Preferential precipitation of C70 over C60 with p-halohomooxacalix[3]arenes
Received
19th August 2002
, Accepted 21st October 2002
First published on 27th November 2002
Abstract
Preferential precipitation of C70 from a toluene solution of C60 and C70 was accomplished with p-trihalohomooxacalix[3]arenes (13·X·X·X) prepared by the reductive coupling of diformylphenols. Heavy halogens, Br and/or I, are essential at the para position of 13·X·X·X for obtaining good yields and selectivities. C70 with up to 92% purity was obtained after the preferential precipitation.
Introduction
Encapsulation of fullerenes is a subject of current interest in the fields of supramolecular and fullerene chemistry.1–8 A practical goal in this area of chemistry is to develop an efficient method for the separation of specific size and/or isomers of fullerenes from mixtures.9 Actually, selective complexation of fullerenes has so far been reported with calixarenes,10–22 cyclodextrins,15,23–25 cyclotriveratrylenes26–28 and metalloporphyrins.29 In 1992, selective extraction of C60 into a water layer was accomplished using γ-cyclodextrin25 and water soluble calix[8]arene22 by Wennerström and Verhoeven and their co-workers, respectively. Selective precipitation from an organic solution of a fullerene mixture, which is a more practical method by which to separate fullerenes, was reported in 1994 using p-tert-butylcalix[8]arene19,20 and cyclotriveratrylene28 by Shinkai and Atwood and their co-workers. More recently, a few bridged or cyclic dimers were found to encapsulate C70 preferentially to C60 in solution.14,17,26,29 However, no host molecules preferentially precipitating C70 have been reported except for one example using p-tert-butylcalix[6]arene.16,20 Herein is described a selective precipitation of C70 over C60 with p-halohomooxacalix[3]arenes.
Results
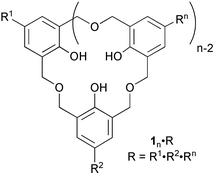
Synthesis of homooxacalix[n]arenes (1n·R)
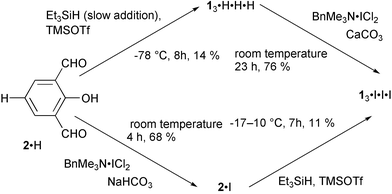
The homooxacalix[n]arenes (1n·R) were prepared according to the recent report based on the reductive coupling of 4-substituted-2,6-diformylphenols (2·R).30p-Triiodohomooxacalix[3]arene (13·I·I·I) was prepared stepwise from 2,6-diformylphenol (2·H)31 through a combination of iodination32,33 and reductive coupling30
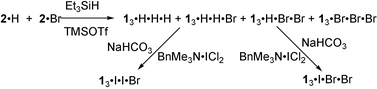
(Scheme 1) because diformylation of p-iodophenol34 gave 2·I in very low yield. p-Trihalohomooxacalix[3]arenes with different halogens, 13·I·I·Br and 13·I·Br·Br, were prepared through the iodination33 of the corresponding 13·H·H·Br and 13·H·Br·Br as shown in Scheme 2. Other p-halohomooxacalix[3]arenes with different substituents, 13·I·I·Bz (Bz = CH2Ph), 13·I·Bz·Bz, 13·I·I·Octt
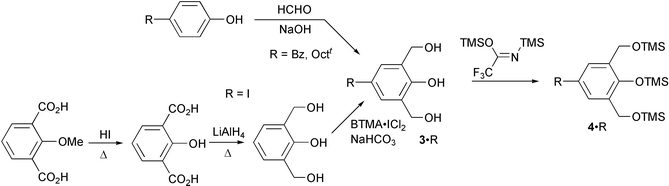
(Octt
= 1,1,3,3-tetramethylbutyl) and 14·I·I·I·Octt, were prepared by reductive heterocoupling between 4-substituted-2,6-diformylphenol (2·R) and the tris(trimethylsilyl) ether of 4-substituted-2,6-bis(hydroxymethyl)phenol (4·R)30 which was prepared via3·R as shown in Scheme 3. The 2,6-bis(hydroxymethyl)phenols with benzyl and tert-octyl substituents (3·Bz and 3·Octt) were prepared according to the reported method.35 Since the direct hydroxymethylation of p-iodophenol was reported to fail, 3·I was prepared in a similar indirect route starting from the commercial compound to that described in ref. 36. The trimethylsilylation of 3 was carried out with N,O-bis(trimethylsilyl)trifluoroacetamide (BTSTA) in acetonitrile to give 4 in high yields after rapid column chromatography with hexane and careful evaporation of the solvents.37,38
 |
| | Scheme 1 | |
 |
| | Scheme 2 | |
 |
| | Scheme 3 | |
Precipitation of C70 with homooxacalix[n]arenes
Thus prepared homooxacalix[n]arenes with a variety of para-substituents were first examined for precipitation of C70
(Table 1). About 70% of C70 was precipitated by 13·I·I·I from a toluene solution of C70
(ca. 1 mg cm−3) as a complex (runs 1 and 2). Under similar conditions, however, no precipitate was observed for C60 with 13·I·I·I (run 3). These contrasting results clearly show a potential of 13·I·I·I for separation of C70 from a mixture of C60 and C70.
Table 1 Precipitation of C70 with p-halohomooxacalix[n]arenes with various substituentsa
| Run |
Host (1n·R) |
C70/mg (µmol) |
Toluene/cm3 |
Precipitate |
|
n
|
R |
Weight/mg (µmol) |
Weight/mg |
Yield (%) of C70b |
|
Mixtures were stirred at room temperature overnight unless otherwise noted.
Calculated from recovery (%) of C70 from the filtrate.
C60.
Bz = CH2Ph.
Octt
= 1,1,3,3-tetramethylbutyl.
At −30 °C.
|
| 1 |
3 |
I·I·I |
24.1 (31) |
51.3 (61) |
48 |
52.5 |
71 |
| 2 |
3 |
I·I·I |
52.8 (67) |
111.6 (130) |
96 |
109.2 |
67 |
| 3 |
3 |
I·I·I |
4.3 (5.5) |
3.8 (5.3)c |
4.5 |
0 |
0c |
| 4 |
3 |
I·I·Bzd |
12.7 (17) |
30.2 (36) |
21.5 |
9.6 |
22 |
| 5 |
3 |
I·I·Octte |
24.0 (31) |
54.8 (65) |
43 |
16.1 |
20 |
| 6 |
4 |
I·I·I·Octte |
15.8 (15) |
31.5 (37) |
21.5 |
9.2 |
20 |
| 7 |
3 |
I·Bz·Bz |
5.1 (7.1) |
4.7 (5.6) |
3.5 |
trace |
∼0 |
| 8f |
3 |
I·Bz·Bzd |
5.6 (7.8) |
4.7 (5.6) |
5.5 |
trace |
∼0 |
| 9 |
3 |
Br·Br·Br |
45.9 (71) |
111.1 (130) |
96 |
52.5 |
33 |
The number of alkyl groups and the nature of the halogens at the para position of p-halohomooxacalix[n]arenes (n
= 3 or 4) remarkably affected the yield of C70
(runs 4–9). p-Iodohomooxacalix[n]arenes (n
= 3 or 4) with one alkyl group afforded C70 precipitates in low yields (runs 4–6), while only a trace amount of precipitate was obtained using p-iodohomooxacalix[3]arene with two alkyl groups (runs 7–8). p-Tribromohomooxacalix[3]arene gave C70 complex in 33% yield (run 9), which is less than half of the yield with p-triiodohomooxacalix[3]arene (runs 1 and 2). A similar precipitation experiment with a more concentrated toluene solution of C70
(2.5 mg cm−3) has been carried out with p-tert-butylcalix[6]arene which gives C70 complex in 31% yield.20
Preferential precipitation of C70 over C60 with homooxacalix[3]arenes
Selective precipitation of C70 from fullerene mixtures was carried out under more concentrated conditions than those in Table 1.39 The results are summarized in Table 2. The selectivity and the yield of C70 strongly depend on the nature of the halogens at the para position of the host molecules. The order of efficiency in the purification of C70 followed the size of the halogens, I > Br > Cl > F (runs 2, 7, 13 and 14). As compared to 13·I·I·I and 13·Br·Br·Br, 13·Cl·Cl·Cl gave low selectivity (run 12) or low yield of C70
(run 13), and 13·F·F·F afforded low yield of C70 with the highest purity (run 14). When 13·I·I·I or 13·Br·Br·Br was added to the toluene solution of a fullerene mixture (C60
∶ C70
= 1 ∶ 1, w/w), the ratios of C60
∶ C70 and the yields of C70 of the precipitates were 21 ∶ 79–38 ∶ 62 and 75–91%, respectively (runs 2, 7 and 9–11). The purity of C70 in the precipitates increased to 92% and 88% from 83% and 80%, respectively, after the selective precipitation (runs 1 and 8). The attempt to obtain pure C70 by this method was not successful; 92% purity is the upper limit of this purification. p-Iodohomooxacalix[3]arenes with one or two Br groups, 13·I·I·Br and 13·I·Br·Br (runs 3 and 6), afforded almost the same results as that of 13·I·I·I (run 2), while the ones with one alkyl group, 13·I·I·Bz and 13·I·I·Octt
(Octt
= 1,1,3,3-tetramethylbutyl), gave much lower yields of C70
(runs 4 and 5). These results show that three heavy halogens are essential for obtaining both good yield and selectivity. The selectivity seems to be similar to the reported ones with tert-butylcalix[6]arene.16,20 When other solvent systems were used instead of toluene, similar results were obtained (runs 10 and 11). The precipitate from a toluene solution of the fullerene mixture (C60
∶ C70
∶ C>70
= 2 ∶ 6 ∶ 2) with 13·I·I·I included a considerable amount of higher fullerenes (C>70) along with C70; that is, 13·I·I·I cannot discriminate C70 and C>70, which is another limitation of this method for the purification of fullerenes. When a toluene solution of 13·I·I·I was added to a toluene solution of raw fullerene mixture (fullerene extract, C60
∶ C70
= 8 ∶ 2), precipitation was not observed to give a dark brown homogeneous solution.
Table 2 Selective precipitation of C70 with p-halohomooxacalix[3]arenes with various substituentsa
| Run |
Host (13·R) |
Fullerene mixture |
Toluene/cm3 |
Precipitate |
Filtrate |
| R |
Weight/mg (µmol) |
C60(mg)/C70(mg) (ratio) |
C60
∶ C70b |
Yield (%)c |
C60
∶ C70b |
|
Mixtures were stirred at room temperature overnight unless otherwise noted.
Weight ratio based on area ratio in HPLC.
Yield of C70 in the precipitates was calculated from the weights of C60 and C70, and the C60
∶ C70 ratios of the precipitates and filtrates.
At −30 °C for 2 days.
Bz = CH2Ph.
Octt
= 1,1,3,3-tetramethylbutyl.
Toluene (10 cm3)
+ hexane (5 cm3).
1,1,2,2-Tetrachloroethane (5 cm3)
+ hexane (5 cm3).
|
| 1 |
I·I·I |
6.9 (8.8) |
2.6/13.0 (17 ∶ 83) |
7 |
8 ∶ 92 |
93 |
63 ∶ 37 |
| 2 |
I·I·I |
12.8 (16) |
20.3/20.4 (50 ∶ 50) |
11 |
24 ∶ 76 |
88 |
86 ∶ 14 |
| 3 |
I·I·Br |
17.0 (23) |
20.2/20.2 (50 ∶ 50) |
11 |
24 ∶ 76 |
93 |
91 ∶ 9 |
| 4d |
I·I·Bze |
11.6 (15) |
10.2/9.9 (51 ∶ 49) |
11 |
14 ∶ 86 |
18 |
55 ∶ 45 |
| 5d |
I·I·Octtf |
41.3 (53) |
20.9/21.7 (49 ∶ 51) |
17 |
11 ∶ 89 |
20 |
55 ∶ 45 |
| 6 |
I·Br·Br |
15.3 (22) |
19.6/20.1 (49 ∶ 51) |
11 |
24 ∶ 76 |
90 |
87 ∶ 13 |
| 7 |
Br·Br·Br |
15.3 (24) |
20.1/19.8 (50 ∶ 50) |
11 |
38 ∶ 62 |
91 |
84 ∶ 16 |
| 8 |
Br·Br·Br |
11.5 (18) |
4.0/16.2 (20 ∶ 80) |
7 |
12 ∶ 88 |
84 |
45 ∶ 55 |
| 9 |
Br·Br·Br |
8.0 (12) |
10.0/9.8 (50 ∶ 50) |
6 |
23 ∶ 77 |
75 |
76 ∶ 24 |
| 10 |
Br·Br·Br |
7.4 (11) |
9.8/10.0 (50 ∶ 50) |
10 + 5g |
22 ∶ 78 |
79 |
78 ∶ 22 |
| 11 |
Br·Br·Br |
7.3 (11) |
10.2/9.8 (51 ∶ 49) |
5 + 5h |
21 ∶ 79 |
76 |
78 ∶ 22 |
| 12 |
Cl·Cl·Cl |
11.6 (23) |
9.7/9.7 (50 ∶ 50) |
5 |
48 ∶ 52 |
93 |
67 ∶ 33 |
| 13 |
Cl·Cl·Cl |
12.1 (24) |
20.2/20.1 (50 ∶ 50) |
11 |
37 ∶ 63 |
63 |
63 ∶ 37 |
| 14 |
F·F·F |
10.9 (24) |
9.8/9.8 (50 ∶ 50) |
5 |
6 ∶ 94 |
19 |
55 ∶ 45 |
In order to recover the fullerene and reuse the host molecules, liberation of the complexes was carried out. Since the binding of the C70 complexes with 13·I·I·I, 13·I·I·Br and 13·I·Br·Br was very tight, it was very difficult to liberate the fullerene by a simple way like that used for the tert-butylcalix[8]arene-C60 complex.19,20 Eventually, C70-13·I·I·I was dissolved in o-dichlorobenzene (ODCB), and the host molecules were extracted with basic aqueous solution.
Discussion
The host ∶ guest ratio of the 13·I·I·I–C70 and 13·Br·Br·Br–C70 complexes is calculated to be about 2 ∶ 5 and 1 ∶ 3 from the results of the elemental analyses.† The ratio was reported to be 1 ∶ 2 in p-tert-butylcalix[6]arene–C70,20p-tert-butylcalix[6]arene–C60 and calix[6]arene–C70 complexes,16 1 ∶ 1 in 13·Br·Br·Br–C60,4013·But·But·But–C60,41p-tert-butylcalix[5]arene–C6041 and p-methylcalix[5]arene–C60 complexes,42 and 2 ∶ 1 in 13·Bz·Bz·Bz–C60
(Bz = CH2Ph),43p-phenylcalix[5]arene–C60,11 hexahomotrioxacalix[3]naphthalene–C60,13 and p-diiodotrimethylcalix[5]arene–C60 complexes.42,44 Such an unprecedented large capacity of p-trihalohomooxacalix[3]arenes for C70 may be attributed to its shallow cavity, intermolecular π–π interactions among the guest molecules,45 the large van der Waals radii of the heavy halogens, and strong van der Waals interactions between the heavy halogens and the guests.44,46 Unfortunately, a crystal structure of the complexes has not been determined yet.
The time-course of the change in absorption spectra of C70 was measured in the presence of 13·I·I·I in toluene at 20 °C as shown in Fig. 1. The brown color of C70 discharged gradually as the precipitation of C70-13·I·I·I proceeded with time. Although a monotonous decrease in the absorption spectra was observed, there was no significant change in the shape of the spectra, indicating that no stable complex existed in solution. On the other hand, no spectroscopic change occurred in the solution of C60 and 13·I·I·I, indicating that no complex or precipitate formed under the conditions.
![Time-course of the absorption spectra of C70
(2.0 × 10−4 M)–p-triiodohomooxacalix[3]arene (13·I·I·I, 8.4 × 10−3M) in toluene at 20 °C.](/image/article/2003/OB/b208107e/b208107e-f1.gif) |
| | Fig. 1 Time-course of the absorption spectra of C70
(2.0 × 10−4 M)–p-triiodohomooxacalix[3]arene (13·I·I·I, 8.4 × 10−3M) in toluene at 20 °C. | |
The results mentioned above imply that most of the starting compounds, 13·I·I·I and the fullerenes, exist separately in solution, and that the complexes with two or three C70, which should exist in a small amount in solution, form precipitates (Scheme 4b), while the C60 complexes do not precipitate at all (Scheme 4a). On the preferential precipitation of the fullerene mixture, at least one of the complexes, 13·I·I·I–C60C70, 13·I·I·I–(C70)2C60, and 13·I·I·I–(C60)2C70, is considered to precipitate along with the C70 complexes, because there is always some C60 in the precipitate (Scheme 4c).
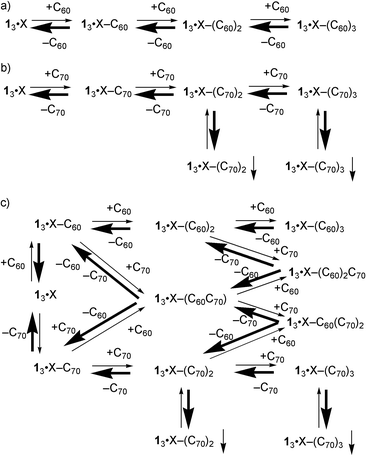 |
| | Scheme 4 | |
Experimental
1H (400 MHz) and 13C (100 MHz) NMR spectra were obtained on a JEOL JNM-FX400 in CDCl3 with Me4Si as internal standard. Chemical shifts are reported in δ and the coupling constants (J) are in Hz. MALDI-TOF-MS analyses were performed by a Shimadzu/Kratos KOMPACT MALDI II using an ethanol–water solution of 2,5-dihydroxybenzoic acid or α-cyano-4-hydroxycinnamic acid as a matrix. High resolution mass spectra were recorded on an Applied Biosystems, Marier (ESI-MS). Absorption spectra were measured in toluene on a Hitachi UV2001. HPLC analyses were performed at 35 °C on a Shimadzu LC-10 equipped with an Imtakt Cadenza CD-C18 column (100 × 4.6 mm) with hexane–propan-2-ol = 4 ∶ 6 (v/v) as an eluent at 0.8 cm3 min−1 using UV detection at 285 nm. Melting points were determined with a Yanaco MP-J3 apparatus and are uncorrected. Flash chromatography was performed with a Wakogel C-300. CH2Cl2 was freshly distilled from CaH2 under argon.
Synthesis of 4-substituted-2,6-diformylphenols (2·R)
Compounds 2·R were prepared according to the literature method34 except for 4-iodo-2,6-diformylphenol.
2,6-Diformyl-4-fluorophenol (2·F).
Yield 20 %; mp 108–109 °C (Found: C, 57.05; H, 2.97. Calc. for C8H5FO3: C, 57.15; H, 3.00%); δH 7.69 (2H, d, J 7.2, Ar–H), 10.22 (2H, s, CHO), 11.39 (1H, s, OH); δC 123.3, 123.8, 153.1, 157.9, 159.8, 191.0.
4-Benzyl-2,6-diformylphenol (2·Bz).
Yield 47 %; mp 105–107 °C (Found: C, 75.04; H, 4.94. Calc. for C15H12O3: C, 74.99; H, 5.03%); δH 4.00 (2H, s, CH2), 7.16–7.34 (5H, m, Ph–H), 7.78 (2H, s, Ar–H), 10.19 (2H, s, CHO), 11.50 (1H, s, OH); δC 40.4, 123.0, 126.7, 128.7, 128.8, 133.1, 137.7, 139.5, 162.1, 192.1.
2,6-Diformyl-4-iodophenol (2·I).
Since the diformylation of 4-iodophenol gave the desired product only in 3% yield, 2,6-diformylphenol (2·H)31 was iodinated with benzyltrimethylammonium dichloroiodate (BTMA·ICl2) in the presence of NaHCO3.33
To a CH2Cl2–methanol (40 cm3
∶ 16 cm3) solution of 2·H (0.60 g, 4.0 mmol) was added BTMA·ICl2
(1.6 g, 4.6 mmol) and NaHCO3
(2.3 g, 27 mmol), and the resulting yellow suspension was stirred at room temperature for 9 hours. After filtration through a Celite-SiO2 bed, the filtrate was concentrated and submitted to flash column chromatography on silica gel to give 2·I (0.77 g, 68%). Mp 145–148 °C (Found: C, 35.09; H, 1.73. Calc. for C8H5IO3: C, 34.81; H, 1.83%); δH 8.22 (2H, s, Ar–H), 10.16 (2H, s, CHO), 11.55 (1H, s, OH); δC 80.8, 125.0, 154.6, 162.9, 190.8.
Synthesis of 4-substituted-2,6-bis(hydroxymethyl)phenols (3·R)
Compounds 3·R were prepared according to the literature method35 except for 4-iodo-2,6-bis(hydroxymethyl)phenol.
4-Benzyl-2,6-bis(hydroxymethyl)phenol (3·Bz).
Mp 92–93 °C (ethyl acetate)
(Found: C, 73.76; H, 6.57. Calc. for C15H16O3: C, 73.74; H, 6.61%); δH 2.47 (2H, s, OH), 3.87 (2H, s, CH2Ph), 4.75 (4H, d, J 2.4, CH2O), 6.89 (2H, s, Ar–H), 7.14–7.29 (5H, m, CH2Ph), 7.92 (1H, s, ArOH).
4-tert-Octyl-2,6-bis(hydroxymethyl)phenol (3·Octt).
Mp 63–66 °C (ethyl acetate)
(Found: C, 69.76; H, 9.74. Calc. for C16H30O4(C16H26O3
+
½ethyl acetate): C, 69.64; H, 9.74%); δH 0.72 (9H, s, C(CH3)3), 1.32 (6H, s, C(CH3)2), 1.68 (2H, s, CH2), 4.81 (4H, s, CH2O), 7.05 (2H, s, Ar–H).
4-Iodo-2,6-bis(hydroxymethyl)phenol (3·I)36.
A commercial 2-methoxyisophthalic acid was demethylated in aqueous HI solution at reflux temperature,47 reduced to dimethanol with lithium aluminum hydride,48 and iodinated with BTMA·ICl2 in the presence of NaHCO3.36
Typical procedure for the synthesis of 4-substituted-2,6-bis(trimethylsilyloxymethyl)phenol trimethylsilyl ethers (4·R)
To an acetonitrile solution (30 cm3) of 3·Octt was added BTSTA (4.5 cm3, 17 mmol) dropwise over 5 minutes at room temperature under Ar. The resulting pale yellow solution was stirred for 22 hours, the mixture was concentrated on a rotary evaporator and rapidly passed through a short column using hexane as eluent. After careful concentration on a rotary evaporator, the desired compound, 4·Octt, was obtained as a colourless oil containing a small amount of hexane in almost quantitative yield, and used immediately in the next coupling reaction.
4-tert-Octyl-2,6-bis(trimethylsilyloxymethyl)phenol trimethylsilyl ether (4·Octt).
δ
H 0.14 (18H, s, TMS), 0.23 (9H, s, TMS), 0.69 (9H, s, C(CH3)3), 1.36 (6H, s, C(CH3)2), 1.71 (2H, s, CH2), 4.65 (4H, s, CH2O), 7.27 (2H, s, Ar–H).
4-Benzyl-2,6-bis(trimethylsilyloxymethyl)phenol trimethylsilyl ether (4·Bz).
δ
H 0.11 (18H, s, TMS), 0.22 (9H, s, TMS), 3.94 (2H, s, CH2Ph), 4.61 (4H, s, CH2O), 7.09 (2H, s, Ar–H), 7.17–7.29 (5H, m, Ph).
4-Iodo-2,6-bis(trimethylsilyloxymethyl)phenol trimethylsilyl ether (4·I).
δ
H 0.16 (18H, s, TMS), 0.24 (9H, s, TMS), 4.57 (4H, s, CH2O), 7.60 (2H, s, Ar–H).
Synthesis of homooxacalix[n]arenes via reductive coupling
7,15,23-Triiodo-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (p-triiodohomooxacalix[3]arene, 13·I·I·I).
As shown in Scheme 1, two synthetic routes were followed from 2·H. The reductive homocoupling of 2·H and 2·I afforded 13·H·H·H and 13·I·I·I in 14% and 11% yields, respectively, following the reported procedure.30 The 13·H·H·H obtained was iodinated according to the following procedure: a yellow solution of 13·H·H·H (0.13 g, 0.32 mmol) and BTMA·ICl2 in dichloromethane (30 cm3)–methanol (12 cm3) was stirred at room temperature for 0.5 hour. Then, CaCO3
(0.20 g, 2.0 mmol) was added to the mixture. After being stirred for 19 hours, the suspension was filtered through Celite, and the filtrate was concentrated to about a half in volume and washed with 5% NaHSO3
(30 cm3) twice. The combined water layer was extracted with dichloromethane (30 cm3) once, and the dichloromethane layers were combined, concentrated and chromatographed on silica gel to give 13·I·I·I (0.19 g, 76%) as a white solid. Mp >300 °C (Found: C, 36.72; H, 2.64. Calc. for C24H21I3O6: C, 36.67; H 2.69%); δH 4.62 (12H, s, CH2), 7.42 (6H, s, Ar–H), 8.67 (3H, s, OH); δC 70.3, 81.1, 126.4, 138.3, 155.6; MALDI-TOF-MS (pos): calcd. for C24H21I3NaO6 809.12, found 809.06 (M + Na+).
7,15,23-Trifluoro-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (p-trifluorohomooxacalix[3]arene, 13·F·F·F).
1
3
·F·F·F was prepared according to the reported procedure.30 Yield 29%; mp 218–220 °C (Found: C, 62.06; H, 4.63. Calc. for C24H21F3O6: C, 62.34; H, 4.58%); δH 4.66 (12H, s, CH2), 6.86 (6H, d, Ar–H), 8.55 (3H, s, OH); δC 70.8, 115.9, 116.2 (d, J 23), 125.1 (d, J 7.1), 151.7 (d, J 2.3), 155.6 (d, J 240); MALDI-TOF-MS (pos): calcd. for C24H21F3NaO6 485.41, found 485.40 (M + Na+).
7,15-Diiodo-23-benzyl-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (13·I·I·Bz).
1
3
·I·I·Bz was prepared according to the reported procedure30 using 2,6-diformyl-4-iodophenol (0.19 g, 0.72 mmol), 2,6-bis(trimethylsilyloxymethyl)-4-benzylphenol trimethylsilyl ether (0.17 g, 0.31 mmol), trimethylsilyl trifluoromethanesulfonate (0.26 cm3, 1.4 mmol), triethylsilane (0.24 cm3, 1.5 mmol) and dichloromethane (20 cm3). Yield 6%; δH 3.86 (2H, s, CH2Ph), 4.61 (4H, s, CH2), 4.62 (4H, s, CH2), 4.65 (4H, s, CH2), 6.94 (2H, s, Bz–Ar–H), 7.11–7.26 (5H, m, Bz), 7.41 (2H, s, I–Ar–H), 8.45 (1H, s, Bz–Ar–OH), 8.76 (2H, s, I–Ar–OH); δC 40.82, 70.23, 70.30, 71.27, 80.97, 123.84, 126.04, 126.42, 126.65, 128.41, 128.77, 130.40, 132.39, 138.18, 138.26, 141.09, 153.85, 155.69; MALDI-TOF-MS (pos): calcd. for C31H28I2NaO6 772.81, found 773.14 (M + Na+).
15,23-Dibenzyl-7-iodo-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (13·I·Bz·Bz).
1
3
·I·Bz·Bz was prepared according to the reported procedure30 using 4-benzyl-2,6-diformylphenol (0.10 g, 0.42 mmol), 2,6-bis(trimethylsilyloxymethyl)-4-iodophenol trimethylsilyl ether (0.11 g, 0.22 mmol), trimethylsilyl trifluoromethanesulfonate (0.14 cm3, 0.80 mmol), triethylsilane (0.14 cm3, 0.88 mmol) and dichloromethane (12 cm3). Yield 26%; δH 3.84 (4H, s, CH2Ph), 4.59 (2H, s, CH2), 4.63 (2H, s, CH2), 4.63 (2H, s, CH2), 6.92 (4H, s, Bn–Ar–H), 7.07–7.26 (10H, m, Ph), 7.38 (2H, s, I–Ar–H), 8.54 (2H, s, Bn–Ar–OH), 8.85 (1H, s, I–Ar–OH); δC 40.82, 70.24, 71.23, 80.86, 123.90, 124.13, 126.01, 126.72, 128.39, 128.77, 130.30, 130.37, 132.26, 138.14, 141.16, 153.95, 155.80.
7,15-Diiodo-23-tert-octyl-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (13·I·I·Octt).
1
3
·I·I·Octt was prepared according to the reported procedure30 using 2,6-diformyl-4-iodophenol (0.20 g, 0.73 mmol), 2,6-bis(trimethylsilyloxymethyl)-4-tert-octylphenol trimethylsilyl ether (0.15 g, 0.31 mmol), trimethylsilyl trifluoromethanesulfonate (0.26 cm3, 1.4 mmol), triethylsilane (0.24 cm3, 1.5 mmol) and dichloromethane (20 cm3). Yield 17%; δH 0.71 (9H, s, C(CH3)3), 1.30 (6H, s, C(CH3)2), 1.67 (2H, s, –CH2–But), 4.62 (4H, s, CH2), 4.63 (4H, s, CH2), 4.70 (4H, s, CH2), 7.09 (2H, s, Octt–Ar–H), 7.43 (4H, s, I–Ar–H), 8.40 (1H, s, Octt–Ar–OH), 8.79 (2H, s, I–Ar–OH); δC 31.54, 31.84, 32.32, 37.83, 56.80, 70.16, 70.31, 71.77, 80.95, 122.99, 126.44, 126.75, 127.56, 138.12, 138.26, 141.67, 152.99, 155.70; MALDI-TOF-MS (pos): calcd. for C32H38I2NaO6 795.49, found 795.48 (M + Na+).
31-tert-Octyl-7,15,23-triiodo-2,3,10,11,18,19,26,27-octahomo-3,11,19,27-tetraoxacalix[4]arene-33,34,35,36-tetraol (14·I·I·I·Octt).
This compound was obtained in a small amount (3.8 mg) as a byproduct in the preparation of 13·I·I·Octt; δH 0.71 (9H, s, C(CH3)3), 1.29 (6H, s, C(CH3)2), 1.67 (2H, s, –CH2–But), 4.63 (8H, s, CH2), 4.64 (4H, s, CH2), 4.69 (4H, s, CH2), 7.09 (2H, s, Octt–Ar–H), 7.41–7.44 (6H, m, I–Ar–H), 8.01 (1H, s, Octt–Ar–OH), 8.30 (1H, s, I–Ar–OH), 8.37 (2H, s, I–Ar–OH); δC 31.56, 31.85, 32.33, 37.80, 56.73, 70.27, 70.38, 70.38, 71.87, 80.90, 80.90, 123.02, 126.41, 126.46, 126.82, 127.72, 138.33, 138.43, 138.54, 141.60, 153.17, 155.76, 155.88; MALDI-TOF-MS (pos): calcd. for C40H45I3NaO8 1057.55, found 1057.79 (M + Na+).
Synthesis of p-trihalohomooxacalix[3]arenes (13·I·I·Br and 13·I·Br·Br, Scheme 2)
p-Bromohomooxacalix[3]arenes (13·H·H·Br and 13·H·Br·Br) were obtained in 3% yield after the purification of the mixture from the reductive coupling of 2,6-diformylphenol (2·H) and 4-bromo-2,6-diformylphenol (2·Br).30 The 13·I·I·Br and 13·I·Br·Br were obtained by iodination of 13·H·H·Br and 13·H·Br·Br with BTMA·ICl2
(2.2 and 1.1 equiv.) in the presence of NaHCO3
(4.6 and 2.3 equiv.), respectively, in dichloromethane–methanol (5 ∶ 2 v/v) followed by a similar work-up to that in the synthesis of 13·I·I·I.
23-Bromo-7,15-diiodo-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (13·I·I·Br).
δ
H 4.62 (2H, s, CH2), 4.63 (2H, s, CH2), 4.64 (2H, s, CH2), 7.25 (2H, s, Ar–H), 7.42 (4H, s, Ar–H), 8.65 (1H, s, OH), 8.67 (2H, s, OH); δC 70.33, 70.34, 70.51, 81.07, 111.26, 125.77, 126.27, 132.27, 138.18, 154.62, 155.45; HRMS (EI): calcd. for C24H20BrI2O6 736.8527, found 736.8541.
15,23-Dibromo-7-iodo-2,3,10,11,18,19-hexahomo-3,11,19-trioxacalix[3]arene-25,26,27-triol (13·I·Br·Br).
δ
H 4.63 (2H, s, CH2), 4.64 (2H, s, CH2), 4.64 (2H, s, CH2), 7.25 (4H, s, Ar–H), 7.43 (2H, s, Ar–H), 8.66 (2H, s, OH), 8.68 (1H, s, OH); δC 70.34, 70.53, 81.06, 111.27, 125.79, 126.29, 132.22, 132.28, 132.33, 138.14, 138.19, 138.25, 154.64, 155.46; HRMS (EI): calcd. for C24H20Br2IO6 688.8677, found 688.8683.
General procedure for the precipitation of the complex of fullerene with homooxacalix[n]arene (Tables 1 and 2)
To a toluene solution of fullerene was added a toluene solution of homooxacalix[n]arene, and the mixture was stirred at room temperature. After filtration of the formed precipitate, the filtrate was concentrated and washed with acetone thoroughly to give the recovered fullerene. The ratios of C60
∶ C70 in both precipitate and filtrate were determined by HPLC.
Recovery of fullerene from the C60/C70–13·I·I·I complex
To an o-dichlorobenzene solution (12 cm3) of the complex (12.4 mg) was added 1 M NaOH (10 cm3), and the resulting heterogeneous solution was vigorously stirred at room temperature for an hour. After separation of the two layers, the organic layer was washed with 1 M NaOH (15 cm3) twice. The combined water extracts were made acidic with conc. HCl and extracted with dichloromethane (20 cm3) three times and toluene (30 cm3) once. Concentration of the combined dichloromethane extracts gave pure 13·I·I·I (3.7 mg). On the other hand, the o-dichlorobenzene layer was combined with toluene extract, dried over MgSO4 and concentrated in vacuo to give liberated fullerenes (9.0 mg).
Acknowledgements
The author is grateful to Professors Keiji Maruoka, Atsuhiro Osuka and Kazumi Matsushige (Kyoto University) for their encouragement, Dr Tomonari Wakabayashi (Kyoto University) for his helpful suggestions, Mr Itaru Yazawa (Imtakt Co.) for his kind offering of a HPLC column (Cadenza CD-C18), Mr Koji Sasaki for his assistance of measuring HRMS, and Messrs Takashi Onozawa and Susumu Kosugiyama (Tokyo Chemical Industry Co., Ltd.) for their assistance with experiments and their kind donation of some reagents. A part of this work was supported by a Grant-in-Aid for Research for Young Researchers from Kyoto University-Venture Business Laboratory (KU-VBL).
References
- S. Shinkai and A. Ikeda, Pure Appl. Chem., 1999, 71, 275 CAS.
- A. L. Balch and M. M. Olmstead, Coord. Chem. Rev., 1999, 185–186, 601 CrossRef CAS.
- S. Shinkai and A. Ikeda, Gazz. Chim. Ital., 1997, 127, 657 Search PubMed.
- T. Braun, Fullerene Sci. Technol., 1997, 5, 615 CAS.
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713 CrossRef CAS.
-
C. L. Raston, in Comprehensive Supramolecular Chemistry, ed. G. W. Gokel, Pergamon, Oxford, 1996, vol. 1, pp. 777–787 Search PubMed.
- P. Lhotak and S. Shinkai, J. Synth. Org. Chem., Jpn., 1995, 53, 963 Search PubMed.
- E. C. Constable, Angew. Chem., Int. Ed. Engl., 1994, 33, 2269 CrossRef.
- J. Theobald, Sep. Sci. Technol., 1995, 30, 2783 Search PubMed.
- T. Haino, Y. Yamanaka, H. Araki and Y. Fukazawa, Chem. Commun., 2002, 402 RSC.
- M. Makha, M. J. Hardie and C. L. Raston, Chem. Commun., 2002, 1446 RSC.
- T. Haino, H. Araki, Y. Yamanaka and Y. Fukazawa, Tetrahedron Lett., 2001, 42, 3203 CrossRef CAS.
- S. Mizyed, M. Ashram, D. O. Miller and P. E. Georghiou, J. Chem. Soc., Perkin Trans. 2, 2001, 1916 RSC.
- J. Wang, S. G. Bodige, W. H. Watson and C. D. Gutsche, J. Org. Chem., 2000, 65, 8260 CrossRef CAS.
- K. Komatsu, K. Fujiwara, Y. Murata and T. Braun, J. Chem. Soc., Perkin Trans. 1, 1999, 2963 RSC.
- J. L. Atwood, L. J. Barbour, C. L. Raston and I. B. N. Sudria, Angew. Chem., Int. Ed., 1998, 37, 981 CrossRef CAS.
- T. Haino, M. Yanase and Y. Fukazawa, Angew. Chem., Int. Ed., 1998, 37, 997 CrossRef CAS.
- T. Suzuki, K. Nakashima and S. Shinkai, Tetrahedron Lett., 1995, 36, 249 CrossRef CAS.
- T. Suzuki, K. Nakashima and S. Shinkai, Chem. Lett., 1994, 699 CAS.
- J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229 CrossRef CAS.
- R. M. Williams, J. M. Zwier and J. W. Verhoeven, J. Am. Chem. Soc., 1994, 116, 6965 CrossRef CAS.
- R. M. Williams and J. W. Verhoeven, Recl. Trav. Chim. Pays-Bas, 1992, 111, 531 Search PubMed.
- T. Andersson, M. Sundahl, G. Westman and O. Wennerström, Tetrahedron Lett., 1994, 35, 7103 CrossRef CAS.
- T. Andersson, G. Westman, O. Wennerström and M. Sundahl, J. Chem. Soc., Perkin Trans. 2, 1994, 1097 RSC.
- T. Andersson, K. Nilsson, M. Sundahl, G. Westman and O. Wennerström, J. Chem. Soc., Chem. Commun., 1992, 604 RSC.
- H. Matsubara, S. Oguri, K. Asano and K. Yamamoto, Chem. Lett., 1999, 431 CrossRef CAS.
- H. Matsubara, A. Hasegawa, K. Shiwaku, K. Asano, M. Uno, S. Takahashi and K. Yamamoto, Chem. Lett., 1998, 923 CrossRef CAS.
- J. W. Steed, P. C. Junk, J. L. Atwood, M. J. Barnes, C. L. Raston and R. S. Burkhalter, J. Am. Chem. Soc., 1994, 116, 10346 CrossRef CAS.
- J.-Y. Zheng, K. Tashiro, Y. Hirabayashi, K. Kinbara, K. Saigo, T. Aida, S. Sakamoto and K. Yamaguchi, Angew. Chem., Int. Ed., 2001, 40, 1858.
- N. Komatsu, Tetrahedron Lett., 2001, 42, 1733 CrossRef CAS.
- C. Zondervan, E. K. van den Beuken, H. Kooijman, A. L. Spek and B. L. Feringa, Tetrahedron Lett., 1997, 38, 3111 CrossRef CAS.
- B. Krenke and W. Friedrichsen, J. Chem. Soc., Perkin Trans. 1, 1998, 3377 RSC.
- S. Kajigaeshi, T. Kakinami, H. Yamasaki, S. Fujisaki, M. Kondo and T. Okamoto, Chem. Lett., 1987, 2109 CAS.
- L. F. Lindoy, G. V. Meehan and N. Svenstrup, Synthesis, 1998, 1029 CrossRef CAS.
- P. D. Hampton, Z. Bencze, W. Tong and C. E. Daitch, J. Org. Chem., 1994, 59, 4838 CrossRef CAS.
- G. T. Crisp and P. D. Turner, Tetrahedron, 2000, 56, 407 CrossRef CAS.
- P. Chirakul, P. D. Hampton and E. N. Duesler, Tetrahedron Lett., 1998, 39, 5473 CrossRef CAS.
- P. D. Hampton, C. E. Daitch and E. N. Duesler, New. J. Chem., 1996, 20, 427 Search PubMed.
- A. N. Ponomarev, B. M. Aksel'rod, V. T. Barchenko, V. P. Belousov, Z. S. Egorova, I. V. Igonchenkov, A. Y. Isakov, O. Y. Kut'eva, V. A. Nikitin, D. V. Nikitin, S. V. Ruzaev, A. V. Rumyantsev, O. S. Tulyakov, N. A. Charykov, M. E. Yudovich and A. L. Yurin, Russ. J. Phys. Chem., 2000, 74, 1942 Search PubMed.
- K. Tsubaki, K. Tanaka, T. Kinoshita and K. Fuji, Chem. Commun., 1998, 895 RSC.
- A. Ikeda, Y. Suzuki, M. Yoshimura and S. Shinkai, Tetrahedron, 1998, 54, 2497 CrossRef CAS.
- T. Haino, M. Yanase and Y. Fukazawa, Tetrahedron Lett., 1997, 38, 3739 CrossRef CAS.
- J. L. Atwood, L. J. Barbour, P. J. Nichols, C. L. Raston and C. A. Sandoval, Chem. Eur. J., 1999, 5, 990 CrossRef CAS.
- T. Haino, M. Yanase and Y. Fukazawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 259 CrossRef CAS.
- L. A. Bulavin, I. I. Adamenko, V. M. Yashchuk, T. Y. Ogul'chansky, Y. I. Prylutskyy, S. S. Durov and P. Scharff, J. Mol. Liq., 2001, 93, 187 CrossRef CAS.
- S. Talukdar, P. Pradhan and A. Banerji, Fullerene Sci. Technol., 1997, 5, 547 CAS.
- G. R. Sprengling and J. H. Freeman, J. Am. Chem. Soc., 1950, 72, 1982 CrossRef CAS.
- J. H. Freeman, J. Am. Chem. Soc., 1952, 74, 6257 CrossRef CAS.
Footnote |
| † Anal. Calcd for C398H42I6O12
(2 ∶ 5 complex): C 82.76, H 0.73, I 13.18; found: C 83.14, H 0.84, I 12.61%. Anal. Calcd for C234H21Br3O6
(3 ∶ 1 complex): C 88.71, H 0.67, Br 7.57; found: C 88.66, H 0.65, Br 7.67%. |
|
| This journal is © The Royal Society of Chemistry 2003 |
Click here to see how this site uses Cookies. View our privacy policy here.