Transformations of cyclic nonaketides by Aspergillus terreus mutants blocked for lovastatin biosynthesis at the lovA and lovC genes
John L.
Sorensen
a,
Karine
Auclair
b,
Jonathan
Kennedy
c,
C.
Richard Hutchinson
c and
John C.
Vederas
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: john.vederas@ualberta.ca; Fax: 00 1 780 492 5475; Tel: 00 1 780 492 8231
bDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 2K6, Canada. E-mail: karine.auclair@mcgill.ca; Fax: 00 1 514 398 3797; Tel: 00 1 514 398 2822
cKosan Biosciences Inc., 3832 Bay Center Place, Hayward, California 94545, USA. E-mail: hutchinson@kosan.com; Fax: 00 1 510 732 8401; Tel: 00 1 510 732 8400
First published on 21st November 2002
Abstract
Two mutants of Aspergillus terreus with either the lovC or lovA genes disrupted were examined for their ability to transform nonaketides into lovastatin 1, a cholesterol-lowering drug. The lovC disruptant was able to efficiently convert dihydromonacolin L 5 or monacolin J 9 into 1, and could also transform desmethylmonacolin J 15 into compactin 3. In contrast, the lovA mutant has an unexpectedly active β-oxidation system and gives only small amounts of 1 upon addition of the immediate precursor 9, with most of the added nonaketide being degraded to heptaketide 22. Similarly, the lovA mutant does not accumulate the polyketide synthase product 5 and rapidly degrades any 5 added as a precursor via two cycles of β-oxidation and hydroxylation at C-6 to give 20. The possible involvement of epoxides 21a and 21b in the biosynthesis of 1 was also examined, but their instability in fermentation media and fungal cells will require purified enzymes to establish their role.
Introduction
Lovastatin 1 (also known as mevinolin, monacolin K, and Mevacor™) is a potent cholesterol-lowering agent1 and a precursor for synthesis of the widely-prescribed drug, simvastatin 2 (Zocor™).2 Its 6-desmethyl analogue, compactin 3 (also designated as ML-236B or mevastatin) can be converted by microbial oxidation to the 6β-hydroxy derivative, pravastatin 4 (Pravachol™).3,2b These compounds and certain fully synthetic mimics block (3S)-hydroxy-3-methylglutaryl-coenzyme A reductase, the rate-limiting enzyme in the biosynthesis of cholesterol.1–4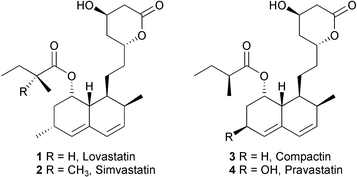
Biosynthetic studies on lovastatin 1 with fungi, such as Aspergillus terreus, show that its assembly via a polyketide pathway involves intial construction of dihydromonacolin L 5. The cooperation of polyketide synthases (PKSs) encoded by the lovB and lovC genes (Fig. 1) accomplish the ca. 35 steps necessary to generate 5 (Scheme 1).5,6 In the absence of the LovC protein, which imparts enoyl reductase activity to the complex, the LovB iterative type I PKS enzyme generates truncated pyranones 6 and 7.5 During normal biosynthesis, oxidative transformations of the PKS product 5 introduce a second double bond in the decalin (decahydronaphthalene) system and add the C-8 hydroxyl to form monacolin J 9.7 The 2-methylbutyryl side chain is produced by a separate PKS encoded by lovF, and further used by LovD to directly acylate 9 and yield 1.5 The function of LovA, a protein essential for the formation of 1, is not fully understood, but it has sequence homology to P450 enzymes and is probably involved in the oxidative conversion of 5 to introduce the diene system present in 9. Biosynthetic studies on compactin 3 using the fungus Penicillium aurantiogriseum indicate a labelling pattern analogous to that seen in lovastatin 1,8a but the genetic machinery required for the formation of 3 has not yet been reported.†
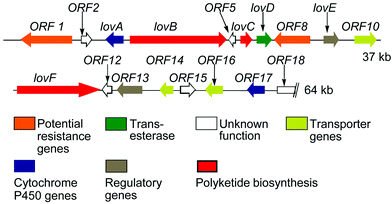 | ||
| Fig. 1 Lovastatin biosynthesis genes. | ||
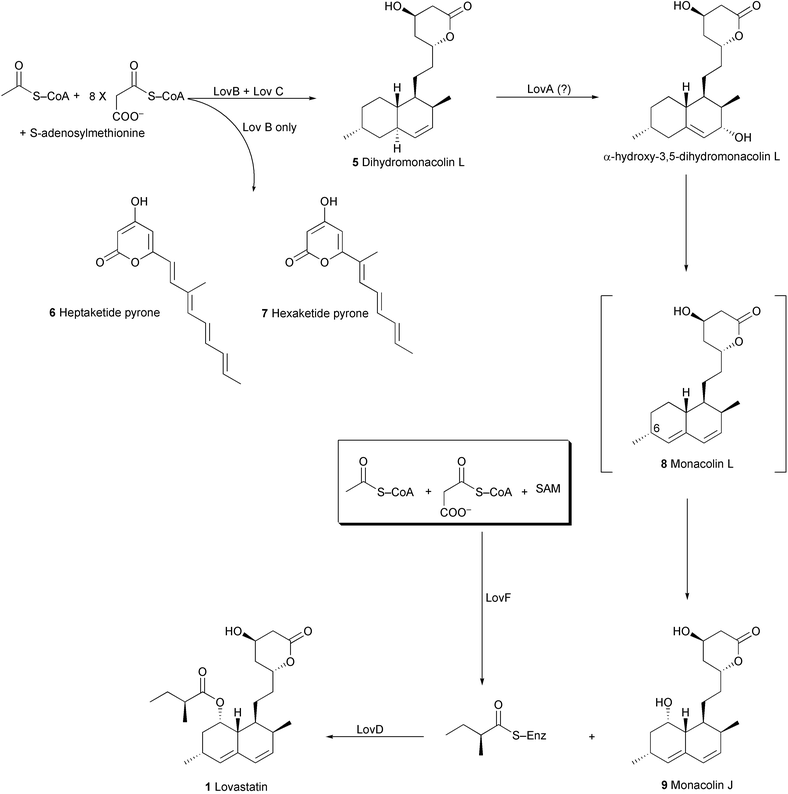 | ||
| Scheme 1 Biosynthesis of Lovastatin. | ||
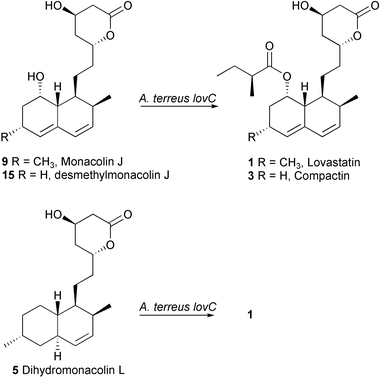
Preliminary studies using an A. terreus mutant with the lovC gene disrupted demonstrated that cyclic nonaketide precursors such as dihydromonacolin L 5 and monacolin J 9 could be transformed to lovastatin 1 by this organism.9 We now report full details of this work as well as transformations of other cyclic nonaketides by this mutant and by an A. terreus mutant having a disrupted lovA gene. The results suggest that gene disruption can alter the profile of secondary metabolites in ways that cannot be easily predicted based solely on their function in a biosynthetic pathway.
Results and discussion
In order to probe the role of the lovA and lovC genes in the biosynthetic pathway, two A. terrus mutants were examined in which one of these genes was disrupted, thereby blocking the production of lovastatin 1. In the first instance we employed an A. terreus mutant with its lovC gene disrupted.‡ As mentioned above, the lovC gene and the LovC protein produced by it have previously been demonstrated to be necessary for the biosynthesis of dihydromonacolin L 5.5,6 It has also been shown that heterologous expression of the lovastatin biosynthesis gene lovB in Aspergillus nidulans results in production of the truncated polyketide metabolites 6 and 7.5 These appear to arise after failure to complete the proper enoyl reductase step at the triketide stage. However, expression of both the lovC and lovB genes in A. nidulans results in complete biosynthesis of dihydromonacolin L 5, thereby indicating that LovC is responsible for providing the required enoyl reductase function to the LovB PKS system. The structure of 5 produced by these cultures has been confirmed by spectroscopic techniques and X-ray crystallography (Fig. 2).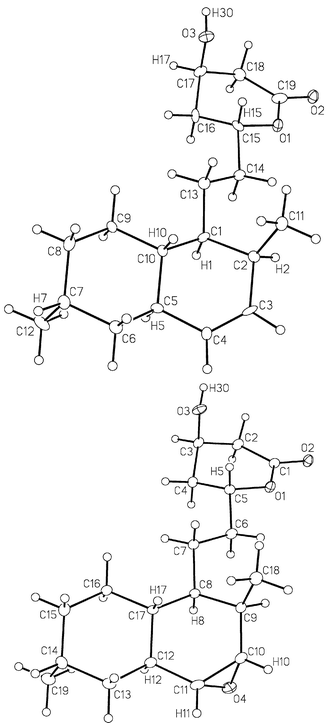 | ||
| Fig. 2 Crystal structure of 5 (top) and 21a (bottom). | ||
Biotransformations by A. terreus lovC mutant
Careful HPLC analysis of fermentation extracts of the A. terreus mutant deficient in the lovC gene confirmed that it did not produce any 5 or 1. However the entire post-PKS machinery required to transform 5 into 1 would still be expected to be intact and functional. In order to test this, a series of biotransformation experiments were conducted. Initially, formation of the diketide side chain (under the control of lovF gene) and its esterification onto monacolin J 9 (under the control of lovD gene) to afford 1 were examined. Monacolin J 9 was added (EtOH solution) to fermentation cultures of A. terreus lovC mutant in mid-log phase. Ten hours later, the culture broth was extracted and examined for the presence of 1. HPLC analysis revealed that about 70% of 9 had been converted into 1. This demonstrates that the lovastatin diketide synthase (LDKS) is present and fully functional, and unlike the lovastatin nonaketide synthase (LNKS i.e. LovB), does not require LovC for proper assembly of the diketide side chain.The specificity of the late stage enzymes was probed by examining whether 6-desmethylmonacolin J 15 could be transformed into compactin 3 by the A. terreus lovC mutant. When 15 (obtained by the basic hydrolysis of 3) was added to fermentation cultures of this mutant, 3 was isolated from the culture broth in 45% yield after purification. This demonstrates that the acyltransferase enzyme (LovD) can tolerate the lack of a methyl group at the C-6 position of monacolin J 9. In a separate experiment ML-236C, the 6-desmethyl analogue of monacolin L 8, was added to cultures of the A. terreus lovC mutant. However, no compactin 3 could be detected by HPLC in the extract of the culture broth. This result suggests that either the enzyme responsible for hydroxylation at C-8 has a stringent requirement for the C-6 methyl group, or that 8 and ML-236C are not intermediates on the biochemical route to 1 and 3, respectively. The latter possibility may be more probable, as occurrence of 8 on the direct lovastatin biosynthetic pathway has been disputed and is still uncertain.10
To help determine if the A. terreus lovC disruptant could be used effectively to make dihydrolovastatin analogues, possible biotransformation of dihydromonacolin J 12 was examined. Compound 12 could be synthesised in 4 steps from 1 as shown in Scheme 2. Basic hydrolysis of 1 gave 9, which could be selectively protected with TBDMSCl in DMF to afford 10. Selective hydrogenation of 10 using Ir(cod)py(Pcy3)PF6 catalyst11 in MeOH generated 11 in 86% yield. Removal of the protecting group with buffered TBAF produced 12 in 34% overall yield from 1. Intermediate 11 could also be acylated with (S)-2-methylbutyric anhydride12 yielding 13 which, with subsequent removal of the protecting group, affords the target dihydrolovastatin 14 in 35% overall yield from 1.13 Compound 12 was added to cultures of the A. terreus lovC disruptant, but formation of 14 could not be detected by extensive HPLC analysis of the culture broth using synthetic 14 as a standard. Dihydrolovastatin 14 has been previously isolated from the fermentations of wild type A. terreus in 8% yield relative to lovastatin 1, as determined by HPLC.14 This earlier study in combination with the current results suggests that either disruption of lovC unexpectedly alters the capability of the organism to transform 12 to 14, or that this conversion does occur, but with alteration of the metabolism which further transforms 14 and brings its concentration below our detection limit.
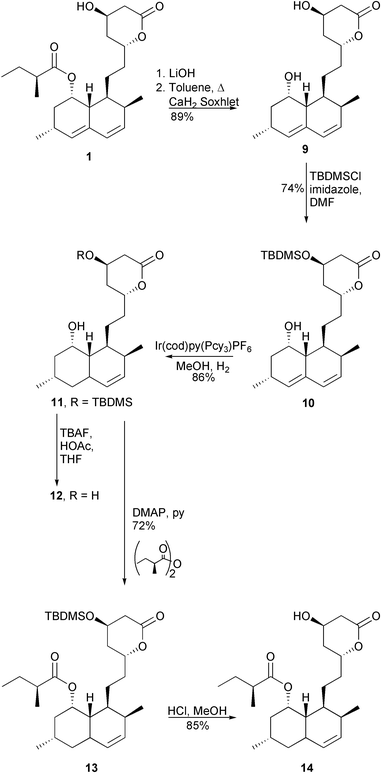 | ||
| Scheme 2 Syntheses of dihydromonacolin J 12 and dihydrolovastatin 14. | ||
To verify that the post-PKS enzyme system required to oxidatively introduce the second double bond of the diene moiety is intact in the A. terreus lovC mutant, dihydromonacolin L 5 (isolated from A. nidulans lovB + lovC) was added to a fermentation of this organism and the broth extract was examined by HPLC for the presence of 1. Precursor 5 was transformed into 1 in 40% isolated yield, thereby demonstrating that all the necessary post-PKS steps for lovastatin biosynthesis are present in this mutant (Scheme 3).
 | ||
| Scheme 3 A. terreus lovC biotransformations. | ||
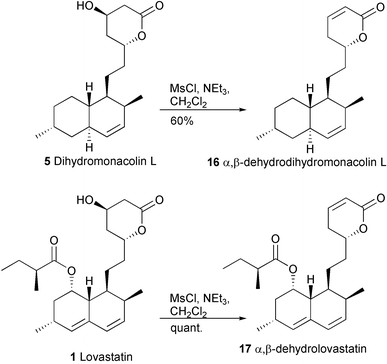
The substrate specificity of the post-PKS machinery could be further tested by attempted transformation of a dihydromonacolin L derivative 16 having an unsaturation in the upper ring (Scheme 4). Elimination from the methanesulfonates of 5 or 1 gave α,β-dehydrodihydromonacolin L 16 and α,β-dehydrolovastatin 17, respectively.15 Addition of 15 to cultures of the A. terreus lovC disruptant followed after 10 hours by HPLC analysis of the culture showed no detectable trace of 17, suggesting that changes in the upper ring are not tolerated. The results of successful biotransformation experiments with A. terreus lovC mutant are summarised in Scheme 3.
 | ||
| Scheme 4 Synthesis of unsaturated analogues. | ||
Biotransformations by A. terreus lovA mutant
The transformation of 5 to 8 requires at least one oxidative enzyme, and it seemed that the lovA gene product, whose sequence is similar to cytochrome P450 enzymes, could be involved in this process. In order to investigate this, the biotransformations performed by an A. terreus mutant disrupted at the lovA gene were examined. This organism would be expected to accumulate dihydromonacolin L 5, as it has an intact PKS system (i.e.LovB + LovC enzymes) and it should lack the ability to produce 1. HPLC analysis of cultures of A. terreus lovA disruptant did not reveal the presence of any 1 (as expected), but 5 could not be detected either. This unexpected result suggests that either the A. terreus lovA mutant does not have the ability to produce 5, or that 5 is somehow further transformed by the fungus.In order to determine the fate of 5, 14C-dihydromonacolin L (produced by addition of sodium 14C-acetate to A. nidulans lovB + lovC cultures), was fed to cultures of A. terreus lovA mutant. The extract of the culture was fractioned by thin layer chromatography, and the band containing most of the radioactivity was isolated to yield a chromatographically homogeneous compound. Full structural assignment reveals that 20 is the major product of 5 in the A. terreus lovA disruptant. This compound presumably arises as a result of two cycles of β-oxidation,16 first yielding the octaketide 18 followed by the heptaketide 19. This is then terminated by a P450 type hydroxylation at C-6 to produce 20 (Scheme 5). This process for metabolising 5 does not appear to be unique to the A. terreus lovA mutant, as 20 was also isolated from extracts of the cultures of A. nidulans lovB + lovC, along with a small amount of the intermediate heptaketide 19. Careful examination of the radioactive 5 obtained from this A. nidulans strain and used as the precursor for the biotransformation confirmed that it was not contaminated by 19 or 20. Since β-oxidation is ubiquitous in fungi such as Aspergillus,16 and hydroxylation at C-6 is common in the metabolism of lovastatin 1,1720 appears to arise from a widespread metabolic pathway in Aspergillus which is unrelated to lovastatin biosynthesis. Although this explains the lack of dihydromonacolin L 5 in the A. terreus lovA mutant cultures, it is perhaps surprising that blockage of a key oxidative enzyme on the route to lovastatin 1 should shunt the metabolism so completely.
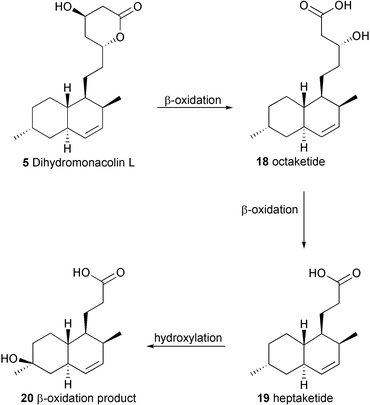 | ||
| Scheme 5 Proposed pathway to 20. | ||
Another well established mode of reaction of P450 enzymes is the epoxidation of a double bond such as that present in 5.18 Hydroxylation with allylic rearrangement to directly form the proposed intermediate, α-hydroxy-3,5-dihydromonacolin L19 (Scheme 1), would actually be an unexpected reaction for this type of enzyme,20 which alternatively may be capable of dehydrogenation of 5 directly to the diene system. In order to determine if the LovA enzyme could form an epoxide during biosynthesis of lovastatin, the two diastereomeric epoxides 21a and 21b were prepared from 5 by treatment with MCPBA, followed by separation of the stereoisomers by HPLC (Scheme 6). The stereochemical assignment of the two epoxides was confirmed by X-ray crystallography of the α-isomer (Fig. 2). In a separate experiment, 14C labelled 5 was used in the synthesis so that the transformation of 21a and 21b could be followed by tracer analysis. Each 14C-labelled diastereomer was fed to separate cultures of A. terreus lovA mutant, which were subsequently analysed by thin layer chromatography with radioisotopic scanning (radio-TLC). If an epoxide were the product of the LovA enzyme and therefore a true intermediate on the pathway to lovastatin, then either 21a or 21b should presumably be converted through the rest of the post-PKS pathway to yield 1. However, no radioactive 1 could be detected by radio-TLC analysis. Analogous experiments wherein 21a and 21b were fed separately to cultures of A. terreus lovC mutant, yielded similar results: no radioactive 1 could be detected. In all cases, none of the original epoxide could be detected or recovered. Control experiments show that the epoxides are quite sensitive to the culture media, and decompose to complex mixtures of polar compounds that are not easily extracted into organic solvents. Treatment of 21a with the methyl ester of glycine results in rapid nucleophilic opening of the epoxide ring by the amino group (data not shown). Hence, it is likely that these epoxides can react with nucleophiles either in the media or in the fungal cells prior to exposure to post-PKS enzymes in the lovastatin pathway. Further experiments to determine the intermediacy of 21a or 21b will require purified enzymes.
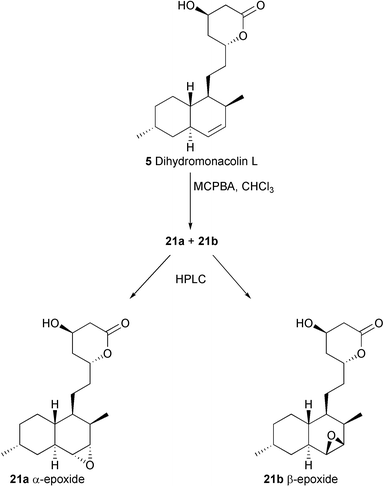 | ||
| Scheme 6 Synthesis of 21a and 21b. | ||
To verify that the late stages of the post-PKS machinery are still functional in the A. terreus lovA mutant, monacolin J 9 was added to cultures of this organism. Examination of the extracts by HPLC allowed isolation of a trace amount (ca. 1%) of lovastatin 1 along with an additional metabolite. The latter was identified as 22. Similar to the fate of 5, compound 9 is primarily metabolised by β-oxidation enzymes to a truncated product. However in this case, the additional double bond in the decalin system appears to hinder hydroxylation at C-6. The biotransformations performed by A. terreus lovA mutant are summarised in Scheme 7.
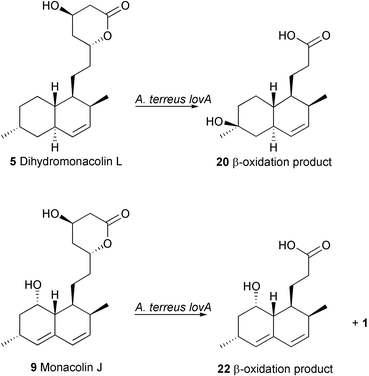 | ||
| Scheme 7 Transformations by A. terreus lovA mutant. | ||
Conclusions
Biotransformations of nonaketides accomplished by mutants of A. terreus deficient in two lovastatin biosynthesis genes (lovA or lovC) have been examined, and the structures of the resulting metabolites were determined. The mutant disrupted at the lovC gene lacks the ability to produce dihydromonacolin L 5 or lovastatin 1, but still contains a highly functional set of post-PKS enzymes. This is established by the ability of the A. terreus lovC mutant to effectively transform either 5 or monacolin J 9 into 1. In addition, this organism can transform 15 into 3, thereby demonstrating that it can accept a slightly modified substrate. This work suggests that it may be possible to use the A. terreus lovC mutant to convert a variety of precursors into lovastatin analogues. Studies to accomplish this are in progress. As different substrate analogues may exhibit varying transport properties and diverse efficiency of transformation, cell-free studies with purified enzymes will be critical for detailed understanding of the biochemical conversions.The mutant of A. terreus disrupted at the lovA gene displays quite different characteristics. In contrast to the lovC mutant, its β-oxidation system is highly active, with the consequence that there is rapid degradation of exogenously-added nonaketides which are known to be intermediates on the biosynthetic route to lovastatin 1. For example, only a small amount of monacolin J 9, the immediate precursor of 1, is converted into the target drug, with most of the added compound being oxidatively degraded to 22. This lovA mutant also does not accumulate dihydromonacolin L 5 as expected, but rather metabolises it rapidly to 20. The LovA protein, although resembling a P450 enzyme and clearly required for production of lovastatin 1, may also be involved in generating compounds which exercise control over expression or operation of the β-oxidation system in A. terreus.
Experimental
General Experimental
All chemicals were purchased from Aldrich Chemical company (Madison, WI) or Sigma Chemicals (St. Louis, MO). All solvents, unless otherwise indicated, were HPLC grade and used as such. Dry CH2Cl2 was prepared by distilling under Ar from CaH2. Diazomethane was prepared as a diethyl ether solution from DIAZALD (N-methyl-N-nitrosotoluene-p-sulfonamide) according to Aldrich technical bulletin AL-113. Evaporation refers to removal of solvent under reduced pressure on a rotary evaporator. Preparative TLC was done on glass plates (20 × 20 cm) pre-coated (0.25, 0.5, 1.0 mm) with silica gel (EM Science, Kieselgel 60 F254). Analytical TLC was done on glass plates (5 × 1.5 cm) pre-coated (0.25 mm) with silica gel (EM Science, Kieselgel 60 F254). Compounds were visualized by exposure to UV light and by dipping the plates in a 1% Ce(SO4)2·4H2O 2.5% (NH4)Mo7O24·4H2O in 10% H2SO4 followed by heating on a hot plate. Flash column chromatography was performed with Silicycle silica gel (60 Å, 230–400 mesh).Preparative HPLC was performed on a RANIN Dynamax high performance liquid chromatograph employing a manual injector, fraction collector and single wavelength detector. Reversed phase HPLC was conducted utilizing a Waters radial compression module with a Bondapak C18 25 × 100 mm column (15–20 µm particle size) with a guard column of similar packing. The mobile phase for RP-HPLC typically consisted of 100% H2O to 70% CH3CN–30% H2O over 25 min, linear gradient. Normal phase HPLC was done on the same system using a Waters radial compression Porasil silica gel column (125 Å, 25 × 100 mm). The NP-HPLC mobile phase employed was 30% EtOAc–70% hexane to 70% EtOAc–30% hexane over 27 min.
NMR spectra were recorded on a Varian Inova 600, Inova 300 or Unity 500 spectrometer. For 1H (300, 500 or 600 MHz) δ values are referenced to CHCl3 (7.24 ppm) and for 13C (125.8 or 150.9 MHz) referenced to CDCl3 (77.0 ppm).
Melting points are uncorrected and were determined on a Kauffman Block microscope or a Thomas-Hoover apparatus using open capillary tubes. Optical rotations were measured on a Perkin-Elmer 241 polarimeter with a micro cell (100 mm path length, 1 mL). IR spectra were recorded on a Nicolet Magna-IR 750 with a Nic-Plan microscope FT-IR spectrometer. Mass spectra were recorded on a Krator AEI MS–50 (HREIMS) and a ZabSpec Isomass VG (HRESMS).
Radioactivity was determined using standard liquid scintillation procedures in plastic 10 ml scintillation vials with a Beckman Ready-gel scintillation cocktail. The scintillation counter used was a Beckman LS 5000TD with automatic quench control to directly determine decompositions per minute (dpm) in the labelled samples against a quench curve prepared from Beckman 14C quenched standards. Radiolabelled compounds were also detected by a TLC assay, using a Berthold LB2760 2D-TLC scanner.
Aspergillus nidulans growth media was prepared by dissolving 20 g of glucose, 20 g of yeast extract, 1 g of peptone and 1 mL of a 1 mg per mL solution of p-aminobenzoic acid in 1 L of distilled deionized water. Aspergillus nidulans production media was prepared by dissolving in 1 L of distilled deionized water 1 mL trace element solution (1.0 g FeSO4·7H2O, 8.8 g ZnSO4·7H2O, 0.4 g CuSO4·5H2O, 0.15 g MnSO4·4H2O, 0.1 g Na2B7O7·10H2O, 0.05 g (NH4)6Mo7O24·4H2O, 0.5 mL conc. HCl dissolved in 1 L H2O), 100 mL of 10 × AMM (Aspergillus Minimal Media) salts (60 g NaNO3, 5.2 g KCl, 15.2 g KH2PO4 dissolved in 1 L of H2O and adjusted to pH 6.5), 1 mL of a 1 mg per mL p-aminobenzoic acid solution, and 0.9 mL of cyclopentanone and autoclaving. After cooling to room temperature 2.5 mL of a sterile solution of 20% MgSO4·7H2O and 25 mL of a sterile 40% lactose solution was added to the above flask. All biological media solutions and equipment were autoclaved at 121 °C and 15 psi of steam pressure for 15 min.
4a,5-Dihydromonacolin L 5
4a,5-Dihydromonacolin L 5 was isolated from a strain of Aspergillus nidulans expressing the lovastatin lovB and lovC biosynthesis genes following the cloning procedure described elsewhere.5 The typical isolation procedure is described below. 10 µL of a concentrated spore suspension was used to inoculate 1 L of A. nidulans growth media and the culture was incubated at 30 °C and 200 rpm for 4 days. The mycelia were harvested by filtering through sterile miracloth (Calbiochem) and were thoroughly rinsed with 2 L of 1% lactose solution. The wet mycelia were divided into 4 equal portions and used to inoculate 4 × 1 L of A. nidulans production media. Cultures were incubated at 30 °C and 200 rpm for 2 days, after which point the growth was supplemented with 1 g L−1 sodium acetate added each day for an additional 5 days. The cultures were harvested by removing the mycelia by vacuum filtration and the combined broth (4 L) was acidified with 2 M HCl (pH < 2) and extracted with CH2Cl2 (2 × 1.5 L). The combined organic layers were dried (Na2SO4), filtered and evaporated to provide ca. 500 mg of crude extract which was fractionated by flash column chromatography to provide 300 mg of pure 4a,5-dihydromonacolin L 5 (typical yields, 50–70 mg L−1 of production media).14C-4a,5-Dihydromonacolin L 5 was produced from 1 L of A. nidulans production culture fermented as described above, except that 250 µCi of sodium [1-14C]acetate (fed as a 5 mL aqueous solution 4 times per day) was added in addition to the unlabelled sodium acetate. The culture was harvested and extracted as above and the crude extract (97 mg) was fractionated by preparative TLC to give 22.5 mg of pure 14C-4a,5-dihydromonacolin L 5 with a specific activity of 0.185 µCi mg−1. The 1H NMR of the 14C-4a,5-dihydromonacolin L 5 was identical to that of the unlabelled compound.
For unlabelled 5: mp 162–163°C, lit. mp 163–164 °C;21 [α]25D = + 115 (c 0.22, methanol), lit. [α]25D = + 123.9 (c 0.5, methanol); UV (MeCN solution) λmax 228 nm (br 210–234); IR (microscope) 3391 (br m), 3018 (w), 2910 (s), 1713 (s), 1254 (s), 1062 (s), 1047 (s) cm−1; HREIMS 306.21902 (306.21948 calcd. for C19H30O3) (M+, 27%), 288.20876 (M − H2O, 18%), 161.13299 (M − C7H13O3, 100%), 105.07044 (C8H9+, 99%). 1H NMR (600 MHz, CDCl3) δ 5.57 (ddd, 1H, J = 9.8, 4.9, 2.7 Hz, H-3), 5.28 (d, 1H, J = 9.8 Hz, H-4), 4.67 (m, 1H, H-11), 4.38 (m, 1H, H-13), 2.72 (dd, 1H, J = 17.6, 5.13 Hz, H-14), 2.60 (ddd, 1H, J = 17.6, 3.7, 1.6 Hz, H-14), 2.21 (m, 1H, H-2), 2.00 (m, 1H, H-6), 1.95 (m, 1H, H-12), 1.89 (m, 1H, H-4a), 1.81–1.73 (m, 2H, H-10 and H-12), 1.62 (m, 2H, H-9), 1.58–1.42 (m, 5H, H-5, H-7, H-8a and H-10), 1.07 (dq, 1H, J = 11.9, 3.7 Hz, H-8), 0.97 (dq, 1H, J = 11.9, 3.0 Hz, H-8), 0.96 (d, 3H, J = 7.3 Hz, 6-CH3), 0.82 (d, 3H, J = 6.9 Hz, 2-CH3); 13C NMR (125 MHz, CDCl3) δ 170.4 (C-15), 132.6 (C-3), 131.6 (C-4), 76.1 (C-11), 62.8 (C-13), 41.4 (C-8a), 40.0 (C-8), 38.9 (C-5), 38.7 (C-14), 37.3 (C-4a), 36.1 (C-12), 33.2 (C-10), 32.3 (C-7), 32.0 (C-2), 27.5 (C-6), 23.7 (C-1), 23.6 (C-9), 18.2 (6-CH3), 14.9 (2-CH3).
Crystal data for 4a,5-dihydromonacolin L 5
Data were acquired on a Bruker P4/RA/SMART 1000 CCD diffractometer. All intensity measurements were performed using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). 4a,5-Dihydromonacolin L 5 (C19H30O3) was obtained as white crystals, space group P212121 (No. 19), a = 5.5947 (9), b = 9.6504 (17), c = 31.930 (6) Å, V = 1723.9 (5) Å3, Z = 4, T = −80 °C, ρcalcd = 1.181 g cm−3, μ = 0.078 mm−1. 10870 reflections were measured, 3287 unique which were used in all least squares calculations, R1(F) = 0.0620 (for 899 reflections with Fo2 ≥ 2σ(Fo2)), wR2(F2) = 0.2159 for all unique reflections. The absolute stereochemistry cannot be directly determined from the X-ray data, but it is correct as shown based on transformation of 5 to 1. CCDC reference number 191453. See http://www.rsc.org/suppdata/ob/b2/b207721c/ for crystallographic files in CIF or other electronic format.Monacolin J 9
Monacolin J 9 was produced by the basic hydrolysis of lovastatin as follows. 200 mg of lovastatin 1 was placed in a 25 mL round bottomed flask and suspended in 10 mL of aqueous 1 M LiOH and heated at reflux overnight. The reaction was cooled to room temperature, acidified with 2 M HCl (pH < 2) and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried (Na2SO4), filtered and evaporated to give 243 mg crude residue which was dissolved in 25 mL toluene and lactonized by heating at reflux with a CaH2 Soxhlet apparatus for one hour. After cooling to room temperature the solvent was evaporated and the residue (208 mg) was fractionated by flash column chromatography (40% EtOAc in hexanes) to give 140 mg (89% yield) pure monacolin J 9.[α]25D = +17.3 (c 0.14, CH3OH); IR (microscope) 3236 (br s), 2927 (s), 2879 (s), 1707 (m), 1645 (m), 1451 (s), 1318 (s), 1093 (s), 1075 (s), 1060 (s), 1049 (s), 1026 (s), 973 (s), 858 (s) cm−1; HREIMS [M]+ 320.19827 (320.19876 calcd. for C19H28O4) 320.2 (2%), 302.2 (9%), 198.1 (58%), 157.1 (100%), 105.1 (41%); 1H NMR (500 MHz, CD3OD) δ 5.93 (d, 1H, J = 10.0 Hz, H-4), 5.75 (dd, 1H, J = 10.0, 6.2 Hz, H-3), 5.45 (br s, 1H, H-5), 4.23 (br dd, 1H, J = 6.5, 2.8 Hz, H-8), 9.92 (m, 1H, H-11), 3.79 (m, 1H, H-11), 3.79 (m, 1H, H-11), 3.79 (m, 1H, H-13), 3.68 (br t, 2H, J = 6.5 Hz, H-14), 2.38 (m, 2H, H-2, H-6), 2.13 (br dd, 1H, J = 12.1, 2.8 Hz, H-13), 1.92–1.68 (m, 4H, H-7, Ha-9, Ha-12), 1.67–1.50 (m, 3H, H-1, Ha-10, Hb-12), 1.41 (m, 1H, Hb-10), 1.31 (m, 1H, H-9), 1.18 (d, 3H, J = 7.4 Hz, 6-Me), 0.89 (d, 3H, J = 6.9 Hz, 2-Me); 13C NMR (125 MHz, CD3OD) δ 190.0 (C-15), 134.1 (C-4), 133.2 (C-4a), 130.6 (C-3) 130.0 (C-5), 71.7 (C-8), 69.2 (C-11), 65. 9 (C-13), 60.1 (C-14), 45.4 (C-7), 41.0 (C-10), 39.8 (C-8a), 37.6 (C-1), 37.1 (C-12), 35.6 (C-9), 32.1, 29.1 (C-2, C-6), 23.65 (6-Me), 14.3 (2-Me).
tert-Butyldimethylsilyloxymonacolin J 10
tert-Butyldimethylsilyloxymonacolin J 10 was prepared according to the procedure of Willard and Smith.12 100 mg (0.312 mmol) monacolin J and 106.2 mg (1.56 mmol, 5 eq.) imidazole were placed in a 10 mL round bottomed flask and dissolved in 4 mL dimethylformamide (DMF). To this was added 117 mg (0.78 mmol, 2.5 eq.) tert-butyldimethylsilyl chloride as a solution in 1mL DMF. The reaction was stirred at room temperature for 18 hours, then diluted with 30 mL CH2Cl2 and extracted with 0.2 M HCl (1 × 10 mL). The organic layer was washed with H2O (3 × 10 mL), dried (Na2SO4), filtered and evaporated to give 197.5 mg crude residue. The product was purified by flash column chromatography (20% EtOAc in hexanes) to give 100 mg (74% yield) tert-butyldimethylsilyloxymonacolin J 10.[α]25D = −10.1 (c 1.35, CH2Cl2); FTIR (cast) 3446 (br), 2954 (s), 1734 (s), 1254 (s), 1082 (s), 735(s) cm−1; HREIMS [M]+ 434.28466 (434.28525 calcd. for C25H42O4Si), 434.3 (7%), 359.2 (17%), 284.2 (24%), 159.1 (100%), 101.0 (59%); 1H NMR (600 MHz, CDCl3) δ 5.95 (d, 1H, J = 9.5 Hz), 5.77 (dd, 1H, J = 6, 9.5 Hz), 5.52 (s, 1H), 4.65 (m, 1H), 4.27 (q, 1H, J = 3.5 Hz), 4.21 (s, 1H), 2.58 (dd, 1H, J = 17.5, 4 Hz), 2.53 (ddd, 1H, J = 17.5, 3, 1.5 Hz), 2.42 (m, 1H), 2.35 (s, 1H, J = 6.5), 2.14 (dd, 1H, J = 12, 2.5 Hz), 1.91–1.72 (m, 6H), 1.69 (ddd, 1H, J = 15, 3 Hz), 1.45 (m, 2H), 1.17 (d, 3H, J = 7.5 Hz), 0.88 (d, 3H, J = 7 Hz), 0.86 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 170.5, 133.7, 131.3, 130.0, 128.4, 76.5, 65.3, 63.5, 39.3, 38.8, 36.9, 36.5, 35.8, 33.0, 30.8, 27.4, 25.7, 24.3, 23.8, 18.0, 14.0, −4.8, −4.9.
4a,5-Dihydro(tert-butyldimethylsilyloxy)monacolin J 11
The iridium complex Ir(cod)py(Pcy3)PF6 was synthesized according to the procedure of Stork and Kahne.22 The selective reduction of tert-butyldimethylsilyloxymonacolin J 10 was achieved by modification of the procedure of DeCamp et al.11 To a solution of tert-butyldimethylsilyloxymonacolin J 10 (0.103 g, 0.24 mmol) in CH2Cl2 (1.7 mL) was added MeOH (0.1 mL) and the Ir complex (2 mg, 2.5 µmol). The mixture was hydrogenated for 10 min. Analytical TLC displayed the presence of starting material, therefore more Ir complex (2 mg, 2.5 µmol) was added and the mixture hydrogenated for an additional 10 min. This process was repeated until no starting material remained by TLC (a total of 8 mg Ir complex was added). The mixture was passed through a pad of Fluorosil to remove the catalyst and the solvent was evaporated under reduced pressure to give 4a,5-dihydro(tert-butyldimethylsilyloxy)monacolin J 11 (105 mg, quant.) as a mixture (86 ∶ 14) of the desired olefin and over-reduced tetrahydro material. The tetrahydro product was removed by flash column chromatography (50% EtOAc in hexanes) to give pure 4a,5-dihydro(tert-butyldimethylsilyloxymonacolin J 11.[α]25D = +76 (c 1.20, CH2Cl2); FTIR (cast) 3463 (br), 1733 (s), 1253 (s) cm−1; HREIMS [M]+ 436.30047 (436.30090 calcd. for C25H44O4Si) 436 (1%), 418 (6%), 361 (28%), 343 (8%) and 75 (100%); 1H NMR (300 MHz, CDCl3) δ 5.61 (ddd, 1H, J = 9.7, 4.9, 2.6 Hz), 5.36 (d, 1H, J = 9.8 Hz), 4.66 (m, 1H,), 4.27 (m, 1H), 4.16 (m, 1H), 2.59 (dd, 1H, J = 17.5, 4.3 Hz), 2.56 (ddd, 1H, J = 17.5, 3.5, 1.4 Hz), 2.52–1.01 (m, 20H), 1.19 (d, 1H, J = 7.4 Hz), 0.87 (m, 9H), 0.81 (d, 3H, J = 7.1 Hz), 0.06 (3 H, s) and 0.05 (3 H, s); 13C NMR (125 MHz, CDCl3) δ 174.0, 170.3, 132.6, 131.0, 76.3, 69.8, 63.6, 41.8, 41.7, 39.3, 38.6, 37.6, 36.8, 35.6, 33.2, 31.3, 30.9, 26.7, 26.7, 25.7, 23.1, 21.0, 16.5, 14.9, 11.7, −4.8, −4.9.
4a,5-Dihydromonacolin J 12
A sample of 30.0 mg (0.071 mmol) 4a,5-dihydro(tert-butyldimethylsiloxy)monacolin J 11 was dissolved in 4 mL THF in a 10 mL round bottomed flask and cooled to 0 °C. 20 µL (0.355 mmol, 5 eq.) glacial acetic acid was added, followed 10 min later by 100 µL of a 1 M solution of tetrabutylammonium flouride (TBAF) in THF (0.100 mmol, 1.5 eq.). The reaction was stirred for 2 h at 0 °C, then warmed to room temp. and stirred for an additional 2 h. Analytical TLC displayed mostly starting material so an additional 100 µL of 1 M TBAF in THF was added and the reaction stirred at room temp. overnight. The solvent was evaporated and the residue was dissolved in 20 mL CH2Cl2 and extracted with 0.4 M CaCl2 (1 × 10 mL), washed with H2O (3 × 10 mL), dried (Na2SO4), filtered and evaporated to give 20.0 mg crude product. 4a,5-Dihydromonacolin J 12 was purified by flash column chromatography over silica gel (1–5% i-PrOH in CH2Cl2) to yield 13.6 mg (60% yield) pure product.[α]25D = +137.4 (c 0.38, MeOH); FTIR (cast), 3361 (br), 2906 (s), 1703 (s), 1259 (s), 1039 (s) cm−1; HREIMS [M]+ 322.21394 (322.2144 calcd. for C19H30O4) 322.2 (1%), 286.2 (10%), 200.1 (18%), 159.1 (79%), 105.1 (100%); 1H NMR (600 MHz, CDCl3) δ 5.60 (ddd, 1H, J = 9.7, 4.8, 2.6 Hz), 5.35 (d, 1H, J = 10.1 Hz), 4.68 (m, 1H), 4.36 (quintet, 1H, J = 3.7 Hz), 4.14 (d, 1H, J = 2.8 Hz), 2.71 (dd, 1H, J = 18.0, 5.1 Hz), 2.60 (ddd, 1H, J = 18.0, 3.8, 1.8 Hz), 2.01–1.90 (m, 3H), 1.82–1.20 (m, 16 H), 1.18 (d, 3H, J = 7.5 Hz), 0.83 (d, 3H, J = 7.0 Hz); 13C NMR (150 MHz, CDCl3) δ 170.4, 132.3, 131.5, 76.1, 67.1, 62.8, 42.7, 39.3, 39.0, 38.6, 37.0, 36.3, 32.8, 31.4, 29.7, 27.0, 23.0, 21.6, 15.0.
4a,5-Dihydro(tert-butyldimethylsilyloxy)lovastatin 13
A sample of 14.0 mg (0.032 mmol) 4a,5-dihydro(tert-butyldimethylsilyloxy)monacolin J 11 and 5 mg (0.041 mmol) 4-dimethylaminopyridine were placed in a 1 mL React-vial and dissolved in 0.5 mL pyridine. To this mixture was added 100 µL (93.4 mg, 0.50 mmol) (S)-2-methylbutyric anhydride and the reaction was stirred at room temperature for 72 hours. The reaction was diluted with 30 mL CH2Cl2 and washed with 1 M HCl (1 × 10 mL). The organic layer was washed with H2O (3 × 10 mL), dried (Na2SO4), filtered and evaporated to give ca. 50 mg of crude residue which was purified by flash column chromatography to give 11.9 mg (72% yield) of pure 4a,5-dihydro(tert-butyldimethylsilyloxy)lovastatin 13.[α]25D = +73 (c 0.20, CHCl3); FTIR (cast) 2295 (s), 1727 (s), 1253 (s), 1079 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 5.62 (ddd, 1H, J = 9.5, 4.5, 2.5 Hz), 5.36 (d, 1H, J = 9.5 Hz), 5.15 (d, 1H, J = 2.5 Hz), 4.55 (m, 1H), 4.25 (quintet, 1H, J = 3.5 Hz), 2.57 (dd, 1H, J = 17, 4.5 Hz), 2.52 (dd, 1H, J = 17, 1 Hz), 2.45 (m, 1H), 2.41 (m, 1H), 2.32 (q, 1H, J = 7 Hz), 2.27 (m, 1H), 2.02 (m, 1H), 1.89 (d, 1H, J = 15 Hz), 1.83–1.80 (m, 2H), 1.72–1.44 (m, 5H), 1.32–1.22 (m, 2H), 1.17 (d, 2H, J = 7 Hz), 1.10 (d, 3H, J = 7 Hz), 1.07 (d, 3H, J = 7.5 Hz), 0.93 (t, 3H, J = 7.5 Hz), 0.86 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 175.0, 170.1, 132.6, 131.0, 76.3, 69.8, 63.6, 41.8, 41.7, 39.3, 38.6, 37.6, 36.8, 35.6, 33.2, 31.3, 30.9, 26.8, 26.7, 26.6, 25.6, 23.1, 21.0, 17.9, 16.5, 16.4, 14.9, 11.7, −4.8, −4.9.
4a,5-Dihydrolovastatin 14
7.7 mg (0.0148 mmol) of 13 was dissolved in 1 mL of 10% HCl in MeOH, and stirred at room temp. for 1 hour. The reaction was diluted with 30 mL CH2Cl2 and extracted with NaHCO3 (3 × 10 mL). The organic layer was washed with H2O (3 × 10 mL), dried and evaporated to give 5.0 mg of 14.1H NMR (300 MHz, CDCl3) δ 5.62 (m, 1H), 5.35 (d, 1H, J = 9.6 Hz), 5.17 (m, 1H), 4.55 (m, 1H), 4.35 (m, 1H), 2.74 (d, 1H, J = 5.0 Hz), 2.68 (d, 1H, J = 5.0 Hz), 2.61 (dd, 1H, J = 3.8, 1.5 Hz), 2.52 (dd, 1H, J = 4.2, 1.5 Hz), 2.43 (d, 1H, J = 6.0 Hz), 2.4–2.2 (m, 2H), 2.05–1.75 (m, 6H), 1.70–1.40 (m, 10H), 1.15 (d, 3H, J = 7.0 Hz), 1.08 (d, 3H, J = 7.4 Hz), 0.88 (t, 3H, J = 8 Hz), 0.82 (d, 3H, J = 7.0 Hz).
6-Desmethylmonacolin J 15
A solution of compactin 3 (55 mg, 0.13 mmol) and LiOH (60 mg, 1.3 mmol) in H2O (15 mL) was heated at reflux for one day. The reaction mixture was cooled in an ice bath, acidified with 2 M HCl (pH < 1) and extracted with EtOAc (5 × 30 mL) and CHCl3 (4 × 20 mL). The combined organic extracts were dried (Na2SO4), filtered and evaporated. The resulting residue was taken up in toluene (60 mL) and heated at reflux in a Sohxlet containing CaH2 for one hour. After cooling the toluene was evaporated and the residue fractionated by flash column chromatography (EtOAc, 100%) to yield 30 mg of 15 as a colorless oil (75%).[α]25D = +78 (c 0.15, CHCl3); FTIR (cast) 3407 (br s), 2928 (s), 1710 (s), 1256 (s), 1075 (s), 754 (s); HREIMS [M]+ 306.18245 (306.18311 calcd. for C18H26O4); 1H NMR (500 MHz, CDCl3) δ 5.92 (d, 1H, J = 9.6 Hz), 5.71 (dd, 1H, J = 9.6, 6.0 Hz), 5.52 (br d, 1H, J = 2.1 Hz), 4.69 (m, 1H), 4.52 (m, 1H), 4.21 (br s, 1H), 2.67 (dd, 1H, J = 17.7, 5.0 Hz), 2.59 (ddd, 1H, J = 17.7, 3.7, 1.7 Hz), 2.31 (m, 2H), 2.14 (m, 2H), 1.96 (m, 2H), 1.82–1.62 (m, 5H), 1.53–1.39 (m, 2H), 0.88 (d, 3H, J = 7.0 Hz); 13C NMR (75 MHZ, CDCl3) δ 170.8, 133.3, 133.0, 128.3, 123.6, 76.2, 64.4, 62.6, 38.8, 38.5, 36.4, 36.1, 32.6, 30.8, 29.1, 23.8, 20.3, 13.9.
α,β-Dehydro-4a,5-dihydromonacolin L 16
A sample of 18.4 mg (0.060 mmol) 4a,5-dihydromonacolin L 5 was dissolved in 0.5 mL CH2Cl2. To this solution was added 7.1 µL (10.3 mg, 0.90 mmol, 1.5 eq.), methanesulfonyl chloride and 2.5 µL (18.2 mg, 0.18 mmol, 3 eq.) triethylamine and the reaction was stirred at room temp. 1 hour. The reaction mixture was diluted with 30 mL CH2Cl2 and extracted with 0.2 M HCl (1 × 10 mL) and satd. NaHCO3 (1 × 10 mL). The organic layer was washed with H2O (3 × 10 mL), dried (Na2SO4), filtered and evaporated to give 21.0 mg crude product. The product was fractionated by preparative TLC (hexane–EtOAc, 1 ∶ 1) to give 10 mg (60%) pure α,β-dehydro-4a,5-dihydromonacolin L 16.[α]25D = +8.6 (c 1.00, CH2Cl2); FTIR (cast) 2915 (s), 1717 (s), 1389 (s), 1248 (s), 818 (s) cm−1; HREIMS [M]+ 288.20866 (288.20892 calcd. for C19H28O2) (19%), 273.2 (17%), 228.2 (22%), 176.1 (85%), 161.1 (71%), 105.1 (100%); 1H NMR (600 MHz, CDCl3) δ 6.85 (ddd, 1H, J = 9.5, 9.5, 4.4 Hz), 6.00 (d, 1H, J = 9.8 Hz), 5.57 (ddd, 1H, J = 9.7, 4.7, 2.8 Hz), 5.28 (d, 1H, J = 9.8 Hz), 4.41 (dddd, 1H, J = 12.3, 12.3, 7.6, 4.7 Hz), 2.33 (m, 2H), 2.21 (m, 1H), 2.11 (m, 1H), 1.91–1.87 (m, 2H), 1.62–1.41 (m, 8H), 1.34–1.22 (m, 2H), 1.08 (m, 1H), 0.99 (dd, 1H, J = 10.3, 2.6 Hz), 0.96 (d, 3H, J = 7.3 Hz), 0.83 (d, 3H, J = 7.0); 13C NMR (150 MHz, CDCl3) δ 164.3, 144.8, 132.5, 131.6, 121.3, 78.0, 41.1, 39.6, 38.3, 38.2, 37.0, 32.0, 31.8, 31.5, 29.0, 27.1, 23.2, 17.7, 14.6.
α,β-Dehydrolovastatin 17
33.0 mg (0.082 mmol) lovastatin 1 was placed in a 25 mL round bottomed flask and dissolved in 3 mL CH2Cl2. To this solution was added 34.7 µL (25 mg, 0.249 mmol, 3 eq.) triethylamine, followed by 9.6 µL (14 mg, 0.123 mmol, 1.5 eq.) methanesulfonyl chloride and the reaction was stirred at room temp. for one hour. The reaction was diluted with CH2Cl2 and extracted with 0.1 M HCl (1 × 10 mL) and satd. NaHCO3 (1 × 10mL), washed with H2O (3 × 10 mL), dried (Na2SO4) and filtered to give 40.4 mg of crude residue which was fractionated by flash column chromatography to give 33.7 mg pure α,β-dehydrolovastatin 17.[α]25D = +136.4 (c 3.30, CH2Cl2); FTIR (cast) 2963 (s), 1723 (s), 1383 (s), 1247 (s), 818 (s) cm−1; HREIMS [M]+ 386.24530 (386.24570 calcd. for C24H34O4) 386.2 (2%), 284.2 (17%), 198.1 (64%), 159.1 (100%); 1H NMR (600 MHz, CDCl3) δ 6.83 (ddd, 1H, J = 9.8, 6.1, 2.4 Hz), 5.98 (ddd, 1H, J = 9.7, 2.6, 0.9 Hz), 5.97 (d, 1H, J = 10.5 Hz), 5.76 (dd, 1H, J = 9.5, 6.1 Hz), 5.50 (m, 1H), 5.36 (d, 1H, J = 3.2 Hz), 4.31 (dddd, 1H, J = 11.5, 11.5, 5.3, 3.5 Hz), 2.42 (m, 1H), 2.37–2.30 (m, 3H), 2.25–2.19 (m, 2H), 1.96–1.87 (m, 3H), 1.69–1.59 (m, 3H), 1.51–1.28 (m, 4H), 1.08 (d, 3H, J = 7.0 Hz), 1.05 (d, 3H, J = 7.4 Hz), 0.87 (d, 3H, J = 7.0 Hz), 0.85 (t, 3H, J = 7.5 Hz); 13C NMR (125 MHz, CDCl3) δ 176.6, 164.2, 144.7, 133.0, 131.6, 129.7, 128.3, 121.5, 78.5, 67.7, 41.4, 37.3, 36.6, 32.7, 32.4, 30.7, 29.5, 27.4, 26.8, 24.2, 22.8, 16.2, 13.8, 11.7.
Heptaketide 19
Heptaketide 19 was isolated from cultures of A. nidulans lovb + lovC as follows. 4 L of A. nidulans lovB + lovC production cultures, fermented as described above for the isolation of 5, were extracted with CH2Cl2 (2 × 1.5 L) WITHOUT acidification of the culture broth. The organic layer was dried (Na2SO4), filtered and evaporated to give ca. 50 mg of crude residue. Purification by flash column chromatography (EtOAc 100%) gave 6.5 mg of pure 19.[α]20D +120.3 (c 0.10, CHCl3); FTIR (cast) 3020, 2952, 2915, 2860, 1696, 1445, 1411, 1377, 1310, 1297, 1267, 1238, 1211, 720 cm−1; HREIMS [M]+ 236.1777 (236.1776 calcd. for C15H24O2); 1H NMR (500 MHz, CDCl3) δ 5.57 (ddd, 1H, J = 10.0, 5.0, 3.0 Hz), 5.28 (d, 1H, J = 10.0 Hz), 2.42 (ddd, 1H, J = 15.5, 10.0, 5.0 Hz), 2.26–2.19 (m, 2H), 2.00 (m, 1H), 1.96–1.97 (m, 2H), 1.60–1.44 (m, 5H), 1.36 (m, 1H), 1.25 (dt, 1H, J = 13.0, 4.5 Hz), 1.10 (dq, 1H, J = 12.0, 4.0 Hz), 1.02–0.94 (m, 1H), 0.96 (d, 3H, J = 7.5 Hz), 0.82 (d, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 179.9, 132.3, 131.5, 41.0, 39.9, 38.9, 37.3, 32.3, 32.0, 31.9, 27.5, 23.8, 23.6, 18.2, 15.0.
β-Oxidation product 20
β-Oxidation product 20, was isolated from a biotransformation experiment as follows. 1 L of A. nidulans growth media was inoculated with 10 mL of a concentrated suspension of Aspergillus terreus lovA spores and incubated at 30 °C and 200 rpm for 2 days. To the growing culture was added a solution of unlabelled 4a,5-dihydromonaoclin L (30 mg) and 52.5 nCi (ca. 0.5 mg) of 14C-4a,5-dihydromoancolin L in 6 mL EtOH. 0.5 mL of this dihydromonacolin L solution was added to the cultures three times a day until completely fed. After seven days total growth the mycelia were removed by vacuum filtration, the broth acidified with 2 M HCl (pH < 2) and extracted with CH2Cl2 (1 × 1 L, 1 × 0.5 L). The combined organic layers were dried (Na2SO4), filtered and evaporated to give 207.2 mg crude extract. The crude extract was fractionated by preparative TLC to give a fraction (26.3 mg) containing the majority of the radioactivity. A portion of this radioactive fraction was treated with CH2N2 (Et2O solution), and fractionated by preparative TLC to isolate the β-oxidation product as the major radioactive component (1.7 mg, 3.4 nCi).This same product could be isolated from the CH2Cl2 extract of A. nidulans lovB + lovC production cultures grown as described above. A portion (23.2 mg) of the crude CH2Cl2 extract was treated with CH2N2 (Et2O solution) to give 26.1 mg crude methylated product which was fractionated by HPLC to give 2.2 mg pure β-oxidation product 20.
FTIR (cast) 3297 (br), 3008 (s), 1737 (s), 1437 (m), 1223 (s), 1170 (s) cm−1; HREIMS 248.17729 (248.1763 calcd. for C16H24O2) [M − H2O]+, 217.1 (18%), 192.1 (14%), 161.1 (100%), 119.1 (52%), 105.1 (78%); 1H NMR (600 MHz, CDCl3) δ 5.58 (ddd, 1H, J = 9.5, 4.5, 2.5 Hz, H-3), 5.30 (d, 1H, J = 9.5 Hz, H-4), 3.65, (s, 3H, OCH3), 2.37 (ddd, 1H, J = 15, 10, 5 Hz, H-10a/b), 2.24 (m, 1H, H-2), 2.18 (ddd, 1H, J = 15, 9.5, 6.5 Hz, H-10a/b), 1.89 (m, 2H, H-9), 1.80, 1.78 (d, 1H, J = 2.5, H-4a), 1.69, 1.67, 1.47, 1.23 (s, 3H, 6-CH3), 1.02, 0.83 (d, 3H, J = 7 Hz, 2-CH3); 13C NMR (125 MHz, CDCl3) δ 171.3, 132.9, 130.3, 71.2, 51.6, 47.1, 41.1, 41.0, 40.8, 39.2, 32.0, 31.9, 27.0, 26.1, 24.3, 14.8.
4a-5-Dihydromonacolin L epoxide 21a, 21b
A sample of 50.0 mg (0.163 mmol) dihydromonacolin L 5 was placed in a 10 ml round bottomed flask and dissolved in 4 mL of CHCl3 under Ar(g). To this solution was added dropwise 50.1 mg (0.244 mmol, 1.5 eq.) of an 85% preparation of m-chloroperbenzoic acid in benzoic acid dissolved in 1 mL of CHCl3. The reaction was stirred under Ar(g) at room temperature for 90 min. The reaction was diluted with 25 mL of CH2Cl2 and extracted with 5% NaHSO3 (1 × 10 mL) and satd. NaHCO3 (1 × 10 mL), and washed with H2O (3 × 10 mL). The organic layer was dried (Na2SO4), filtered and evaporated to give 54.1 mg crude product which was fractionated by flash column chromatography (40% EtOAc in hexanes) to yield 11.3 mg of 21a and 11.2 mg of 21b (44% total yield of pure 21a and 21b).For 4a,5-dihydromonacolin L α-epoxide 21a: [α]25D = +90 (c 0.95, CHCl3); FTIR (cast) 3392 (br), 2915 (s), 2876 (s), 1709 (s), 1257 (s), 1044 (s), 842 (s); HREIMS [M]+ 322.21451 (322.21442 calcd. for C19H30O4) 322 (5%), 307 (5%), 191 (48%), 179 (100%), 95 (65%); 1H NMR (500 MHz, CDCl3) δ 4.63 (dddd, 1H, J = 11.0, 11.0, 7.6, 3.8 Hz), 4.36 (m, 1H), 2.98 (d, 1H, J = 2.4 Hz), 2.72 (d, 1H, J = 3.7 Hz), 2.69 (d, 1H, J = 5.0 Hz), 2.61 (dd, 1H, J = 3.6, 1.7 Hz), 2.56 (dd, 1H, J = 3.7, 1.7 Hz), 2.30 (m, 1H), 2.05 (m, 2H), 1.93 (m, 2H), 1.8–1.6 (m, 2H), 1.6–1.3 (m, 10H), 0.95 (d, 3H, J = 7.1 Hz), 0.89 (d, 3H, J = 7.2 Hz); 13C NMR (125 MHz, CDCl3) δ 170.4, 76.0, 62.8, 58.8, 57.5, 38.6, 38.8, 37.4, 36.3, 36.2, 36.1, 33.0, 32.0, 30.3, 27.6, 23.9, 23.7, 18.1, 10.2.
For 4a,5-Dihydromonacolin L β-epoxide 21b: 1H MNR (600 MHz, CDCl3) δ 4.65 (dddd, 1H, J = 11.5, 11.5, 7.3, 4.0 Hz), 4.37 (m, 1H), 3.17, (dd, 1H, J = 3.9, 5.5 Hz), 2.91 (d, 1H, J = 3.9 Hz), 2.71 (dd, 1H, J = 17.6, 5.0 Hz), 2.60 (ddd, 1H, J = 17.6, 3.7, 1.7 Hz), 2.15 (m, 1H), 2.05 (m, 1H), 1.90 (m, 2H), 1.75 (m, 2H), 1.65–1.40 (m, 8H), 1.20 (m, 2H), 0.97 (d, 3H, J = 7.2 Hz), 0.92 (d, 3H, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3) δ 170.2, 75.8, 62.8, 58.2, 57.0, 41.5, 38.6, 37.6, 35.9, 35.6, 34.6, 33.2, 31.7, 28.9, 26.9, 23.2, 22.7, 18.0, 9.5.
Crystal data for 4a,5-dihydromonacolin L α-epoxide 21a
Data were acquired on a Bruker P4/RA/SMART 1000 CCD diffractometer. All intensity measurements were performed using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). 4a,5-Dihydromonacolin L α-epoxide 20a (C19H30O4) was obtained as white crystals, space group P212121 (No. 19), a = 5.5318 (11), b = 9.7123 (19), c = 32.163 (7) Å, V = 1728.0 (6) Å3, Z = 4, T = −80 °C, ρcalcd = 1.239 g cm−3, μ = 0.085 mm−1. 9765 reflections measured, 3482 unique which were used in all least squares calculations, R1(F) = 0.0538 (for 2478 reflections with Fo2 ≥ 2σ(Fo2)), wR2(F2) = 0.1044 for all unique reflections. The absolute stereochemistry of 21a cannot be directly determined from the data, but it is correct as shown based on conversion of 5 to 1 and to 21a. CCDC reference number 191454. See http://www.rsc.org/suppdata/ob/b2/b207721c/ for crystallographic files in CIF or other electronic format.β-Oxidation product 22
β-Oxidation metabolite 22 was isolated as follows. A. terreus lovA spores were used to inoculate flasks (2 × 100 mL) of A. nidulans growth media after 4 days the mycelia were harvested by filtration and transferred to A. nidulans production media (2 × 100 mL). After 48 hours, a solution of monacolin J in EtOH (30 mg in 3.0 ml EtOH) was administered in intervals (0.5 mL of the above solution every 12 hours until completely fed). After seven days total growth in the production media, the culture broth was harvested by vacuum filtration. The combined broth was acidified and extracted with EtOAc (2 × 100 ml). The combined organic extracts were dried, filtered and evaporated to give 42.0 mg crude extract. The crude extract was fractionated by preparative TLC (EtOAc–hexane 4 ∶ 1) to give a crude sample of 22. This was further purified after treatment with CH2N2 (Et2O solution) to give 0.1 mg of the methyl ester of 22.1H NMR (600 MHz, CDCl3) δ 5.95 (d, 1H, J = 10.0 Hz), 5.75 (dd, 1H, J = 9.1, 6.2 Hz), 5.55 (m, 1H), 4.28 (m, 1H), 3.68 (s, 3H), 2.50–2.20 (m, 6H), 1.80–1.20 (m, 10H), 1.16 (d, 3H, J = 7.4 Hz), 0.88 (d, 3H, J = 7.0 Hz).
Acknowledgements
The authors thank Dr Robert McDonald (University of Alberta) for crystallographic analyses, Dr Joanna Harris (University of Alberta) for performing the selective reduction of 10 to 11 and Karr-Hong Lee (University of Alberta) for isolating 19. This work was supported by the Natural Sciences and Engineering Research Council of Canada, the Canada Research Chair in Bioorganic and Medicinal Chemistry, the Canada Foundation for Innovation, and the Alberta Science and Research Authority.References
- (a) A. W. Alberts, J. Chen, G. Kuron, V. Hunt, J. Huff, C. Hoffman, J. Rothrock, M. Lopez, H. Joshua, E. Harris, A. Patchett, R. Monaghan, S. Currie, E. Stapley, G. Albers-Schonberg, O. Hensens, J. Hirshfield, K. Hoogsteen, J. Liesch and J. Springer, Proc. Natl. Acad. Sci. USA, 1980, 77, 3957–3961 CAS; (b) A. Endo, J. Antibiot., 1979, 32, 852–854 Search PubMed.
- (a) W. F. Hoffman, A. W. Alberts, P. S. Anderson, J. S. Chen, R. L. Smith and A. K. Willard, J. Med. Chem., 1986, 29, 849–852 CrossRef CAS; V. F. Mauro and J. L. Macdonald, DICP Ann. Pharmacother., 1991, 25, 257–264 Search PubMed; (b) for an overview of statins see M. Igel, T. Sudhop and K. vonBergmann, Eur. J. Clin. Pharmacol., 2001, 57, 357–364 Search PubMed.
- (a) Y. Tsujita, M. Kuroda, Y. Shimada, K. Tanzawa, M. Arai, I. Kaneko, M. Tanaka, H. Masuda, C. Tarumi, Y. Watanabe and S. Fujii, Biochimica. Biophys. Acta, 1986, 877, 50–60 Search PubMed; (b) N. Serizawa, K. Nakagawa, K. Hamano, Y. Tsujita, A. Terahara and H. Kuwano, J. Antibiot., 1983, 36, 604–607 Search PubMed; (c) for a review of pravistatin pharmacology see D. Mctavish and E. M. Sorkin, Drugs, 1991, 42, 65–89 Search PubMed.
- A. Sutherland, K. Auclair and J. C. Vederas, Curr. Opin. Drug Discovery Dev., 2001, 4, 229–236 Search PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J.C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- K. Auclair, A. Sutherland, J. Kennedy, D. J. Witter, J. P. Van den Heever, C. R. Hutchinson and J. C. Vederas, J. Am. Chem. Soc., 2000, 122, 11519–1120 CrossRef CAS.
- (a) For a review see C. R. Hutchinson, J. Kennedy, C. Park, S. Kendrew, K. Auclair and J. C. Vederas, Antonie van Leeuwenhoek, 2000, 78, 287–295 Search PubMed; (b) see also A. Endo, Y. Negishi, T. Iwashita, K. Mizukawa and M. Hirama, J. Antibiot., 1985, 38, 444–448 Search PubMed; (c) A. Endo, K. Hasumi and S. Negishi, J. Antibiot., 1985, 38, 420–422 Search PubMed.
- (a) K. Wagschal, Y. Yoshizawa, D. J. Witter, Y. Q. Liu and J. C. Vederas, J. Chem. Soc., Perkin Trans. 1, 1996, 2357–2363 RSC; (b) Y. Abe, T. Suzuki, C. Ono, K. Iwamoto, M. Hosbuchi and H. Yoshikawa, Mol. Genet. Genomics, 2002, 267, 636–646 Search PubMed.
- K. Auclair, J. Kennedy, C. R. Hutchinson and J. C. Vederas, Bioorg. Med. Chem. Lett., 2001, 11, 1527–1531 CrossRef CAS.
- L. R. Treiber, R. A. Reamer, C. S. Rooney and H. G. Ramjit, J. Antibiot., 1989, 42, 30–36 Search PubMed.
- A. E. Decamp, T. R. Verhoeven and I. Shinkai, J. Org. Chem., 1989, 54, 3207–3208 CrossRef CAS.
- A. K. Willard and R. L. Smith, J. Labelled Compd. Radiopharm., 1982, 19, 337–344 CAS.
- (a) For other examples of the synthesis of dihydrolovastatin see J. Schnaubelt, B. Frey and H. U. Reissing, Helv. Chim. Acta, 1999, 82, 666–676 Search PubMed; (b) C. M. Blackwell, A. H. Davidson, S. B. Launchbury, C. N. Lewis, E. M. Morrice, M. M. Reeve, J. A. R. Roffey, A. S. Tipping and R. S. Todd, J. Org. Chem., 1992, 57, 5596–5606 CrossRef CAS; (c) S. Hanessian, P. J. Roy, M. Petrini, P. J. Hodges, R. Di Fabio and G. Carganico, J. Org. Chem., 1990, 55, 5766–5777 CrossRef CAS.
- G. Albers-Schonberg, H. Joshua, M. B. Lopez, O. D. Hensens, J. P. Springer, J. Chen, S. Ostrove, C. H. Hoffman, A. W. Alberts and A. A. Patchett, J. Antibiot., 1981, 34, 507–512 Search PubMed.
- W. Bartmann, G. Beck, E. Granzer, H. Jendralla, B. V. Kerekjarto and G. Wess, Tetrahhedron Lett., 1986, 27, 4709–4712 Search PubMed.
- (a) M. F. Baltazar, F. M. Dickinson and C. Ratledge, Microbiology (Reading, UK), 1999, 145, 271–278 Search PubMed; (b) S. Valenciano, J. R. DeLucas, A. Pedregosa, I. F. Monistrol and F. Laborda, Arch. Microbiol., 1996, 166, 336–341 CrossRef CAS.
- W. Jacobsen, G. Kirchner, K. Hallensleben, L. Mancinelli, M. Deters, I. Hackbarth, L. Z. Benet, K. Sewing and U. Christians, Drug Metab. Dispos., 1999, 27, 173–179 Search PubMed.
- R. B. Silverman, The Organic Chemistry of Enzyme Catalyzed Reactions, Academic Press, San Diego, 2000, p. 204 Search PubMed.
- T. Nakamura, D. Komagata, S. Murakawa, K. Sakai and A. Endo, J. Antibiot., 1990, 43, 1597–1600 Search PubMed.
- P. R. Ortiz de Montellano and J. J. De Voss, Nat. Prod. Rep., 2002, 19, 477–493 RSC.
- A. Endo and K. Hasumi, J. Antibiot., 1985, 38, 321–327 Search PubMed.
- G. Stork and D. E. Kahne, J. Am. Chem. Soc., 1983, 105, 1072–1073 CrossRef CAS.
Footnotes |
| † The gene cluster for compactin 3 has recently been characterised (see ref. 8b). |
| ‡ For a detailed description of the construction of A. terreus disruptants and A. nidulans transformants see the supporting information for ref. 5. |
| This journal is © The Royal Society of Chemistry 2003 |