d, J
= 20.0 Hz, Ph–CH), 134.97 (d, J
= 20.5 Hz, Ph–CH), 136.33 (d, J
= 14.2 Hz, Ph–C), 138.28 (d, J
= 11.4 Hz, Ph–C), 158.54 (C), 193.25 (CO); 31P NMR: δ
−16.02; MS m/z
(rel%)
= 645 (M+, 58), 617 (100), 559 (15), 483 (30).
(S,Sp)-2-[2-(Dicyclohexylphosphino)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (4)
Phosphine 4 was synthesized as described for compound 3, starting from 8 (692 mg, 1.5 mmol) using chlorodicyclohexylphosphine (535 mg, 2.3 mmol) as nucleophile. Column chromatography on silica gel using petroleum ether–diethyl ether (9 ∶ 1) as eluent afforded 4 as a white foam (650 mg, 66%). [α]D = −18.6 (c = 0.7, CHCl3); anal. calcd for C27H37NO4PRe: C, 49.38; H, 5.68; N, 2.13; found: C, 49.54; H, 5.88; N, 1.78%; IR (KBr):/cm−1
= 3432, 2926, 2851, 2026, 1927, 1664; 1H NMR: δ 0.86 (s, 9H, CH3), 0.77–2.04 (m, 22H, Cy), 3.84 (dd, 1H, J
= 8.8 and 9.6 Hz, CH2), 4.03 (dd, 1H, J
= 8.8 and 8.8 Hz, CH), 4.25 (dd, 1H, J
= 8.8 and 8.5 Hz, CH2), 5.22 (m, 1H, Cp–H), 5.40 (m, 1H, Cp–H), 6.08 (m, 1H, Cp–H); 13C NMR: δ 26.41 (CH3), 26.64 (CH2), 27.28 (d, J
= 9.2 Hz, CH2), 27.84 (d, J
= 7.6 Hz, CH2), 29.34 (d, J
= 13.7 Hz, CH2), 30.15 (d, J
= 11.4 Hz, CH2), 32.25 (d, J
= 16.8 Hz, CH2), 33.73 (d, J
= 15.3 Hz, CH), 34.11 (C), 36.92 (d, J
= 16.0 Hz, CH), 69.21 (CH2), 76.49 (CH), 81.85 (Cp–CH), 90.86 (d, J
= 2.3 Hz, Cp–CH), 92.47 (Cp–CH), 100.31 (d, J
= 44.0 Hz, Cp–C), 103.04 (Cp–C), 159.68 (C), 193.09 (CO); 31P NMR: δ
−7.62; MS (CI)
m/z
(rel%)
= 658 (100), 657 (M+, 20), 574 (33).
(S,Sp)-2-[2-(Trimethylsilyl)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (9)
To a degassed solution of 8 (1.72 g, 3.73 mmol) in diethyl ether (50 mL) was added dropwise n-BuLi (2.65 mL of a 1.7 M solution in hexane, 4.51 mmol) at −78 °C. The reaction mixture was stirred at this temperature for 15 min. Then, chlorotrimethylsilane (0.72 mL, 610 mg, 5.60 mmol) was added. The mixture was allowed to warm to room temperature and stirred for another 3 h. The reaction was quenched with saturated NaHCO3 solution (10 mL), and the organic phase was separated, washed with brine, and dried with MgSO4. The solvent was evaporated and the residue was purified by chromatography on silica gel. Elution with petroleum ether–diethyl ether (19 ∶ 1 → 9 ∶ 1) afforded 9 (1.29 g, 65%) as a white solid. Mp 106 °C; [α]D = −40.8 (c = 0.5, CHCl3); anal. calcd for C18H24NO4ReSi: C, 40.59; H, 4.54; N, 2.63; found: C, 40.79; H, 4.45; N, 2.58%; IR (KBr):/cm−1
= 3119, 2960, 2019, 1931, 1663; 1H NMR: δ 0.00 (s, 9H, CH3), 0.59 (s, 9H, CH3), 3.63 (dd, 1H, J
= 8.8 and 9.0 Hz, CH2), 3.74 (dd, 1H, J
= 8.8 and 8.8 Hz, CH), 3.91 (dd, 1H, J
= 8.8 and 8.3 Hz, CH2), 5.02 (m, 1H, Cp–H), 5.06 (m, 1H, Cp–H), 5.60 (m, 1H, Cp–H); 13C NMR: δ 0.00 (SiCH3), 25.53 (CH3), 33.42 (C), 68.01 (CH2), 76.31 (CH), 84.79 (Cp–CH), 88.56 (Cp–CH), 92.29 (Cp–C), 93.83 (Cp–CH), 97.40 (Cp–C), 158.17 (C), 192.66 (CO); MS m/z
(rel%)
= 533 (M+, 39), 518 (15), 476 (100).
(S,Rp)-2-[2-(Diphenylphosphino)-5-(trimethylsilyl)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (10) and (S,Rp)-2-[2-(diphenylphosphinoyl)-5-(trimethylsilyl)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (11)
To a degassed solution of compound 9 (1.17 g, 2.2 mmol) and TMEDA (302 mg, 0.39 mL, 2.6 mmol) in diethyl ether (25 mL) was added dropwise n-BuLi (1.53 mL of a 1.7 M solution in hexane, 2.6 mmol) at −78 °C. The reaction mixture was stirred at this temperature for 10 min. Then, Ph2PCl (729 mg, 3.3 mol) was added. After 40 min at −78 °C, the reaction was quenched with saturated NaHCO3 solution (10 mL), and the organic phase was separated, washed with brine, and dried over MgSO4. After removal of the solvent, the residue was purified by colmn chromatography (silica gel). Elution with petroleum ether–diethyl ether (9 ∶ 1) afforded phosphine 10 (536 mg, 34%) as a white foam, and further elution with petroleum ether–diethyl ether (1 ∶ 4) gave the unexpected phosphine oxide 11 (871 mg, 54%) as a white solid.Phosphine 10: [α]D
=
+15.6 (c
= 0.5, CHCl3); anal. calcd for C30H33NO4PReSi: C, 50.26; H, 4.64; N, 1.95; found: C, 50.16; H, 4.66; N, 1.71%; IR (CHCl3): /cm−1
= 3055, 2958, 2025, 1934, 1660; 1H NMR: δ 0.24 (s, 9H, CH3), 0.51 (s, 9H, CH3), 3.81 (dd, 1H, J
= 7.5 and 10.3 Hz, CH2), 3.92–4.01 (m, 2H, CH and CH2), 4.70 (m, 1H, Cp–H), 5.19 (m, 1H, Cp–H), 7.16–7.39 (m, 10H, Ph–H); 13C NMR: δ 0.00 (SiCH3), 24.64 (CH3), 28.90 (C), 32.73 (C), 67.52 (CH2), 75.46 (CH), 91.39 (Cp–CH), 92.50 (d, J
= 4.8 Hz, Cp–CH), 95.95 (Cp–C), 101.36 (d, J
= 14.4 Hz, Cp–C), 102.71 (d, J
= 28.2 Hz, Cp–C), 127.30 (d, J
= 7.1 Hz, Ph–CH), 127.35 (Ph–CH), 127.53 (d, J
= 7.2 Hz, Ph–CH), 128.46 (Ph–CH), 131.10 (d, J
= 19.8 Hz, Ph–CH), 134.17 (d, J
= 21.5 Hz, Ph–CH), 135.59 (d, J
= 14.4 Hz, Ph–C), 137.73 (d, J
= 10.7 Hz, Ph–C), 157.56 (d, J
= 3.6 Hz, C), 192.22 (CO); 31P NMR: δ
−15.66; MS m/z
(rel%)
= 717 (M+, 41), 689 (81), 660 (100), 605 (74).
Phosphine oxide 11: mp 62 °C; [α]D
=
−38.4 (c
= 0.5, CHCl3); anal. calcd for C30H33NO5PReSi: C, 49.17; H, 4.54; N, 1.91; found: C, 49.74; H, 5.07; N, 1.65%; IR (KBr): /cm−1
= 3420, 2956, 2028, 1929, 1665; 1H NMR: δ 0.01 (s, 9H, CH3), 0.44 (s, 9H, CH3), 3.15 (dd, 1H, J
= 10.1 and 10.1 Hz, CH2), 3.42–3.54 (m, 2H, CH and CH2), 5.08 (m, 1H, Cp–H), 5.38 (m, 1H, Cp–H), 7.10–7.27 (m, 6H, Ph–H), 7.45–7.52 (m, 2H, Ph–H), 7.62–7.70 (m, 2H, Ph–H); 13C NMR: δ 0.00 (CH3), 25.25 (CH3), 26.18 (C), 32.51 (C), 67.95 (CH2), 75.89 (CH), 91.33 (d, J
= 10.8 Hz, Cp–CH), 94.18 (d, J
= 13.2 Hz, Cp–C), 95.54 (d, J
= 11.3 Hz, Cp–CH), 100.10 (d, J
= 10.77 Hz, Cp–C), 103.69 (d, J
= 36.9 Hz, Cp–C), 127.01 (d, J
= 13.2 Hz, Ph–CH), 127.36 (d, J
= 12.5 Hz, Ph–CH), 130.69 (d, J
= 3.0 Hz, Ph–CH), 130.92 (d, J
= 3.0 Hz, Ph–CH), 131.30 (d, J
= 5.3 Hz, Ph–CH), 131.43 (d, J
= 6.0 Hz, Ph–CH), 133.59 (d, J
= 32.7 Hz, Ph–C), 135.67 (d, J
= 28.1 Hz, Ph–C), 155.16 (C), 191.02 (CO); 31P NMR: δ 23.64; MS m/z
(rel%)
= 733 (M+, 62), 705 (100).
(S,Rp)-2-[2-(Diphenylphosphinoyl)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (12)
To a solution of 11 (700 mg, 0.95 mmol) in THF (5 mL) was added TBAF (5 mL of a 1 M solution in THF), the resulting mixture was stirred at room temperature for 5 min. After removal of the solvent in vacuo, the residue was purified by column chromatography (silica gel). Elution with petroleum ether–ethyl acetate (1 ∶ 1 → 0 ∶ 1) afforded compound 12 (251 mg, 40%) as a white solid. Mp: 158 °C (dec.); [α]D = −21.7 (c = 0.52, CHCl3); anal. calcd for C27H25NO5PRe: C, 49.08; H, 3.81; N, 2.12; found: C, 49.19; H, 3.67; N, 2.00%; IR (KBr):/cm−1
= 3438, 2956, 2028, 1926, 1664; 1H NMR: δ 0.62 (s, 9H, CH3), 3.55–3.65 (m, 2H, CH and CH2), 3.88 (dd, 1H, J
= 7.1 and 7.7 Hz, CH2), 5.26 (m, 1H, Cp–H), 5.45 (m, 1H, Cp–H), 5.95 (m, 1H, Cp–H), 7.35–7.48 (m, 6H, Ph), 7.67–7.73 (m, 2H, Ph), 7.76–7.82 (m, 2H, Ph); 13C NMR: δ 25.96 (CH3), 34.13 (C), 69.30 (CH2), 76.39 (CH), 83.08 (d, J
= 9.2 Hz, Cp–CH), 89.87 (d, J
= 6.9 Hz, Cp–CH), 94.23 (Cp–C), 97.29 (d, J
= 10.7 Hz, Cp–C), 97.39 (d, J
= 10.7 Hz, Cp–CH), 128.29 (d, J
= 13.0 Hz, Ph–CH), 128.62 (d, J
= 12.2 Hz, Ph–CH), 132.01 (d, J
= 9.9 Hz, Ph–CH), 132.03 (d, J
= 19.8 Hz, Ph–C), 132.05 (d, J
= 3.8 Hz, Ph–CH), 132.23 (d, J
= 3.1 Hz, Ph–CH), 132.47 (d, J
= 13.7 Hz, Ph–CH), 133.10 (d, J
= 25.2 Hz, Ph–C), 157.29 (C), 191.26 (CO); 31P NMR: δ 23.21; MS m/z
(rel%)
= 661 (M+, 74), 632 (100), 575 (50), 520 (19).
(S,Rp)-2-[2-(Diphenylphosphino)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (5)
Mp: 151 °C (dec.); [α]D
=
+63.4 (c
= 0.5, CHCl3); anal. calcd for C27H25NO4PRe: C, 50.30; H, 3.91; N, 2.17; found: C, 50.50; H, 4.04; N, 2.00%; IR (KBr): /cm−1
= 3438, 2956, 2028, 1926, 1664; 1H NMR: δ 0.54 (s, 9H, CH3), 3.78 (dd, 1H, J
= 8.6 and 8.3 Hz, CH2), 3.92–4.02 (m, 2H, CH and CH2), 4.61 (m, 1H, Cp–H), 5.16 (m, 1H, Cp–H), 6.01 (m, 1H, Cp–H), 7.22–7.34 (m, 10H, Ph); 13C NMR: δ 25.82 (CH3), 34.21 (C), 69.04 (CH2), 76.35 (CH), 82.92 (Cp–CH), 90.49 (Cp–CH), 92.42 (d, J
= 5.3 Hz, Cp–CH), 94.41 (Cp–C), 101.66 (d, J
= 10.7 Hz, Cp–C), 128.59 (Ph–CH), 128.71 (d, J
= 4.2 Hz, Ph–CH), 128.87 (d, J
= 6.6 Hz, Ph–CH), 129.69 (Ph–CH), 132.65 (d, J
= 19.7 Hz, Ph–CH), 133.65 (d, J
= 20.3 Hz, Ph–CH), 136.10 (d, J
= 12.0 Hz, Ph–C), 138.28 (d, J
= 11.4 Hz, Ph–C), 159.13 (C), 192.72 (CO); 31P NMR: δ
−15.99; MS m/z
(rel%)
= 645 (M+, 46), 617 (100), 560 (19), 483 (20).
(S,Sp)-2-[2-(Diphenylselenophosphinoyl)ferrocenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (13)
Compound 13 was prepared by stirring ferrocenylphosphine 2 (119 mg, 0.2 mmol) with selenium (158 mg, 2 mmol) in CHCl3 (2 mL) at room temperature for 10 min. After filtration of the excess of selenium under Ar, the solvent was removed under vacuum. The resulting product (yellow solid) was directly used for the spectroscopical analysis. Mp: 172 °C (dec.); [α]D = +52.0 (c = 0.25, CHCl3); anal. calcd for C29H30FeNOPSe: C, 60.65; H, 5.26; N, 2.44; found: C, 58.89; H, 5.50; N, 2.16%; IR (KBr):/cm−1
= 3449, 2955, 1664; 1H NMR: δ 0.70 (s, 9H, CH3), 3.57 (br m, 2H, CH2), 3.97 (br m, 2H, CH and Cp–H), 4.41 (br s, 6H, Cp–H), 5.00 (m, 1H, Cp–H), 7.24–7.40 (m, 6H, Ph), 7.66–7.79 (m, 4H, Ph); 13C NMR: δ 26.33 (CH3), 33.97 (C), 68.58 (CH2), 72.44 (5 Cp–CH), 76.54 (CH), 82.92 (Cp–CH), 90.49 (Cp–CH), 92.42 (d, J
= 5.3 Hz, Cp–CH), 94.41 (Cp–C), 101.66 (d, J
= 10.7 Hz, Cp–C), 129.02 (d, J
= 12.6 Hz, Ph–CH), 128.23 (d, J
= 11.9 Hz, Ph–CH), 131.05 (Ph–CH), 131.06 (Ph–CH), 132.43 (d, J
= 10.2 Hz, Ph–CH), 132.83 (d, J
= 10.8 Hz, Ph–CH), 136.10 (d, J
= 12.0 Hz, Ph–C), 138.28 (d, J
= 11.4 Hz, Ph–C), 159.13 (C), 192.72 (CO); 31P NMR: δ 35.26 (J
= 325.0 Hz); MS m/z
(rel%)
= 574 (M+, 14), 511 (100), 454 (23), 410 (68).
(S,Sp)-2-[2-(Diphenylselenophosphinoyl)cyrhetrenyl]-4-tert-butyl-4,5-dihydro-1,3-oxazole (14)
Compound 14 was obtained as a white solid in the same way as compound 13, except for a higher excess of selenium (50 equiv.) and longer reaction time (3 h). Mp: 82 °C (dec.); [α]D = −11.0 (c = 1.0, CHCl3); anal. calcd for C27H25NO4PReSe: C, 44.81; H, 3.48; N, 1.94; found: C, 45,92; H, 4.27; N, 1.61%; IR (KBr):/cm−1
= 3449, 2958, 2013, 1894; 1H NMR (d8-toluene): δ 0.44 (s, 9H, CH3), 3.05 (dd, 1H, J
= 9.2 and 9.2 Hz, CH2), 3.18 (dd, 1H, J
= 9.2 and 8.4 Hz, CH), 3.31 (dd, 1H, J
= 8.4 and 8.2 Hz, CH2), 4.00 (m, 1H, Cp–H), 4.95 (m, 1H, Cp–H), 5.13 (m, 1H, Cp–H), 6.70–6.85 (m, 6H, Ph), 7.61–7.80 (m, 4H, Ph); 13C NMR (d8-toluene): δ 25.87 (CH3), 33.44 (C), 68.74 (CH2), 76.45 (CH), 82.11 (d, J
= 9.0 Hz, Cp–CH), 88.33 (d, J
= 5.9 Hz, Cp–CH), 98.38 (d, J
= 12.0 Hz, Cp–C), 100.19 (Cp–C), 106.00 (Cp–C), 127.06 (d, J
= 13.8 Hz, Ph–CH), 127.30 (d, J
= 12.0 Hz, Ph–C), 127.47 (d, J
= 13.0 Hz, Ph–CH), 127.80 (d, J
= 12.2 Hz, Ph–CH), 131.13 (d, J
= 3.0 Hz, Ph–CH), 132.51 (d, J
= 39.5 Hz, Ph–C), 132.60 (d, J
= 11.4 Hz, Ph–CH), 133.29 (d, J
= 11.4 Hz, Ph–CH), 153.72 (C), 191.82 (CO); 31P NMR: δ 29.01 (J
= 394.0 Hz); MS m/z
(rel%)
= 724 (M+, 14), 695 (38), 561 (82), 633 (100), 575 (45).
General procedure for asymmetric allylic alkylation reactions
[Pd(η3-C3H5)Cl]2 (1.8 mg, 0.005 mmol, 1 mol% Pd) and the ligand (0.01 mmol, 1 mol%) were mixed in degassed CH2Cl2 (1 mL). To this pale yellow solution were subsequently added under argon the substrate [reaction 1: (E)-1,3-diphenylprop-2-en-1-yl acetate (252 mg, 1 mmol); reaction 2: (E)-pent-3-en-2-yl acetate (128 mg, 1 mmol); reaction 3: cyclopent-2-enyl acetate (126 mg, 1 mmol); reaction 4: cyclohex-2-enyl acetate (140 mg, 1 mmol); reaction 5: cyclohept-2-enyl acetate (154 mg, 1 mmol)], dimethyl malonate (390 mg, 0.34 mL, 3 mmol), BSA (610 mg, 0.74 mL, 3 mmol) and a catalytic amount of KOAc. The reaction mixture was degassed and then stirred at a given temperature. The progress of the reaction was monitored by TLC. When the reaction was complete, diethyl ether (15 mL) was added and the organic layer was washed twice with saturated NH4Cl solution and dried over MgSO4. After removal of the solvent, the residue was purified by column chromatography (silica gel).Reaction 1: eluent: petroleum ether–dichloromethane (1 ∶ 1); the enantiomer ratio was determined by chiral HPLC (Chiralcel AD, 250 × 4.6 mm2, 0.5 ml min−1, propan-2-ol–n-heptane (5 ∶ 95), 20 °C, TR = 26.86 min, TS = 37.34 min); specific rotation of optically pure methyl (S,E)-2-methoxycarbonyl-3,5-diphenylpent-4-enoate: [α]25D = −22.4 (c = 1.8, CHCl3).18
Reaction 2: eluent: petroleum ether–diethyl ether (3 ∶ 1); the enantiomer ratio was calculated based on the specific rotation; specific rotation of optically pure methyl (S,E)-2-methoxycarbonyl-3-methylhex-4-enoate: [α]20D = −19.8 (c = 1.1, CHCl3).19
Reaction 3: eluent: petroleum ether–diethyl ether (3 ∶ 1); the enantiomer ratio was calculated based on the specific rotation; specific rotation of optically pure dimethyl (R)-cyclopent-2-enylmalonate: [α]20D = −98.7 (c = 2.27, CHCl3).20
Reaction 4: eluent: petroleum ether–diethyl ether (3 ∶ 1); the enantiomer ratio was calculated based on the specific rotation; specific rotation of optically pure dimethyl (R)-cyclohex-2-enylmalonate: [α]20D = −46.1 (c = 2.86, CHCl3).20
Reaction 5: eluent: petroleum ether–diethyl ether (3 ∶ 1); the enantiomer ratio was calculated based on the specific rotation; specific rotation of optically pure dimethyl (R)-cyclohept-2-enylmalonate: [α]20D = −7.8 (c = 3.04, CHCl3).20
General procedure for the asymmetric allylic amination reaction
[Pd(η3-C3H5)Cl]2 (1.8 mg, 0.005 mmol, 1 mol% Pd) and the ligand (1 mol%) were mixed in degassed CH2Cl2 (1 mL). To this pale yellow solution were subsequently added (E)-1,3-diphenylprop-2-en-1-yl acetate (252 mg, 1 mmol) and benzylamine (129 mg, 1.2 mmol). The reaction mixture was degassed and then stirred at room temperature. The progress of the reaction was monitored by TLC. When the reaction was complete, the solvent was removed, the residue was purified by column chromatography on silica gel with petroleum ether–ethyl acetate (95 ∶ 5) as eluent. The enantiomer ratio was determined by chiral HPLC (Chiralcel OJ, 250 × 4.6 mm2, 1 mL min¬1, ethanol–n-heptane (2 ∶ 98), 20 °C, TS = 17.44 min, TR = 23.76 min); specific rotation of optically pure (R,E)-N-benzyl(1,3-diphenylprop-2-enyl)amine: [α]20D = −24.8 (c = 1.4, CHCl3).21General procedure for the asymmetric transfer hydrogenation reaction
A degassed solution of Ru(PPh3)3Cl2 (4.8 mg, 0.25 mmol%) and ligand (1 mmol%) in anhydrous propan-2-ol (4 mL) was heated to 82 °C for 30 min. After cooling to room temperature a solution of propiophenone (134 mg, 1 mmol) in dry degassed propan-2-ol (6 mL) was added via cannula, followed by KOtBu (1 mL of 0.1 M freshly prepared solution in propan-2-ol, 0.1 mmol). The resulting reaction was stirred at a given temperature for a certain time. For work-up, the reaction mixture was filtered through a pad of silica gel, washed with EtOAc. Concentration and purification by column chromatography using petroleum ether–diethyl ether (3 ∶ 1) as eluent afforded the product as a colorless liquid. The enantiomer ratio was determined by chiral HPLC (Chiralcel OD-H, 250 × 4.6 mm2, 0.5 mL min¬1, propan-2-ol–n-heptane (4 ∶ 96), 20 °C, TR = 18.30 min, TS = 20.11 min).General procedure for the asymmetric hydrosilylation reaction
The catalyst was prepared in situ by stirring [Rh(cod)Cl]2 (1.23 mg, 0.25% mmol) and the ligand (1% mmol) in degassed solvent (1 mL) at room temperature for 1 h. After addition of acetophenone (120 mg, 1 mmol), the reaction flask was dipped into a thermo-regulated bath at a given temperature. Diphenylsilane (276 mg, 0.28 mL, 1.5 mmol) was then slowly added by syringe. The reaction was monitored by TLC. For work-up, methanol (1 mL) was slowly added to the reaction mixture at 0 °C, which was stirred for 0.5 h. After gas evolution ceased, 1 M aqueous HCl (1 mL) was added, and stirring was continued at room temperature for 1 h. The reaction mixture was diluted with diethyl ether, and washed with water; the organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (silica gel) using petroleum ether–diethyl ether (2 ∶ 1) as eluent to afford the product as a colorless oil. The enantiomer ratio was determined by chiral GC (cyclodex β-I/P, 2,3,6-trimethyl-β-cyclodextrin, 0.25 mm × 25 m, 100 kPa N2, column 120 °C, injector 200 °C, split 1 ∶ 1, TR = 17.49 min, TS = 18.75 min).Acknowledgements
We thank the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (DFG) within the Collaborative Research Center (SFB) 380 ‘Asymmetric Synthesis by Chemical and Biological Methods’ for financial support. L. X. is grateful to the Alexander von Humboldt Foundation for a postdoctoral fellowship.References
- (a) Transition Metals in Organic Synthesis, eds. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 1998 Search PubMed; (b) Comprehensive Asymmetric Catalysis, eds. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed; (c) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2000 Search PubMed.
- (a) Ferrocenes, eds. A. Togni and T. Hayashi, VCH, Weinheim, 1995 Search PubMed; (b) A. Togni, Chimia, 1996, 50, 86 Search PubMed; (c) A. Togni, Angew. Chem., 1996, 108, 1581 ( Angew. Chem., Int. Ed. Engl. , 1996 , 35 , 1475 ); (d) H.-U. Blaser, H.-P. Buser, K. Coers, R. Hanreich, H.-P. Jalett, E. Jelsch, B. Pugin, H.-D. Schneider, F. Spindler and A. Wegmann, Chimia, 1999, 53, 275 Search PubMed; (e) T. Sturm, L. Xiao and W. Weissensteiner, Chimia, 2001, 55, 688 Search PubMed.
- (a) C. Bolm and K. Muñiz, Chem. Commun., 1999, 1295 RSC; (b) C. Bolm, N. Hermanns, J. P. Hildebrand and K. Muñiz, Angew. Chem., 2000, 112, 3609 ( Angew. Chem., Int. Ed. , 2000 , 39 , 3465 ) CrossRef.
- C. Bolm, M. Kesselgruber, N. Hermanns, J. P. Hildebrand and G. Raabe, Angew. Chem., 2001, 113, 1536 ( Angew. Chem., Int. Ed. , 2001 , 40 , 1488 ) CrossRef.
- (a) C. J. Richards, T. Damalidis, D. E. Hibbs and M. B. Hursthouse, Synlett, 1995, 74 CrossRef CAS; (b) T. Sammakia, H. A. Latham and D. R. Schaad, J. Org. Chem., 1995, 60, 10 CrossRef CAS; (c) Y. Nishibayashi and S. Uemura, Synlett, 1995, 79 CrossRef CAS.
- (a) C. J. Richards and A. W. Mulvaney, Tetrahedron: Asymmetry, 1996, 7, 1419 CrossRef CAS; (b) K. H. Ahn, C.-W. Cho, H.-H. Beak, J. Park and S. Lee, J. Org. Chem., 1996, 61, 4937 CrossRef CAS.
- (a) K. H. Ahn, C.-W. Cho, J. Park and S. Lee, Tetrahedron: Asymmetry, 1997, 8, 1179 CrossRef CAS; (b) S.-L. You, X.-L. Hou, L.-X. Dai, Y.-H. Yu and W. Xia, J. Org. Chem., 2002, 67, 4684 CrossRef CAS.
- (a) A. J. Hennessy, Y. M. Malone and P. J. Guiry, Tetrahedron Lett., 1999, 40, 9163 CrossRef CAS; (b) W.-P. Deng, X.-L. Hou, L.-X. Dai and X.-W. Dong, Chem. Commun., 2000, 1483 RSC.
- (a) T. Sammakia and E. L. Stangeland, J. Org. Chem., 1997, 62, 6104 CrossRef CAS; (b) Y. Nishibayashi, I. Takei, S. Uemura and M. Hidai, Organometallics, 1999, 18, 2291 CrossRef CAS.
- (a) Y. Nishibayashi, K. Segawa, K. Ohe and S. Uemura, Organometallics, 1995, 14, 5486 CrossRef CAS; (b) Y. Nishibayashi, I. Takei, S. Uemura and M. Hidai, Organometallics, 1998, 17, 3420 CrossRef CAS; (c) I. Takei, Y. Nishibayashi, Y. Arikawa, S. Uemura and M. Hidai, Organometallics, 1999, 18, 2271 CrossRef CAS; (d) I. Takei, Y. Nishibayashi, Y. Mizobe, S. Uemura and M. Hidai, Chem. Commun., 2001, 2360 RSC.
- C. Bolm and M. Kesselgruber, DE 100 40 017.5, 2000.
- D. A. Evans, G. S. Peterson, J. S. Johnson, D. M. Barnes, K. R. Campos and K. A. Woerpel, J. Org. Chem., 1998, 63, 4541 CrossRef CAS.
- Reviews: (a) G. Consiglio and R. M. Waymouth, Chem. Rev., 1989, 89, 257 CrossRef CAS; (b) C. D. Frost, J. Howarth and J. M. J. Williams, Tetrahedron, 1992, 3, 1089 CrossRef CAS; (c) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS; (d) B. M. Trost, Acc. Chem. Res., 1996, 29, 355 CrossRef CAS; (e) A. Pfaltz and M. Lautens, Comprehensive Asymmetric Catalysis, eds. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, ch. 24, p. 833 Search PubMed.
- Reviews: (a) G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051 CrossRef CAS; (b) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (c) M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045 CrossRef CAS; (d) M. Wills, M. Palmer, A. Smith, J. Kenny and T. Walsgrove, Molecules, 2000, 5, 4 Search PubMed; (e) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS.
- Reviews: (a) H. Nishiyama and K. Itoh, Transition Metals in Organic Synthesis, eds. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 1998, ch. 2, p. 111 Search PubMed; (b) H. Nishiyama, Comprehensive Asymmetric Catalysis, eds. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, ch. 6.3, p. 267 Search PubMed.
- (a) L.-X. Dai, X.-L. Hou, W.-P. Deng, S.-L. You and Y.-G. Zhou, Pure Appl. Chem., 1999, 71, 1401 CAS; (b) W.-P. Deng, X.-L. Hou, L.-X. Dai, Y.-H. Yu and W. Xia, Chem. Commun., 2000, 285 RSC; (c) W.-P. Deng, S.-L. You, X.-L. Hou, L.-X. Dai, Y.-H. Yu, W. Xia and J. Sun, J. Am. Chem. Soc., 2001, 123, 6508 CrossRef CAS.
- (a) D. W. Allen and B. F. Taylor, J. Chem. Res. (S), 1981, 220 Search PubMed; (b) D. W. Allen and B. F. Taylor, J. Chem. Soc., Dalton Trans., 1982, 51 RSC; (c) D. W. Allen, L. A. March and I. W. Nowell, J. Chem. Soc., Dalton Trans., 1984, 483 RSC; (d) D. W. Allen and I. W. Nowell, J. Chem. Soc., Dalton Trans., 1985, 2505 RSC; (e) D. W. Allen and B. F. Taylor, J. Chem. Res. (S), 1986, 392 Search PubMed.
- J. Sprinz and G. Helmchen, Tetrahedron Lett., 1993, 34, 1769 CrossRef CAS.
- P. von Matt and A. Pfaltz, Angew. Chem., 1993, 105, 614 ( Angew. Chem., Int. Ed. Engl. , 1993 , 32 , 566 ) CAS.
- P. Sennhenn, B. Gabler and G. Helmchen, Tetrahedron Lett., 1994, 35, 8595 CrossRef CAS.
- T. Hayashi, A. Yamamoto, Y. Ito, E. Nishioka, H. Muira and K. Yanagi, J. Am. Chem. Soc., 1989, 111, 6301 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |