Reactions of 5-mercaptoazoles and pyridine-2-thiones with acetylenic esters. Selectivity of the formation of novel fused thiazin-4-ones and thiazolidin-4-ones
Vasiliy A.
Bakulev
*a,
Vera S.
Berseneva
a,
Natalia P.
Belskaia
a,
Yury Yu.
Morzherin
a,
Andreiy
Zaitsev
a,
Wim
Dehaen
*b,
Ingrid
Luyten
b and
Suzanne
Toppet
b
aDepartment of Technology and Organic Synthesis, The Urals State Technical University, 620002, Ekaterinburg, Russia. E-mail: primavera@ural.org; Fax: +7 3432 420086
bDepartment of Chemistry, K. U. Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: wim.dehaen@chem.kuleuven.ac.be; Fax: +32 16 32 79 90
First published on 5th December 2002
Abstract
A systematic study of the reactions of dimethyl acetylenedicarboxylate (DMAD) and methyl propynoate with 5-mercaptoazoles and pyridine-2-thiones has been carried out and as a result, a number of novel imidazo[1,5-b]thiazin-4-ones 6a,b, pyrazolo[1,5-b] thiazin-4-ones 15a–f, imidazo[1,5-b]thiazol-4-ones 7a,b and thiazolo[3,2-a]pyridines 21a–c have been prepared. The influence of the size of the ring of the starting “cyclic” thioamides on the size of the fused ring in the reaction products has been established. The preferred formation of a six-membered thiazine ring took place in the reactions of 5-mercaptoazoles. In contrast, the five membered thiazolidine ring is formed in reactions of pyridine-2-thiones. In both cases the product is a five-membered ring fused to a six-membered heterocycle.
Introduction
The reactions of thioureas, thiosemicarbazides and thioamides with electron poor acetylenes are known as convenient and effective methods to prepare thiazolidin-4-ones and thiazin-4-ones.1–12 The most probable mechanism of these reactions involves the addition of the sulfur atom of the thiocarbamoyl moiety onto the triple bond of the acetylenes, followed by cyclocondensation of the intermediate vinylthioimidates of type 2via elimination of a molecule of methanol. Reactions of thioamides of various natures with DMAD have been shown to occur via one of two possible mechanisms where generally the α-situated ester group participates in the cyclization leading to a five-membered thiazolidine ring 3.3,4,6,10–12 An alternative possibility for the cyclization of intermediate 2 (R2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) involves the reaction of the β-ester group to afford a thiazin-4-one ring.4,6,10
H) involves the reaction of the β-ester group to afford a thiazin-4-one ring.4,6,10
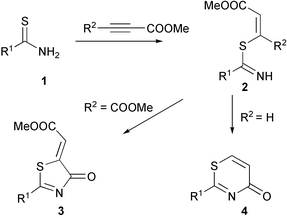
In previous work, we have shown that novel 2,5-dimethylenethiazolidin-4-one derivatives can be prepared by reaction of malonthioamide derivatives with DMAD11 and a new synthetic approach to conjugated bicyclic heterocycles has been elaborated based on a similar reaction of heterocyclic thioamides.12 Reactions of heterocyclic compounds where an endocyclic thioamide group is present with DMAD and methyl propynoate have only been reported once.4 In order to find a general synthetic method to thiazin-4-ones and thiazolin-4-ones fused to azole and azine rings we have studied the reactions of 5-mercaptoimidazoles, -pyrazoles, -1,2,3-triazoles and tetrahydropyridine-2-thiones with DMAD and methyl propynoate.
Results and discussion
Reaction of 5-mercaptoimidazole-4-carboxamide 5a with DMAD in principle could give both thiazine 6a and thiazolidine 7a rings. We have found that this reaction in methanol at room temperature results in the exclusive formation of fused thiazine 6a in 66% yield. However, a mixture of thiazine 6a and thiazolidine 7a in a ratio of 7 to 3 was obtained when sodium methoxide was used as a catalyst. It should be noted that compound 5a did not react with methyl propynoate in either circumstances.The respective thiazin-4-one and thiazolin-4-one structures of products 6a and 7a are confirmed by their 1H and 13C NMR spectral data (Experimental section, Table 1). The 1H NMR spectra for both products are very similar but all signals of thiazine 6a are shifted 0.14–0.18 ppm downfield in comparison with those of 7a. The signals in the 13C NMR spectra are also very similar for both isomeric products 6a and 7a. The 1H coupled 13C NMR spectra of 6a and 7a show doublet signals at 119.0 and 118.8 ppm, respectively, with similar 1JCH coupling constants of 175 Hz, which is typical for CC units. The final decision in favor of the thiazolidine structure for 7a and thiazine for 6a can be made after considering the magnitudes of the 13C–1H coupling constants in the 13C NMR spectra.2,11,12 The C6 and C4 signals of 7a are found in the coupled spectrum as doublets with 2JC6–H5 coupling constants of 1.4 Hz and 3JC4–H5 of 4.8 Hz. This shows the presence of an exocyclic double bond in the structure of thiazolidine 7a. In contrast to this, the coupled spectrum of 6a contains a doublet signal for C4 with a 2JC4–H5 coupling constant of 1.4 Hz, which confirms the structure of 6a as thiazin-4-ones.
| N | Chemical shifts (ppm), multiplicity | Coupling constants/Hz | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C2 | C4 | C5 | C6 | C7 | Others | J C5–H5 | J C4–H5 | J C8–H8 | J C6–H5 | |
| 6a | 124.2 d | 156.4 d | 118.8 d | 143.5 s | 162.5 s | 54.3 q (OCH3), 133.1 s (C9), 133.9 d (C8H), 162.0 s (C10) | 175.0 | <1.0 | 223.0 | |
| 6b | 127.6 d | 156.8 d | 119.0 d | 144.3 s | 162.2 s | 54.3 q (OCH3), 133.2 s (C9), 134.9 d (C8H), 163.5 s (C10) | 174.0 | 1.5 | 224.0 | |
| 7a | 124.2 d | 156.4 d | 118.8 d | 143.5 d | 162.0 s | 54.3 q (OCH3), 127.6 s (C9), 134.0 d (C8H), 186.6 s (C10) | 175.0 | 4.8 | 222.2 | 1.4 |
| 7b | 127.6 d | 158.2 d | 112.7 d | 144.3 s | 160.1 s | 54.3 q (OCH3), 127.6 s (C9), 132.7 d (C8), 187.3 s (C10) | 174.0 | 3.3 | 220.0 | |
| 15a | 124.1 | 154.5 | 122.4 | 141.3 | 162.0 | 11.7 (C14), 54.3 (OCH3), 54.7 (OCH3), 114.8 (C11), 124.1 (C12), 125.4 (C8), 135.6 (C13), 145.9 (C10), 152.8 (C9) | ||||
| 15b | 125.8 | 154.5 | 122.1 | 141.8 | 161.8 | 11.7 (C14), 21.0 (CH3), 54.3 (OCH3), 121.8 (C11), 130.1 (C12), 136.5 (C8), 141.8 (C13), 149.8 (C10), 153.1 (C9) | ||||
| 15c | 127.0 s | 155.3 br s | 122.7 d | 142.1 s | 162.7 s | 12.6 q (Me), 54.2 q (OCH3), 136.5 s, 154.1 s (Cpyrazole), 123.5 d, 130.5 d, 132.3 s, 152.6 s (Ar) | 173.6 | |||
| 15d | 125.9 s | 154.9 d | 122.6 d | 142.1 s | 161.9 s | 12.0 q (Me), 54.3 q (OCH3), 136.5 s, 154.6 s (Cpyrazole), 123.5 d, 129.4 d, 137.2 s, 150.6 s (Ar) | 174.1 | 0.6 | ||
| 15e | 127.2 | 154.6 | 122.9 | 141.8 | 161.9 | 11.8 (C14), 14.2 (CH3), 54.5 (OCH3), 61.2 (OCH2), 123.0 (C11), 130. (C12), 136.0 (C8), 141.6 (C13), 153.8 (C9), 153.8 (C10), 165.2 (COOEt) | ||||
| 15f | 126.4 d | 155.1 d | 118.8 d | 137.9 dd | 12.0 q (Me), 136.5 s, 153.5 s (Cpyrazole), 123.4 d, 129.5 d, 137.0 s, 150.8 s (Ar) | 173.1 | 10.6 | 2.3 | ||
| 19 | 122.6 | 154.6 | 121.8 | 141.2 | 162.2 | 11.8 (C14), 53.9 (OCH3), 119.2 (C8), 151.7 s (C9), 121.8 (C11), 131.4 (C12), 132.4 (C13), 149.8 (10) | ||||
| 21a | 137.7 | 166.1 | 113.7 | 143.1 | 166.2 | 52.3 (OCH3), 66.0 (C10), 109.7 (C8), 119.2 (CN), 127.2, 127.6, 128.6, 144.9 (Ar), 147.6 (C11), 164.5 (C12) | ||||
| 21b | 137.7 | 166.0 | 114.5 | 144.3 | 166.1 | 52.4 (OCH3), 34.6 (C9), 65.7 (C10), 109.6 (C8), 119.1 (CN), 147.9 (C11), 147.3, 125.2, 125.5 d, 127.0 (Cthienyl), 164.5 (C12) | ||||
| 21c | 142.0 d | 164.3 d | 115.8 d | 142.3 s | 165.9 q | 13.7 and 61.2 q (OCH2CH3), 52.5 q (OCH3), 35.1 d, 65.7 m, 106.7 d, 147.6 d (Cpyr), 118.7 s (CN), 147.2 m, 127.0 d, 125.2 d, 125.1 d (Cthienyl) | 172.8 | 5.6 | ||
In contrast to 5a, 5-mercapto-1,2,3-triazole-4-carboxamide does not react with DMAD and methyl propynoate. All our attempts to involve this compound in these reactions failed. We have shown that 5-mercapto substituted imidazole-4-thiocarboxamides can react with DMAD to form 2-imidazolylthiazolidines.12 Therefore, we expected from the reaction of thioamide 5b with DMAD the formation of the products of type 6–9 from the condensation with both the exocyclic and endocyclic thioamide groups.
We have found that reaction of 5b with DMAD in methanol, either with or without sodium methoxide, affords a mixture of thiazine 6b and thiazolidine 7b in a ratio of 9 to 1 in 50% yield. The 1H and 13C NMR spectra of products 6b, 7b are very similar to those of 7a and 7b (See Table 1). The downfield shift (0.5– 0.6 ppm) of the signals of C8–H of 6b, 7b in comparison with the conjugated imidazolylthiazolidines12 also confirmed the formation of fused imidazoles in this reaction. The 13C NMR spectra of 7a and 7b contain signals at 186.8 and 187.3 ppm corresponding to signals of the unchanged thioamide groups.
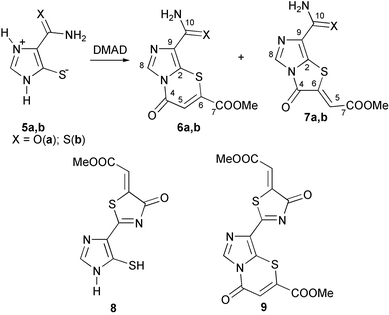
We have found that 5-mercapto-1,2,3-triazole-4-thiocarboxamides 10 are less active in the reaction with DMAD in comparison with 5-mercaptoimidazoles. Triazoles, that are unsubstituted at the ring nitrogen did not react either with DMAD or methyl propynoate. On the other hand 1-R-5-mercapto-1,2,3-triazoles reacted with both DMAD and methyl propynoate to form vinyl sulfide derivatives 11. The presence of two ester groups and a vinyl moiety in compounds 11 was confirmed by the presence of two signals in their 1H NMR spectra at 3.45–3.76 ppm, signals at 6.69 and 6.70 in spectra of 11a,b respectively and of doublet signals at 6.10 and 7.25 with vicinal coupling constant of 8.6 Hz in spectra of 11c. We did not manage to react compounds 11a–c with a further equivalent of DMAD, although 11c smoothly reacted with o-nitrophenacyl bromide to give 2-triazolylthiazole 12 in 74% yield.
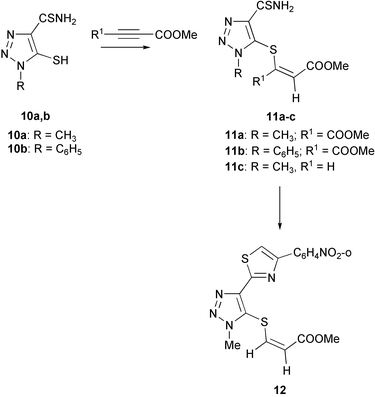
Indeed, reaction of DMAD with 5-mercaptoazole-4-thiocarboxamides 5b,10a,b preferably occurs with participation of the endocyclic thioamide group, similar to the reaction of these compounds with alkylating agents.13,14 This can be rationalized by the existence of 5b,10 in a zwitterionic tautomeric form where the negative charge is located at the 5-sulfur atom which should enhance an electrophilic attack to this center.
In the reaction of 4-arylhydrazonopyrazole-5-thiones 13 with esters of acetylenecarboxylic acids one can theoretically expect the formation at least 4 products 15–18 because both thioamide and hydrazono groups of 13 are known to react with DMAD.10–12,15 If we suppose that the most nucleophilic sulfur atom reacts first with the acetylene to form an intermediate vinylthioimidate of type 14 then the direction of its cyclization will govern the composition of the products 15–18.
We have found that compounds 13a–f readily react with DMAD and methyl propynoate to form 15a–f as the only products in good yields. The assignments of the structures of compounds 15 as pyrazolo[1,5-b]thiazin-4-ones follows from the analysis of their 1H and 13C NMR spectra and from the reaction of 15d with sodium dithionite. Mass spectra, and decoupled 1H and 13C NMR spectra did not allow one to make a difference between structures 15–18. At the same time, the similarity of the NMR spectra for the products obtained in the reaction of DMAD and methyl propynoate allowed us to assume that the same type of product was formed in both types of reactions. The spectrum of 15f shows two doublets as an AB system with a coupling constant of 10.6 Hz which is typical for a CHCH moiety. The C4 and C7 signals in the coupled spectrum of 15d are found at δ
= 154.9 ppm as doublets with a 2JC4–H5 coupling constant of 0.6 Hz and at 161.9 ppm with 3JC7–H5 of 3.8 Hz, respectively. These data allowed us to rule the structures 16 and 17 out of consideration, since one should observe an AB system in their spectra with a geminal constant of 18–22 Hz and a higher magnitude (2–5 Hz) for the coupling constant JC4–H5.
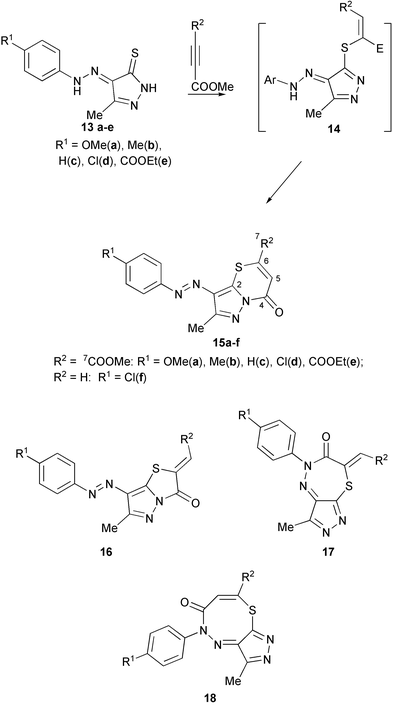
An additional argument in favor of structure 15 was made from reduction of compound 15d by sodium dithionite. The arylhydrazino derivative 19 was obtained, and hydrazines are typical reduction products of arylazo compounds. This finally eliminates structures 17 and 18.
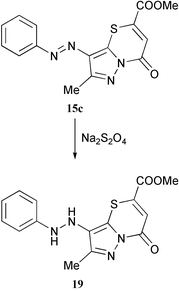
Thus, the novel imidazo[1,5-b]thiazin-4-ones 6a,b, pyrazolo[1,5-b]thiazin-4-ones 15a-f and imidazo[1,5-b]thiazole-4-ones 7a,b have been prepared by reactions of 5-mercaptoazoles with DMAD and methyl propynoate.
To study whether the size of the ring formed in the reaction of endocyclic thioamides with acetylene carboxylic esters depends on the type of ring in the starting compounds, we have carried out the reaction of 3,4-dihydropyridine-2(1H)-thiones 20a–c with DMAD.
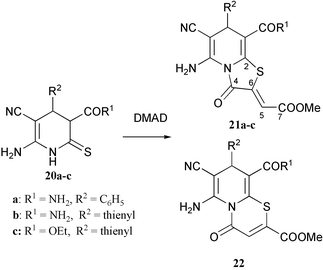
Reaction of 2(1H)-pyridinethiones with DMAD is known to give thiazolo[3,2-a]pyridinium salts. At the same time, the reaction of 2(1H)-pyridinethiones with methyl propynoate results in the acyclic condensation products.16–18 3,4-Dihydropyridin-2(1H)-ones have not been reacted with acetylene carboxylic acids so far.
By analogy with the reactions of malonthioamides and with the chemistry of the 5-mercaptoazoles mentioned above, one can expect the formation of both pyridothiazoles of type 21 and pyridothiazines 22 from the reaction of compounds 20 with DMAD. We have found that the reaction of pyridinethiones 20a–c with DMAD in chloroform in the presence of triethylamine selectively affords thiazolo[3,2-a]pyridines 21a–c in good yields. The structure assignment of the compounds prepared follows from their NMR spectra. 1H NMR spectra of 21a–c show signals at 6.65–6.78 ppm and the coupled 13C NMR spectrum of 21c contains doublets of C4 with a 3JCH coupling constant of 5.6 Hz with the vinyl proton, and of C7 with a 2JCH coupling constant of less than 1.0 Hz with the same vinyl proton. This is in accordance with the presence of an exocyclic double bond in the structures of compounds 21a–c. Furthermore, the magnitude of the constant 3JC4–H5 confirms the (Z)-configuration of this bond.
Conclusion
The data obtained allows us to make some conclusions. The size of the rings formed in the reactions of cyclic thioamides with acetylenecarboxylic esters depends on the size of the starting heterocycle. Thus, a five-membered thiazolidine ring condenses onto the pyridine ring and a six-membered thiazine ring is fused onto a five-membered azole ring. In contrast to all reactions of thioamides of various structures,9–12 including reactions of pyridinethiones with DMAD where the thiazolidin-4-ones are formed exclusively, the reactions of 5-mercaptoazoles with acetylenecarboxylic esters lead to the preferred formation of a six-membered thiazine ring. This exception to the general rule can be explained by the increased ring strain for two fused five-membered rings compared to a situation where a six-and five-membered ring are fused. This also implies that the reactions are under thermodynamic rather than kinetic control.Experimental
General
1H and 13C spectra were recorded at 400 and 100 MHz, respectively on a Bruker AMX 400 with SiMe4 as an internal reference in ether DMSO-d6 + CCl4 or CDCl3 solutions. Mass spectra were obtained on a Varian MAT 311A instrument using the electron impact ionization technique (40–200 °C, 70 eV). Reactions were monitored by TLC (Silufol®) on aluminium foil plates) in CHCl3–EtOH (9 : 1), CHCl3–EtOH–NH4OH (15 : 8 : 1), acetone–hexane (3 : 5) visualized under UV light. All solvents were distilled prior to use. C(5)H), 7.63 (1H, s, NH), 7.83 (1H, s, NH), 8.80 (1H, s, C(8)H).
C(5)H), 7.45 (1H, s, NH), 7.65 (1H, s, NH), 8.63 (1H, s, C(8)H).
C(5)H), 8.86 (1H, s, C(8)H), 9.41 (1H, s, NH), 9.70 (1H, s, NH); 7bδH
(DMSO-d6) 3.76 (3H, s, OCH3), 6.93 (1H, s, C(5)H), 8.70 (1H, s, C(8)H), 9.42 (1H, s, NH), 9.70 (1H, s, NH).
CH), 9.50 (1H, s, NH), 9.72 (1H, s, NH).
CH), 7.20–7.45 (5H, m, Ph), 9.53 (1H, s, NH), 9.79 (1H, s, NH).
CH), 7,25 (1H, d, 8.6, CH), 9.56 (1H, s, NH), 9.80 (1H, s, NH).
CH), 7.40 (1H,d, J
= 9.6 Hz, CH), 7.73 (1H, t, ArH), 8.15–8.22 (1H, m, ArH), 8.35–8.41 (1H, m, ArH), 8.50 (1H, s, Hthiazole), 8.77 (1H, t, ArH).
C(5)H), 6.92–6.94 (1H, m, Hthienyl), 7.03–7.04 (1H, m, NH), 7.28–7.31 (2H, m, Hthienyl), 7.33 (1H, s, NH), 7.43 (2H, s, NH2).
C(5)H), 6.90–6.95 (2H, m, Hthienyl), 7.25–7.30 (1H, m, Hthienyl), 7.35 (2H, s, NH2).
General method for the synthesis of arylazopyrazolethiones 13a-e
Arylazopyrazolones23 (0.004 mol) were refluxed about 30 min in toluene (50 mL) with Lawesson's reagent (0.0022 mol). The solution was cooled and the solid filtered off. The product was crystallized from ethanol.Acknowledgements
W. D. thanks the F. W. O.-Vlaanderen, the Ministerie voor Wetenschapsbeleid and the University of Leuven for financial support. V. S. B. and V. A. B. thank the Russian Foundation for Basic Research (grant 01-03-33173) and CRDF (grant Rec 005).References
- E. I. Greenblart and I. Ya. Postovskii, Zh. Obshch. Khim., 1961, 31, 394 Search PubMed.
- U. Vogeli, W. Von Philipsborn, K. Nagarajan and M. D. Nair, Helv. Chim. Acta, 1978, 61, 607 CrossRef.
- M. D. Nair, K. Nagarajan and J. H. Desai, Tetrahedron Lett., 1979, 20, 53 CrossRef.
- R. M. Acheson and J. D. Wallis, J. Chem. Soc., Perkin Trans. 1, 1981, 415 RSC.
- L. I. Giannola, G. Giammona, S. J. Palasso and L. Lamatrina, J. Chem. Soc., Perkin Trans. 1, 1984, 2707 RSC.
- S. Coen, B. Ragonnet, C. Vieillescares and J. P. Roggero, Heterocycles, 1985, 23, 1225 Search PubMed.
- N. D. Abramova and B. V. Trzhtsinskaya, Khim. Geterotsikl. Soedin., 1988, 12, 1587 Search PubMed.
- V. Ya. Kauss, E. E. Liepinsh, I. Ya. Kalvinsh and E. Lukevits, Khim. Geterotsikl. Soedin., 1990, 120 Search PubMed.
- G. Giammona, M. Neri, A. Pazzo and C. La Rosa, J. Heterocycl. Chem., 1991, 28, 325 Search PubMed.
- V. S. Berseneva, N. Yu. Biryucheva and V. A. Bakulev, Khim. Geterotsikl. Soedin., 1993, 1688 Search PubMed.
- V. S. Berseneva, A. V. Tkachev, Yu. Yu. Morzherin, W. Dehaen, I. Luyten and V. A. Bakulev, J. Chem. Soc., Perkin Trans. 1, 1998, 2133 RSC.
- V. S. Berseneva, Yu. Yu. Morzherin, W. Dehaen, I. Luyten and V. A. Bakulev, Tetrahedron, 2001, 57, 2179 CrossRef CAS.
- V. A. Bakulev, V. S. Mokrushin, A. N. Grishakov and Z. V. Pushkareva, Khim. Geterotsikl. Soedin., 1982, 957 Search PubMed.
- E. D. Shtephan and V. Yu. Vvedenskii, Uspekhi Khimii, 1996, 65, 326 Search PubMed.
- A. Reliquet, R. Besbes, F. Reliquet and J. C. Meslin, Synthesis, 1991, 543 CrossRef CAS.
- J. Becher and C. C. Stidsen, Sulfur Reports, 1988, 8, 105 Search PubMed.
- R. Lie and K. Undheim, Acta Chem. Scand., 1973, 27, 1756 Search PubMed.
- K. Undheim and L. A. Riege, J. Chem. Soc., Perkin Trans. 1, 1975, 1493 RSC.
- V. I. Ofitserov, V. S. Mokrushin, V. I. Nifontov, Z. V. Pushkareva, N. V. Nikiforova and L. N. Lych, Khim. Geterotsikl. Soedin., 1975, 1550 Search PubMed.
- V. A. Bakulev, A. T. Lebedev, E. F. Dankova, V. S. Mokrushin and V. S. Petrosyan, Tetrahedron, 1989, 45, 7329 CrossRef CAS.
- L. A. Rodinovskaya, A. M. Shestopalov and V. N. Nesterov, Khim. Geterotsikl. Soedin., 1996, 1376 Search PubMed.
- V. P. Litvinov, Izvest. AN, Seriya Khim., 1998, 11, 2123 Search PubMed.
- C. Kashima, H. Harada, I. Kita, I. Fukuchi and A. Hosomi, Synthesis, 1994, 61 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |