Bisubstrate inhibitors for the enzyme catechol-O-methyltransferase (COMT): influence of inhibitor preorganisation and linker length between the two substrate moieties on binding affinity
Christian
Lerner
a,
Birgit
Masjost
a,
Armin
Ruf
b,
Volker
Gramlich
c,
Roland
Jakob-Roetne
b,
Gerhard
Zürcher
b,
Edilio
Borroni
b and
François
Diederich
*a
aLaboratorium für Organische Chemie, ETH-Hönggerberg, HCI, CH-8093 Zürich, Switzerland
bPharma Division, Präklinische Forschung, F. Hoffmann-La Roche AG, CH-4070 Basel, Switzerland
cLaboratorium für Kristallographie, ETH-Zentrum, Sonneggstrasse 5, CH-8092 Zürich, Switzerland
First published on 26th November 2002
Abstract
Inhibition of the enzyme catechol-O-methyltransferase (COMT) is an important approach in the treatment of Parkinson's disease. A series of new potent bisubstrate inhibitors for COMT, resulting from X-ray structure-based design and featuring adenosine and catechol moieties have been synthesised. Biological results show a large dependence of binding affinity on inhibitor preorganisation and the length of the linker between nucleoside and catechol moieties. The most potent bisubstrate inhibitor for COMT has an IC50 value of 9 nM. It exhibits competitive kinetics for the SAM and mixed inhibition kinetics for the catechol binding site. Its bisubstrate binding mode was confirmed by X-ray structure analysis of the ternary complex formed by the inhibitor, COMT and a Mg2+ ion.
Introduction
Inhibitors, where a substrate analogue and a cofactor analogue are covalently linked to form a bisubstrate inhibitor, may offer opportunities for specificity and activity in blocking therapeutically important enzymes that are not available to simple substrate analogues.1 In principle, any enzyme that catalyses chemical transformations between two or more bound substrates or a substrate and a cofactor is a plausible candidate for the design of multisubstrate inhibitors. Recent examples of enzymes targeted with bisubstrate inhibitors are the insulin receptor tyrosine kinase,2 farnesyltransferase,3,4 serotonin-N-acetyltransferase5 and acetyl-CoA-carboxylase.6 Other important targets are methyltransferases that depend on S-adenosylmethionine (SAM).The enzyme catechol-O-methyltransferase (COMT) catalyses the methyl group transfer from the cofactor SAM to the phenolic hydroxy group of catechol-type substrates, such as L-dopa and dopamine, in the presence of magnesium ions. The inhibition of COMT is of significant interest in the treatment of Parkinson's disease.7 The symptoms of Parkinson's disease are a consequence of a low level of the neurotransmitter dopamine in the brain. Current therapies increase the dopamine level by oral administration of L-dopa, which is decarboxylated to dopamine by the enzyme aromatic amino acid decarboxylase (AAD). L-Dopa is able to cross the blood brain barrier, whereas dopamine is not. To avoid decarboxylation in the periphery, L-dopa is given in combination with an inhibitor for the decarboxylase which cannot cross the blood brain barrier. When AAD is inhibited, deactivation by COMT becomes the major metabolic pathway for L-dopa in the periphery. Inhibition of COMT increases the elimination half-time of L-dopa, ensuring that a larger quantity of orally administered L-dopa reaches its target in the brain.7
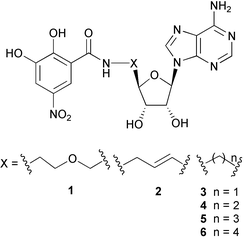
Nitro-substituted catechols were found to be potent COMT inhibitors, and two derivatives, tolcapone (Tasmar®)8 and entecapone (Comptan®)9 have been introduced into the marketplace. Using rational design based on the X-ray crystal structure of COMT,10,11 we developed the first effective bisubstrate inhibitor 1 for COMT (IC50 = 2 µM; IC50 = concentration of inhibitor at which 50% Vmax is observed), featuring both nucleoside and catechol moieties for occupancy of the SAM and substrate binding sites, respectively.11,12 Here, we report the synthesis and biological evaluation of a new series of bisubstrate inhibitors 2–6 reaching binding activities in the single-digit nanomolar range (compound 2).13 We show that affinity for COMT strongly depends on the size and shape of the linker between the nucleoside and catechol moieties. The bisubstrate inhibition mode of 2 was confirmed by kinetic analysis and the principles of molecular recognition governing complex formation elucidated by X-ray crystal structure analysis of the ternary complex formed between 2, COMT and a Mg2+ ion.
Results and discussion
Inhibitor design
For a cytosine analogue of 1, 1H NMR conformational analysis in D2O had revealed a strong nuclear Overhauser effect (NOE) between the catechol protons and H–C(5) of the nucleobase, indicating hydrophobic collapse of the unbound inhibitor.14 In the collapsed conformation, the catechol moiety does not adopt the right orientation to bind to the active site of COMT and, therefore, upon binding significant reorganisational energy would be required. Additionally, the solvation of a collapsed conformation of the inhibitor is particularly favourable, which further reduces the measurable binding free energy. Therefore, we were interested in increasing its degree of preorganisation15 prior to complexation. To prevent hydrophobic collapse in the unbound state, the new inhibitors 2–6 with shorter and conformationally less flexible linkers between nucleoside and catechol moieties were designed using the molecular modeling program MOLOC16 with the MAB force field. For the exploration of their geometric and electronic complementarity to the enzyme active site, the proposed inhibitors were docked manually into this site by overlaying their catechol and nucleoside parts on 3,5-dinitrocatechol and SAM of the X-ray crystal structure of COMT,10,11 and subsequently energy-minimized, while the coordinates of COMT and the Mg2+ ion were constrained. Within the series of inhibitors 3–6 with various linker sizes, the three-carbon bridge in 5 was predicted to provide the best geometric fit for bisubstrate binding. A particularly favourable binding mode was predicted for 2 with a spacer rigidified by introduction of a trans double bond.Synthesis
The activated ester 711 reacted selectively with the primary amino group of 5′-amino-2′,3′-O-isopropylidene-5′-deoxyadenosine 817 to give inhibitor 3 (Scheme 1). Generally, attempts to obtain pure inhibitors by recrystallisation were not reliable and gave poor yields. Due to the highly polar nature of the nitrocatechol derivatives, purification by column chromatography on SiO2 was also not possible. Therefore, reverse phase chromatography on SiO2 100 C18 became the method of choice for purification of the target compounds, using MeCN/1% aq. HCOOH mixtures as eluent.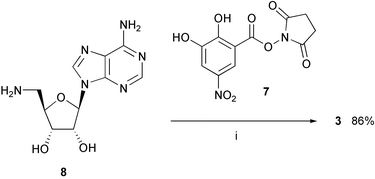 | ||
| Scheme 1 Synthesis of inhibitor 3. Reagents and conditions: (i) Et3N, DMF, 20 °C. | ||
For the synthesis of inhibitor 4, 2′,3′-O-isopropylideneadenosine 918 was transformed in high yield (89%) into nitrile 1019 using the Mitsunobu–Wilk procedure,20 with acetone cyanohydrin as the cyanide ion source (Scheme 2). This method gave much better yields than the previously reported19 reaction of 2′,3′-O-isopropylidene-5′-O-tosyladenosine with potassium cyanide in the presence of [18]crown-6. Reduction of nitrile 10 was accomplished by hydrogenation using platinum oxide in acetic acid; attempts using other catalysts or solvents failed. Subsequent deprotection afforded amine 11,21 purified by ion exchange chromatography, which reacted with the activated ester 7 to give inhibitor 4 in excellent yield (98%) after purification by reversed-phase chromatography.
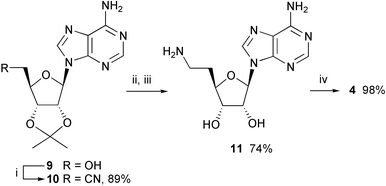 | ||
| Scheme 2 Synthesis of inhibitor 4. Reagents and conditions: (i) acetone cyanohydrin, PPh3, DEAD, THF, 0 °C → 20 °C; (ii) PtO2, AcOH, H2, 20 °C; (iii) TFA–H2O (5∶2), 20 °C, then Dowex® 50 Wx4 (NH4+); (iv) Et3N, DMF, 20 °C. | ||
In the synthesis of inhibitors 2 and 5, we initially used published procedures22,23 to synthesize the unsaturated ester 12 starting from 9. However, oxidation to the aldehyde under the conditions of Pfitzner and Moffat,24 followed by a Wittig reaction, gave only low yields. One-pot 5′-oxidation with ortho-iodoxybenzoic acid (IBX) followed by olefination with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Et25 was more effective (Scheme 3). Reduction with DIBAL-H gave the allylic alcohol 13 in very good yield (98%), which was transformed into phthalimide 14 by a Mitsunobu reaction. The X-ray crystal structure of 1413 showed a large spatial separation of the nucleobase and phthalimide moieties, indicating that an alkene spacer would effectively prevent a hydrophobic collapse of the free bisubstrate inhibitor 2
(Fig. 1).
CHCO2Et25 was more effective (Scheme 3). Reduction with DIBAL-H gave the allylic alcohol 13 in very good yield (98%), which was transformed into phthalimide 14 by a Mitsunobu reaction. The X-ray crystal structure of 1413 showed a large spatial separation of the nucleobase and phthalimide moieties, indicating that an alkene spacer would effectively prevent a hydrophobic collapse of the free bisubstrate inhibitor 2
(Fig. 1).
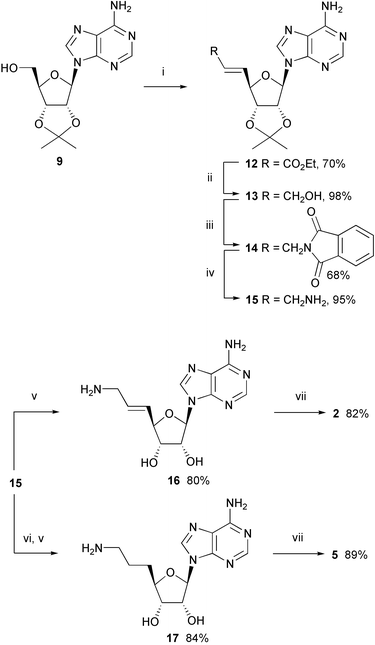 | ||
| Scheme 3 Synthesis of inhibitors 2 and 5. Reagents and conditions: (i)
o-iodoxybenzoic acid (IBX), Ph3PCHCO2Et, Me2SO, 20 °C; (ii) DIBAL-H, hexane, CH2Cl2, −78 °C; (iii) phthalimide, DEAD, PPh3, THF, 20 °C; (iv) MeNH2, EtOH, 20 °C; (v) TFA–H2O (5∶2), 20 °C, then Dowex® 50 Wx4 (NH4+); (vi) H2, Pd/C, EtOH, 20 °C; (vii) Et3N, DMF, 20 °C. | ||
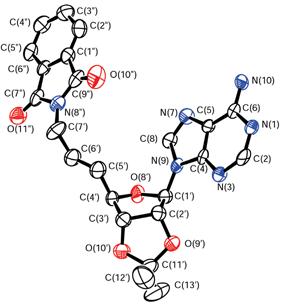 | ||
| Fig. 1 X-Ray crystal structure of 14. Arbitrary numbering. Thermal ellipsoids are shown at the 50% probability level. | ||
Cleavage of phthalimide 14 with methylamine26 gave amine 15 in better yield than the reaction using toxic hydrazine. Deprotection and purification of 15 by ion exchange chromatography led to 16, which reacted with 7 to provide bisubstrate inhibitor 2 with a rigidified olefinic linker.
Catalytic hydrogenation of allylic amine 15, followed by deprotection with TFA–H2O (5∶2) and removal of TFA by ion exchange chromatography gave 17 which was coupled to 7 to give the target compound 5.
In the preparation of 6, catalytic hydrogenation of allylic alcohol 13 followed by Mitsunobu–Wilk cyanation20 gave nitrile 18 which was reduced and deprotected (Scheme 4). Purification of the resulting free primary amine proved problematic, therefore the crude product was eventually coupled to 7 to provide 6.
 | ||
| Scheme 4 Synthesis of inhibitor 6. Reagents and conditions: (i) H2, Pd/C, EtOH, 20 °C; (ii) acetone cyanohydrin, PPh3, DEAD, THF, 0 °C → 20 °C; (iii) PtO2, AcOH, H2, 20 °C; (iv) TFA–H2O (5∶2), 20 °C; (v) Et3N, DMF, 20 °C. | ||
Biological results
The IC50 values were determined in a radiochemical assay as previously reported11,27 and are listed in Table 1. In the assay, the enzyme is preincubated with varying concentrations of inhibitor in a buffered aqueous solution containing Mg2+ ions for 15 min at 37 °C giving the system time to establish an equilibrium. The reaction is started by adding the cofactor SAM which carries a tritylated methyl group and the substrate benzene-1,2-diol. After incubation for 15 min at 37 °C, the reaction is quenched by addition of acetic acid and an organic scintillation fluid is added. The product (3H)-2-methoxyphenol is extracted into the organic phase, whereas (3H)-SAM remains in the aqueous phase. The product concentration is measured by counting the decays per min (dpm) in the organic phase. The dpm are proportional to the concentration of the reaction product, which is a measure for the enzymatic activity.| Compound | IC50/µM |
|---|---|
| 1 | 2 |
| 2 | 0.009 |
| 3 | 90 |
| 4 | 0.06 |
| 5 | 0.2 |
| 6 | 5 |
As expected, the linker of 3 was too short to provide effective bisubstrate binding (IC50 = 90 µM). Somewhat unexpectedly, compound 4 with a two-carbon linker showed a lower IC50 value (60 nM) than inhibitor 5 with a three-carbon bridge (200 nM). The four-carbon linker used in 6 (IC50 = 5 µM), similar to the four-atom linker in 1 (IC50 = 2 µM), seems to be too long. Clearly, the spacers in 4 and 5 have the best length to allow docking in both the SAM and the catechol binding sites. Further rigidification of the spacer by introduction of a trans double bond had a tremendous effect on binding affinity. Compound 2, with an IC50 value of 9 nM, is the most potent bisubstrate inhibitor for COMT to date.
To investigate the mechanism of enzyme inhibition by 2, kinetic studies were performed.11 Initial velocities were determined without preincubation. Ki values were calculated by globally fitting the data to eqn. (1) or (2), corresponding to linear competitive or mixed inhibition, respectively. In these equations, Kic and Kiu are competitive and uncompetitive inhibition constants, respectively.
| v = V[S]/(Km(1 + [I]/Kic) + [S]) | (1) |
| v = V[S]/(Km(1 + [I]/Kic) + [S](1 + [I]/Kiu)) | (2) |
Inhibitor 2 showed a competitive inhibition mechanism with regard to the SAM binding site (Fig. 2), and had a Kic value of 28 ± 2 nM. With respect to the catechol binding site, 2 showed a more complex, mixed inhibition mechanism (Fig. 3). The calculated inhibition constants were Kic = 23 ± 4 nM and Kiu = 13 ± 4 µM. The competitive inhibition pattern with respect to the SAM binding site and the mixed inhibition pattern with respect to the catechol site probably indicate an ordered mechanism of inhibition in which occupation of the SAM binding site is first required to allow effective binding to the catechol binding site.11
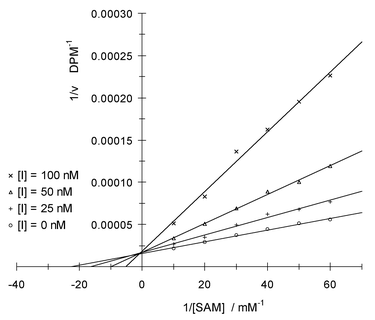 | ||
| Fig. 2 Lineweaver–Burk plot of reciprocal enzymatic activity vs. reciprocal SAM concentration for varying concentrations of inhibitor 2 at saturating benzene-1,2-diol concentration; DPM = decays per minute. | ||
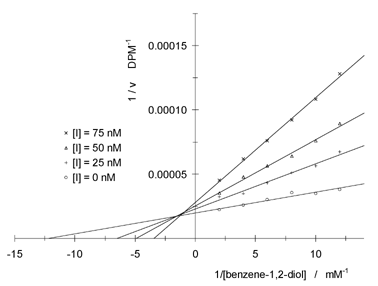 | ||
| Fig. 3 Lineweaver–Burk plot of reciprocal enzymatic activity vs. reciprocal benzene-1,2-diol concentration for varying concentrations of inhibitor 2 at saturating SAM concentration. | ||
Details of the bisubstrate binding mode as revealed by X-ray crystallography
For structural characterisation, COMT was co-crystallised with inhibitor 2 and Mg++ ions. The X-ray crystal structure of the ternary complex was refined at 2.6 Å resolution using the program CNX.13 Bond distances to the Mg ion were not restrained during refinement. A total of 88.9% of all residues are located in the most favoured regions of the Ramachandran plot with Tyr68 being the only residue in a disallowed region. Atomic coordinates are available from the Brookhaven Protein Databank, with the PDB entry code 1JR4. The protein structure in the ternary complex resembles that of the quaternary complex of COMT10,11 with SAM, 3,5-dinitrocatechol, and a Mg2+ ion, with an overall root mean square (r.m.s.) deviation for the Cα atoms of 0.4 Å. The electron density reveals crystallographic disorder about the side chain of Trp143. As shown in Fig. 4, the bisubstrate inhibitor fills both the nucleoside and the catechol binding pockets. Fig. 5 shows a schematic representation of the hydrogen bonding in the ternary complex.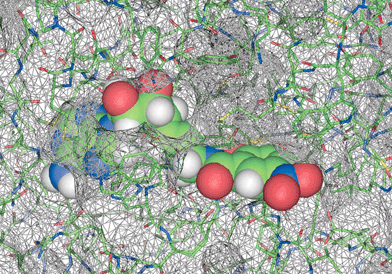 | ||
| Fig. 4 The active site region in the X-ray crystal structure of the ternary complex between COMT, Mg2+ ion and inhibitor 2. Space filling representation of 2 and solvent-accessible surface of COMT. It is clearly seen how the indole ring of Trp143 shields the adenosine and linker binding from exterior solvent. | ||
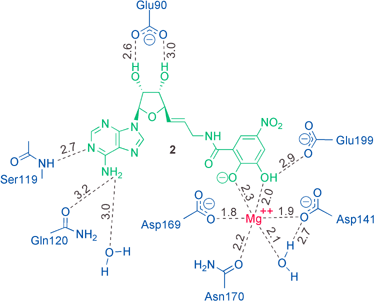 | ||
| Fig. 5 Schematic drawing of the H-bonding interactions (dashed lines) in the ternary complex. Distances are given in Ångstroms (Å). | ||
The adenine moiety has a favourable edge-to-face contact (shortest C⋯C distance: 3.5 Å) with the indole moiety of Trp143 which is located on the surface of COMT and shields the nucleobase and the linker from exterior bulk solvent (Fig. 6). The nucleobase is sandwiched between the imidazole ring of His142, deeply located in the enzyme, and the side chain of Met91 (Fig. 6). Whereas the imidazole ring undergoes a favourable CHis–H⋯π interaction with the adenine nucleus (centre-to-centre distance between the two ring systems: 5.1 Å), the methionine side chain stacks atop the six-membered ring of the nucleobase, with a closest intermolecular S⋯C contact of 3.6 Å. Close intermolecular interactions between methionine side chains, with their highly polarisable S-atom, and nucleobases are frequently observed in kinases28 and other nucleobase-binding enzymes.29
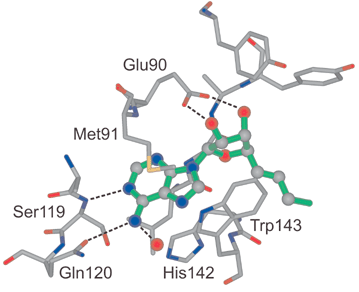 | ||
| Fig. 6 The nucleoside binding region in the X-ray crystal structure of the ternary complex. Hydrogen bonds are represented as dashed lines. | ||
The NH2 group of adenine forms hydrogen bonds to the side chain amide CO of Gln120 and a fixed H2O molecule, whereas N(1) undergoes hydrogen-bonding to the backbone N–H of Ser119 (for distances see Fig. 5). The two short ionic H-bonds of the 2′,3′-OH groups of the ribose moiety to the carboxylate of Glu90 seem to be quite essential in the recognition of the inhibitor by the enzyme. We found that removal of one of these OH-groups in 2 dramatically decreases the inhibitory affinity.30
In the catechol binding pocket (Fig. 7), the Mg2+ ion is coordinated octahedrally to the side chains of Asp141, Asp169 and Asn170, one localized H2O molecule and the two OH groups of the catechol moiety. The OH group in para position to the electron withdrawing nitro group is most probably deprotonated (measured pKa in aqueous solution = 4.4; it can be expected that coordination to the Mg2+ ion leads to a further increase in acidity) whereas the other one forms a H-bond to the carboxylate of Glu199. The nitro group has favourable van der Waals contacts to Pro174. The side chain of Met40 shields the catechol and parts of the linker from the exterior, and its methyl group is rotated by 79° relative to the conformation of Met40 found in the quaternary complex of COMT.
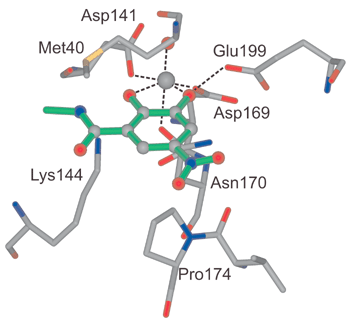 | ||
| Fig. 7 The catechol binding region in the X-ray crystal structure of the ternary complex. Hydrogen bonds are represented as dashed lines. | ||
Conclusions
Bisubstrate inhibition is a powerful concept to enhance binding affinity and selectivity for therapeutically relevant enzymes that catalyse reactions of two or more substrates or of a substrate and a cofactor. This is clearly demonstrated in the present study, which reports the versatile synthesis of a very potent bisubstrate inhibitor (2) for COMT with an IC50 value of 9 nM. The bisubstrate binding mode of 2 was proven in kinetic studies and by X-ray crystal structure analysis. This breakthrough could lead to novel effective drugs against the symptoms of Parkinson's disease.In order to accommodate in a strain-free way both the nucleoside and the catechol moieties at their respective binding sites, the size of the linker between them requires an optimal length. In this particular study, linkers consisting of one (too short; 3) or four carbon atoms (too long; 6) only led to inhibitors with micromolar activities, whereas linkers with two (4) or three (5) carbon atoms are active in the nanomolar range. The olefinic three-carbon spacer in 2 leads to further enhanced binding affinity since the inhibitor is highly preorganised prior to complexation, being unable to undergo hydrophobic collapse in the unbound state. The results of this investigation strongly advise to consider both the unbound and bound state in the optimisation of a new inhibitor. Thus the present findings are of considerable interest for the development of inhibitors for kinases and other methyltransferases that also require, in addition to a substrate, a cofactor for function and are popular targets in modern medicinal chemistry. We are now focusing on structural variations of the ribose, catechol and nucleobase moieties in next-generation bisubstrate inhibitors for COMT and are exploring the extension of the gained knowledge to other enzymes.
Experimental
General details
Solvents and reagents were purchased reagent-grade and used without further purification. All reactions were carried out under an Ar atmosphere. The following compounds were prepared according to literature procedures: N-hydroxysuccinimide ester 7,11 5′-amino-2′,3′-O-isopropylidene-5′-deoxyadenosine (8),17 2′,3′-O-isopropylideneadenosine (9),18 and o-iodoxybenzoic acid (IBX).31 THF was freshly distilled from sodium benzophenone ketyl, CH2Cl2 from CaH2. Anhydrous Me2SO and DMF, stored over molecular sieves, were purchased from Fluka. Evaporation in vacuo was done at ca. 20 Torr. All products were dried under high vacuum (0.05 Torr) before analytical characterisation. Thin layer chromatography was performed on glass plates coated with Merck SiO2 60 F254. Column chromatography was performed using Fluka SiO2 60 (230–400 mesh, 0.040–0.063 mm). Reversed-phase column chromatography was done using Fluka silica gel 100 C18 (230–400 mesh, 0.040–0.063 mm). Analytical reversed-phase HPLC was performed on a LiChrospher C-18 column (250 × 4 mm, 5 µm, 100 Å) from Merck, eluted with a linear gradient of 5% MeCN in water containing 0.1% TFA to 100% MeCN during 20 min and a flow-rate of 1 cm3 min−1 (UV detection at 254 nm). Melting points were determined with a Büchi B-540 melting point apparatus and are uncorrected. Optical rotations were measured on a Perkin–Elmer 241 polarimeter at λ = 589 nm or λ = 578 nm and are given in 10−1 deg cm2 g−1. Infrared spectra were recorded on a Perkin–Elmer 1600-FT–IR spectrometer. 1H and 13C NMR spectra (δ [ppm]; J [Hz]) were recorded on Varian Gemini 300 and Varian Mercury 300 spectrometers. MALDI mass spectra were recorded on an IonSpec Ultima instrument using 2,5-dihydroxybenzoic acid or 2,4,5-trihydroxyacetophenone–diammonium citrate (2∶1) as a matrix. The nomenclature was generated with the computer program ACD-Name (ACD/Labs).N-{[(2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl]methyl}-2,3-dihydroxy-5-nitrobenzene-1-carboxamide (3)
To 8 (89 mg, 0.34 mmol) in dry DMF (5 cm3), Et3N (0.1 cm3, 1 mmol) and 7 (110 mg, 0.372 mmol) were added and the resulting red solution was stirred at 20 °C for 15 h. Water (100 cm3) was added and the mixture adsorbed on a column with reversed-phase SiO2 (1 cm × 25 cm). After washing with 1% aq. HCOOH (200 cm3), 3 was eluted with CH3CN–1% aq. HCOOH (2∶8 → 3∶8). Evaporation to dryness by lyophilisation and re-precipitation of the residue from MeOH–Et2O afforded 3 (130 mg, 86%) as an orange powder, mp 198 °C (MeOH–Et2O); HPLC tR 7.02 min; [α]20D −27 (c. 0.33 in Me2SO); νmax(KBr)/cm−1 3363s, 3204s, 1647s, 1506s, 1473s, 1333s, 1175m, 1129m, 1072m, 855w, 825w, 769w, 743w, 649w; δH(300 MHz; (CD3)2SO + 1 drop of CF3COOD) 3.62–3.75 (2 H, m), 4.15 (1 H, dd, J 10.3, 5.3), 4.24 (1 H, t, J 5.3), 4.67 (1 H, t, J 5.3), 5.97 (1 H, d, J 5.3), 7.72 (1 H, d, J 2.5), 8.38 (1 H, d, J 2.5), 8.43 (1 H, s), 8.73 (1 H, s), 9.31 (1 H, t, J 5.0); δC(75 MHz; (CD3)2SO) 41.7, 71.6, 73.2, 82.9, 88.2, 111.3, 115.1, 115.7, 119.6, 137.7, 140.4, 147.3, 149.6, 152.2, 155.9, 157.3, 168.1; HRMS (MALDI) calcd. for C17H18N7O8+ (MH+): 448.1217; found: 448.1208.2-{(3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl}ethanenitrile (10)
To a well stirred suspension of 9 (758 mg, 2.47 mmol) and PPh3 (1.617 g, 6.166 mmol) in THF (12.4 cm3), acetone cyanohydrin (0.57 cm3, 6.2 mmol) was added. Within 5 min, DEAD (0.98 cm3, 97%, 6.1 mmol) was added at 0 °C. The solution was stirred at 0 °C for 50 min and further at 20 °C for 16 h. Evaporation in vacuo and column chromatography (SiO2; EtOAc–MeOH (96∶4)) afforded 10 (698 mg, 89%) as a brown oil; δH(200 MHz; CDCl3) 1.42 (3 H, s), 1.64 (3 H, s), 2.84–3.08 (2 H, m), 4.51–4.60 (1 H, m), 5.16 (1 H, dd, J 6.2, 3.3), 5.50 (1 H, dd, J 6.2, 2.1), 5.71 (2 H, br s), 6.12 (1 H, d, J 2.1); 7.93 (1 H, s); 8.38 (s, 1 H).(2R,3S,4R,5R)-2-(2-Aminoethyl)-5-(6-amino-9H-purin-9-yl)tetrahydrofuran-3,4-diol (11)
To a 0.1 M solution of 10 in THF (11 cm3, 1.1 mmol), glacial acetic acid (20 cm3) and PtO2 (200 mg) were added. The suspension was hydrogenated (4 bar H2) for 19 h. The catalyst was removed by filtration and the solvent evaporated in vacuo. Water (4 cm3) and TFA (10 cm3) were added to the residue, and the resulting solution was stirred for 2 h at 20 °C. The mixture was diluted with water (300 cm3) and extracted with CHCl3 (3 × 100 cm3). The aqueous phase was evaporated in vacuo to give a residue that was dissolved in water (10 cm3) and loaded onto a column of Dowex® 50 Wx4 (NH4+, 1 cm × 25 cm). Washing with MeOH–H2O (1∶1, 400 cm3) and elution with MeOH–13% aq. NH3 (1∶1), followed by removal of MeOH in vacuo, left an aqueous product-containing solution that was lyophilised to give 11 (227 mg, 74%) as a brown foam; δH(300 MHz; CD3OD + 1 drop of CF3COOD) 2.14–2.18 (2 H, m), 3.10 (2 H, t, J 6.9), 4.10–4.14 (1 H, m), 4.27 (1 H, t, J 4.5), 4.67 (1 H, t, J 4.5), 6.04 (1 H, d, J 4.1), 8.40 (1 H, s), 8.44 (1 H, s).N-{2-[(2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl]ethyl}-2,3-dihydroxy-5-nitrobenzene-1-carboxamide (4)
Starting from 11 (100 mg, 0.357 mmol) and 7 (116 mg, 0.392 mmol), the procedure described for 3 afforded 4 (161 mg, 98%) as an orange powder, mp 150 °C (decomp.) (Et2O–MeOH); HPLC tR 7.89 min; [α]20D −11 (c. 0.5 in Me2SO); νmax(KBr)/cm−1 3332s, 1696s, 1560w, 1516m, 1475w, 1420w, 1333s, 1201s, 1135m, 893w, 838w, 800w, 744w, 723m; δH(300 MHz; (CD3)2SO + 1 drop of CF3COOD) 1.97–2.10 (2 H, m), 3.36–3.48 (2 H, m), 4.03–4.10 (2 H, m), 4.61 (1 H, t, J 4.7), 5.97 (1 H, d, J 4.7), 7.69 (1 H, d, J 2.5), 8.33 (1 H, d, J 2.5), 8.45 (1 H, s), 8.65 (1 H, s); δC(75 MHz; (CD3)2SO) 32.4, 36.3, 73.2, 73.3, 81.9, 87.9, 111.9, 114.1, 114.2, 118.9, 138.2, 141.6, 146.8, 147.6, 148.5, 152.2, 156.1, 168.0; HRMS (MALDI) calcd. for C18H20N7O8+ (MH+): 462.1373; found: 462.1364.Ethyl (E)-3-{(3aR,4R,6R,6aR)-6-(6-amino-9H-purin-9-yl)-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-enoate (12)
To 9 (12.3 g, 40 mmol) in Me2SO (100 cm3), Ph3PCHCO2Et (42.8 g, 100 mmol) and o-iodoxybenzoic acid (IBX)
(27.8 g, 100 mmol) were added. The mixture was stirred at 20 °C for 72 h. Water (500 cm3) was added and the mixture extracted with EtOAc (2 × 500 cm3). The combined organic phases were dried (Na2SO4) and concentrated to a residue which was purified by column chromatography (SiO2; EtOAc–MeOH (96∶4)) to give 12
(10.5 g, 70%) as colorless crystals, mp 102 °C (EtOH)
(lit.,22 104–106 °C); δH(300 MHz; CDCl3) 1.22 (3 H, t, J 7.2), 1.40 (3 H, s), 1.63 (3 H, s), 4.11 (2 H, q, J 7.2), 4.79–4.83 (1 H, m), 5.14 (1 H, dd, J 6.2, 3.4), 5.56 (1 H, dd, J 6.2, 1.9), 5.81 (1 H, dd, J 15.9, 1.7), 6.13 (1 H, d, J 1.9), 6.96 (1 H, dd, J 15.9, 5.6), 7.87 (1 H, s), 8.33 (1 H, s).
(E)-3-{(3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-en-1-ol (13)
To 12 (375 mg, 1 mmol) in CH2Cl2 (3 cm3), a 1 M solution of DIBAL-H in hexane (8 cm3, 8 mmol) was added dropwise. The mixture was stirred at −78 °C for 2 h and then quenched with MeOH (5 cm3). A saturated aqueous solution of potassium sodium tartrate monohydrate (Rochelle salt, 50 cm3) was added, and the resulting suspension was stirred vigorously for 16 h at 20 °C, then extracted with EtOAc (3 × 50 cm3). The combined organic phases were dried (Na2SO4) and evaporated to dryness in vacuo to afford 13 (328 mg, 98%) as a colorless foam; [α]20D 17.8 (c. 1 in CHCl3); νmax(KBr)/cm−1 3339s, 2982m, 2932m, 1646s, 1595m, 1472w, 1370w, 1210m, 1080m; δH(300 MHz; CDCl3) 1.40 (3 H, s), 1.63 (3 H, s), 4.07 (2 H, s), 4.72 (1 H, dd, J 4.1, 2.5), 5.02 (1 H, dd, J 6.2, 4.1), 5.54 (1 H, dd, J 6.2, 2.2), 5.56 (2 H, s), 5.85–5.87 (2 H, m), 6.10 (1 H, d, J 2.2), 7.88 (1 H, s), 8.37 (1 H, s); δC(75 MHz; CDCl3) 25.3, 27.0, 61.7, 84.3, 84.6, 87.8, 90.7, 114.3, 119.8, 126.9, 134.4, 139.9, 149.2, 153.1, 155.6; HRMS (MALDI) calcd. for C15H20N5O4+ (MH+): 334.1515; found: 334.1505.2-(E)-3-{(3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl}prop-2-enyl-2,3-dihydro-1H-isoindole-1,3-dione (14)
DEAD (0.234 cm3, 97%, 1.46 mmol) was added dropwise to a stirred suspension of 13 (490 mg, 1.46 mmol), phthalimide (215 mg, 1.46 mmol) and Ph3P (383 mg, 1.46 mmol) in THF (7 cm3). After stirring for 1.5 h at 20 °C, a colorless solid started to precipitate. Stirring was continued for 1 h, after which the mixture was cooled to 0 °C and filtered. The residue was washed with Et2O and dried invacuo to give 14 (461 mg, 68%) as colorless crystals, mp 215 °C (MeOH); [α]20D 9.0 (c. 1 in CHCl3); νmax(KBr)/cm−1 3304m, 3155m, 1712s, 1676s, 1603m, 1430m, 1401m, 1335m, 1299m, 1207s, 1079s; δH(300 MHz; CDCl3) 1.36 (3 H, s), 1.59 (3 H, s), 4.19 (2 H, d, J 5.3), 4.69 (1 H, dd, J 6.5, 3.1), 4.98 (1 H, dd, J 6.2, 3.1), 5.49 (1 H, dd, J 6.2, 2.0), 5.59 (2 H, br s), 5.73 (1 H, dt, J 15.6, 5.3), 5.84 (1 H, dd, J 15.6, 6.5), 6.07 (1 H, d, J 2.0), 7.71–7.76 (2 H, m), 7.80–7.86 (3 H, m), 8.24 (1 H, s); δC(75 MHz; CDCl3) 25.3, 27.1, 38.7, 84.2, 84.6, 87.2, 90.6, 114.4, 120.2, 123.4, 127.4, 130.4, 132.0, 134.0, 139.8, 149.4, 153.2, 155.6, 167.7; HRMS (MALDI) calcd. for C23H23N6O5+ (MH+): 463.1730; found: 463.1724.9-{(3aR,4R,6R,6aR)-6-[(E)-3-Aminoprop-1-enyl]-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl}-9H-purin-6-amine (15)
To phthalimide 14 (885 mg, 1.91 mmol), a solution of 33% MeNH2 in EtOH (30 cm3) was added and the mixture was stirred at 20 °C for 16 h. After evaporation in vacuo, the residue was dissolved in CHCl3 (25 cm3) and extracted with 10% aqueous AcOH (30 cm3). The aqueous phase was washed with CHCl3 (3 × 25 cm3), adjusted to pH > 12 with 2 N NaOH and then extracted again with CHCl3 (4 × 25 cm3). The combined organic phases were dried (Na2SO4) and evaporated to give 15 (605 mg, 95%) as a colorless foam; [α]20D 30.8 (c. 1 in CHCl3); νmax(KBr)/cm−1 3686m, 3414m, 2999m, 1715m, 1631s, 1469w, 1376m, 1087s; δH(300 MHz; CDCl3) 1.39 (3 H, s), 1.62 (3 H, s), 3.22 (2 H, d, J 5.1), 4.68 (1 H, dd, J 7.3, 3.4), 4.99 (1 H, dd, J 6.2, 3.4), 5.53 (1 H, dd, J 6.2, 2.2), 5.71 (1 H, dd, J 15.6, 7.3), 5.83 (1 H, dt, J 15.6, 5.1), 5.87 (2 H, br s), 6.08 (1 H, d, J 2.2), 7.87 (1 H, s), 8.35 (1 H, s); δC(75 MHz; CDCl3) 25.4, 27.2, 43.3, 84.2, 84.6, 87.7, 90.4, 114.4, 120.2, 126.0, 136.3, 139.7, 149.3, 153.0, 155.4; HRMS (MALDI) calcd. for C15H21N6O3+ (MH+): 333.1675; found: 333.1667.(2R,3S,4R,5R)-2-[(E)-3-Aminoprop-1-enyl]-5-(6-amino-9H-purin-9-yl)tetrahydrofuran-3,4-diol (16)
A solution of 15 (481 mg, 1.45 mmol) in TFA–H2O (5∶2, 14 cm3) was stirred at 20 °C for 1 h and then evaporated to dryness. Purification of the residue by ion-exchange chromatography as described for 11 gave 16 (339 mg, 80%) as a colorless foam; [α]20D 65.0 (c. 1 in MeOH); νmax(KBr)/cm−1 3286br s, 1704s, 1507m, 2429m, 1325w, 1209s, 1142s, 1052m, 975w, 838m, 800m, 724s; δH(300 MHz; D2O + 1 drop of CF3COOD) 3.55 (2 H, d, J 6.2), 4.28 (1 H, dd, J 5.9, 5.0), 4.51 (1 H, t, J 5.0), 4.68 (1 H, dd, J 5.0, 4.1), 5.81–6.01 (2 H, m), 6.05 (1 H, d, J 4.1), 8.32 (1 H, m), 8.33 (1 H, s); δC(75 MHz; D2O) 44.2, 76.2, 76.3, 87.0, 90.4, 121.4, 129.6, 137.5, 142.4, 151.3, 155.4, 158.1; HRMS (MALDI) calcd. for C12H17N6O3+ (MH+): 293.1362; found: 293.1357.N-{(E)-3-[(2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl]prop-2-enyl}-2,3-dihydroxy-5-nitrobenzene-1-carboxamide (2)
Starting from 16 (29 mg, 0.1 mmol) and 7 (30 mg, 0.1 mmol), the procedure described for 3 afforded 2 (39 mg, 82%) as an orange powder, mp 173 °C (2-propanol); HPLC tR 9.31 min; [α]20D −9 (c. 0.5 in Me2SO); νmax(KBr)/cm−1 3378br s, 3115m, 1700s, 1642s, 1514m, 1473s, 1427m, 1351s, 1203s, 1131s, 1049m, 972w; δH(300 MHz; (CD3)2SO) 3.92–4.03 (2 H, m), 4.08–4.16 (1 H, m), 4.35 (1 H, t, J 5.3), 4.64–4.74 (1 H, m), 5.23 (1 H, br s), 5.52 (1 H, br s), 5.79–5.96 (3 H, m), 7.34 (2 H, br s), 7.65 (1 H, d, J 2.5), 8.07 (1 H, s), 8.32 (1 H, s), 8.48 (1 H, d, J 2.5), 9.64 (1 H, br s); δC(125 MHz; (CD3)2SO) 40.3, 72.8, 74.0, 84.0, 87.6, 111.0, 114.2, 115.3, 119.2, 129.0, 129.6, 137.2, 140.0, 147.2, 149.3, 152.3, 155.9, 157.7, 167.7; HRMS (ESI) calcd. for C19H20N7O8+ (MH+): 474.1373; found: 474.1358.(2R,3S,4R,5R)-2-(3-Aminopropyl)-5-(6-amino-9H-purin-9-yl)tetrahydrofuran-3,4-diol (17)
The allylic amine 15 (125 mg, 0.376 mmol) in EtOH (10 cm3) was hydrogenated (4 bar H2) in the presence of 10% Pd/C (100 mg) for 18 h. The catalyst was removed by filtration and the solvent evaporated in vacuo. TFA–H2O (5∶2, 30 cm3) was added and the mixture stirred for 1 h at 20 °C, then evaporated to dryness. Purification by ion-exchange chromatography as described for 11 gave 17 (94 mg, 84%) as a yellow foam; [α]20D −11.3 (c. 1 in MeOH); νmax(KBr)/cm−1 3105br s, 1695s, 1506m, 1431s, 1322m, 1206s, 1128s, 1061m, 839m, 800m, 717m; δH(300 MHz; CD3OD) 1.75–1.87 (4 H, m), 2.97 (2 H, t, J 7.5), 4.00–4.05 (1 H, m), 4.21 (1 H, t, J 5.0), 4.78 (1 H, t, J 5.0), 5.97 (1 H, d, J 5.0), 8.19 (1 H, s), 8.24 (1 H, s); δC(75 MHz; CD3OD) 25.2, 31.1, 40.4, 74.6, 74.8, 84.9, 90.2, 120.5, 141.4, 150.4, 153.7, 157.1; HRMS (MALDI) calcd. for C12H19N6O3+ (MH+): 295.1519; found: 295.1508.N-{3-[(2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl]propyl}-2,3-dihydroxy-5-nitrobenzene-1-carboxamide (5)
Starting from 17 (10 mg, 0.038 mmol) and 7 (11 mg, 0.038 mmol), the procedure described for 3 afforded 5 (16 mg, 89%) as a yellow foam; HPLC tR 9.20 min; [α]20D −55 (c. 1.0 in THF); νmax(KBr)/cm−1 3340br s, 1706s, 1646s, 1475s, 1263s, 1077m, 796w; δH((CD3)2SO + 1 drop of CF3COOD) 1.60–1.73 (4 H, m), 3.30–3.40 (2 H, m), 3.91–3.95 (1 H, m), 4.04 (1 H, t, J 5.0), 4.58 (1 H, t, J 5.0), 5.94 (1 H, d, J 5.0), 7.71 (1 H, d, J 2.5), 8.42 (1 H, d, J 2.5), 8.49 (1 H, s), 8.69 (1 H, s), 9.21 (1 H, br s); δC(75 MHz; (CD3)2SO + 1 drop of CF3COOD) 25.0, 30.3, 73.0, 73.4, 83.9, 87.8, 111.8, 113.9, 114.0, 118.6, 138.2, 142.4, 144.5, 146.5, 148.0, 149.7, 155.8, 167.8, one peak missing due to signal overlap; HRMS (MALDI) calcd. for C19H21N7O8+ ([M + Na]+): 498.1349; found: 498.1345.4-{(3aR,4R,6R,6aR)-6-(6-Amino-9H-purin-9-yl)-2,2-dimethylperhydrofuro[3,4-d][1,3]dioxol-4-yl}butanenitrile (18)
The allylic alcohol 13 (267 mg, 0.8 mmol) in EtOH (20 cm3) was hydrogenated (6 bar H2) in the presence of 10% Pd/C (100 mg) for 3 h. The catalyst was removed by filtration and the solvent evaporated in vacuo. The residue was dissolved in THF (5 cm3), then PPh3 (0.51 g, 2.0 mmol) and acetone cyanohydrin (0.18 cm3, 2.0 mmol) were added. Within 5 min, DEAD (0.3 cm3, 97%, 1.9 mmol) was added at 0 °C. The solution was stirred at 20 °C for 70 h. The orange mixture was evaporated in vacuo to give an oil that was purified by column chromatography (SiO2; CHCl3–MeOH (96∶4)) to provide 18 (131 mg, 49%) as a colorless oil; [α]20D −2.5 (c. 1 in CHCl3); νmax(CHCl3)/cm−1 3412m, 1716m, 1632s, 1588m, 1473m, 1376m, 1156m, 1094s; δH(300 MHz; CDCl3) 1.34 (3 H, s), 1.56 (3 H, s), 1.63–1.88 (4 H, m), 2.24–2.30 (2 H, m), 4.07–4.14 (1 H, m), 4.85 (1 H, dd, J 6.5, 4.0), 5.44 (1 H, dd, J 6.5, 2.3), 5.99 (1 H, d, J 2.3), 6.49 (2 H, br s), 7.84 (1 H, s), 8.28 (1 H, s); δC(75 MHz; CDCl3) 17.0, 21.9, 25.4, 27.2, 32.1, 83.8, 84.0, 85.9, 90.0, 114.6, 119.1, 120.1, 139.6, 149.0, 153.0, 155.8; HRMS (MALDI) calcd. for C16H21N6O3+ (MH+): 345.1675; found: 345.1673.N-{4-[(2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl]butyl}-2,3-dihydroxy-5-nitrobenzene-1-carboxamide (6)
Nitrile 18 (122 mg, 0.8 mmol) was hydrogenated and deprotected as described for 4 but the resulting amine was used without purification in the next step. Reaction with 7 (243 mg, 0.82 mmol) as described for 3 afforded 6 (69 mg, 18%) as an orange foam; HPLC tR 9.72 min; [α]20Hg578 +1.0 (c. 0.8 in MeOH); νmax(KBr)/cm−1 3384br s, 2941m, 1700 s, 1507m, 1473m, 1340s, 1202s, 1136m, 799w 723m; δH(300 MHz; CD3OD) 1.53–1.87 (6 H, m), 3.41 (2 H, t, J 6.2), 4.01–4.07 (1 H, m), 4.14 (1 H, t, J 5.0), 4.69 (1 H, t, J 5.0), 5.99 (1 H, d, J 5.0), 7.72 (1 H, d, J 2.5), 8.27 (1 H, d, J 2.5), 8.33 (1 H, s), 8.38 (1 H, s); δC(75 MHz; CD3OD) 24.2, 30.0, 34.1, 40.5, 75.1, 75.4, 86.0, 90.5, 113.3, 115.5, 116.1, 120.7, 140.7, 143.6, 147.3, 148.2, 150.2, 153.2, 156.5, 169.8; HRMS (MALDI) calcd. for C20H24N7O8+ (MH+): 490.1686; found: 490.1680.Enzymatic studies
Full experimental details of the radiochemical assay to determine IC50 values and the kinetic measurements for determining the enzyme inhibition mechanisms are given in previous publications.11,13,27 The procedure for co-crystallizing COMT with inhibitor 2 and Mg++ ions and solving the X-ray crystal structure has also been reported.13Acknowledgements
We thank F. Hoffmann-La Roche, Basel for generous support of this work. We are grateful to P. Malherbe for the cloning of COMT, P. Caspers for the expression of COMT, A. Cesura for enzyme purification, B. Wipf for fermentation and H. W. Lahm for sequencing.References
- D. Broom, J. Med. Chem., 1989, 32, 2–7 CrossRef.
- (a) W. T. Miller, Nat. Struct. Biol., 2001, 8, 16–18 CrossRef CAS; (b) K. Parang, J. H. Till, A. J. Ablooglu, R. A. Kohanski, S. R. Hubbard and P. A. Cole, Nat. Struct. Biol., 2001, 8, 37–41 CrossRef CAS.
- K. Hinterding, P. Hagenbuch, J. Retey and H. Waldmann, Chem. Eur. J., 1999, 5, 227–236 CrossRef CAS.
- M. Schlitzer, M. Bohm and I. Sattler, Bioorg. Med. Chem., 2002, 10, 615–620 Search PubMed.
- E. Wolf, J. De Angelis, E. M. Khalil, P. A. Cole and S. K. Burley, J. Mol. Biol., 2002, 317, 215–224 Search PubMed.
- K. L. Levert and G. L. Waldrop, Biochem. Biophys. Res. Commun., 2002, 291, 1213–1217 CrossRef CAS.
- (a) P. T. Männistö, I. Ulmanen, K. Lundström, J. Taskinen, J. Tenhunen, C. Tilgmann and S. Kaakkola, Prog. Drug Res., 1992, 39, 291–350 Search PubMed; (b) P. T. Männistö and S. Kaakkola, Pharmacol. Rev., 1999, 51, 593–628 Search PubMed; (c) V. V. Myllylä, M. Jackson, J. P. Larsen and H. Baas, Eur. J. Neurol., 1997, 4, 333–341; (d) M. C. Kurth, C. H. Adler, M. St. Hilaire, C. Singer, C. Waters, P. LeWitt, D. A. Chernik, E. E. Dorflinger and K. Yoo, Neurology, 1997, 48, 81–87 Search PubMed; (e) H. Baas, A. G. Beiske, J. Ghika, M. Jackson, W. H. Oertel, W. Poewe and G. Ransmayr, J. Neurol. Neurosurg. Psychiatry, 1997, 63, 421–428 CAS.
- J. Borgulya, M. Da Prada, J. Dingemanse, R. Scherschlicht, B. Schläppi and G. Zürcher, Drugs of the Future, 1991, 16, 719–721 Search PubMed.
- E. Nissinen, I. B. Linden, E. Schultz and P. Pohto, Naunyn-Schmiedebergs Arch. Pharmacol., 1992, 346, 262–266 Search PubMed.
- J. Vidgren, L. A. Svensson and A. Liljas, Nature, 1994, 368, 354–358 CrossRef CAS.
- B. Masjost, P. Ballmer, E. Borroni, G. Zürcher, F. K. Winkler, R. Jakob-Roetne and F. Diederich, Chem. Eur. J., 2000, 6, 971–982 CrossRef CAS.
- For earlier approaches to the development of bisubstrate inhibitors of COMT, see: E. K. Yau and J. K. Coward, J. Org. Chem., 1990, 55, 3147–3158 Search PubMed.
- For a preliminary communication of parts of this work, see: C. Lerner, A. Ruf, V. Gramlich, B. Masjost, G. Zürcher, R. Jakob-Roetne, E. Borroni and F. Diederich, Angew. Chem., Int. Ed. Engl., 2001, 40, 4040–4042 Search PubMed.
- B. Masjost, doctoral dissertation ETH No. 13719, Zürich, 2000.
- D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020 CrossRef.
- P. R. Gerber and K. Muller, J. Comput.-Aided Mol. Des., 1995, 9, 251–268 Search PubMed.
- M. Kolb, C. Danzin, J. Barth and N. Claverie, J. Med. Chem., 1982, 25, 550–556 CrossRef CAS.
- J. Tomasz, in Nucleic Acid Chemistry, ed. R. S. Tipson and L. B. Townsend, John Wiley & Sons, New York, 1978, vol. 2, 765–769 Search PubMed.
- J. Hollmann and E. Schlimme, Liebigs Ann. Chem., 1984, 98–107 Search PubMed.
- B. K. Wilk, Synth. Commun., 1993, 23, 2481–2484 CAS.
- E. J. Delaney, P. W. Morris, T. Matsushita and D. L. Venton, J. Carbohydr., Nucleosides, Nucleotides, 1981, 8, 445–459 Search PubMed.
- A. Matsuda and T. Ueda, Chem. Pharm. Bull., 1986, 34, 1573–1578 CAS.
- J. A. Montgomery, A. G. Laseter and K. Hewson, J. Heterocycl. Chem., 1974, 11, 211–214 Search PubMed.
- K. E. Pfitzner and J. G. Moffatt, J. Am. Chem. Soc., 1965, 87, 5661–5670 CrossRef CAS.
- D. Crich and X. S. Mo, Synlett, 1999, 67–68 CAS.
- S. Wolfe and S. K. Hasan, Can. J. Chem., 1970, 48, 3572–3579 CAS.
- G. Zürcher and M. Da Prada, J. Neurochem., 1982, 38, 191–195 CAS.
- (a) J. Vogt, R. Perozzo, A. Pautsch, A. Prota, P. Schelling, P. Pilger, G. Folkers, L. Scapozza and G. E. Schulz, Proteins, 2000, 41, 545–553 CrossRef CAS; (b) S. Pakhomova, M. Kobayashi, J. Buck and M. E. Newcomer, Nat. Struct. Biol., 2001, 8, 447–451 CrossRef CAS.
- (a) V. Villeret, S. H. Huang, Y. P. Zhang and W. N. Lipscomb, Biochemistry, 1995, 34, 4307–4315 CrossRef CAS; (b) C. Zubieta, X. Z. He, R. A. Dixon and J. P. Noel, Nat. Struct. Biol., 2001, 8, 271–279 CrossRef CAS; (c) A. Ruf, V. Rolli, G. de Murcia and G. E. Schulz, J. Mol. Biol., 1998, 278, 57–65 CrossRef CAS; (d) E. A. Meyer, R. Brenk, R. K. Castellano, M. Furler, G. Klebe and F. Diederich, ChemBioChem, 2002, 3, 250–253 CrossRef CAS.
- C. Lerner, R. Siegrist and F. Diederich, unpublished results.
- R. E. Ireland and L. B. Liu, J. Org. Chem., 1993, 58, 2899–2899 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |