Synthesis of 1,3-dithiol-2-ones as proligands related to molybdopterin
Ben
Bradshaw
a,
David
Collison
a,
C. David
Garner
b and
John A.
Joule
*a
aChemistry Department, The University of Manchester, Manchester, UK M13 9PL
bSchool of Chemistry, University of Nottingham, University Park, Nottingham, UK NG7 2RD
First published on 27th November 2002
Abstract
The reaction of suitably disubstituted alkynes with diisopropyl xanthogen disulfide gives differentially substituted 4,5-disubstituted-1,3-dithiol-2-ones as proligands for metal complexes related to the molybdenum cofactor.
Introduction
We have described linear1–4 and convergent5,6 routes to differentially 4,5-disubstituted 1,3-dithiol-2-ones 1 (and -2-thione analogues) and shown how these can be used to form metal complexes7–12via hydrolytic release of the masked ene-1,2-dithiolate ligand from 1,3-dithiol-2-one. We have also demonstrated how appropriately substituted 1,3-dithiol-2-ones 1 can serve as key intermediates for the construction of tricyclic pyrano-quinoxalines, and complexes therefrom, which mimic13,14 the structure of molybdopterin 2, and we have constructed molybdopterin itself, in protected form and with the ene-dithiolate masked in this way, 3.15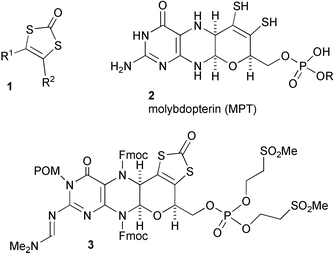
In the context of the synthesis of 1,3-dithiol-2-ones, key intermediates in our strategy for the construction of molybdopterin and its analogues, we were attracted to reports by Gareau16,17 that the reaction of alkynes with diisopropyl xanthogen disulfide†4 (and the corresponding dithiodiisopropyl xanthogen disulfide), in the presence of a radical initiator, leads directly to these five-membered heterocycles. Gareau proposed that homolytic cleavage of the S–S bond produces a radical 5 that adds to the alkyne, generating 6, which cyclises with loss of an isopropyl radical (Scheme 1). At the time it seemed significant that all of the examples (12) given in the two papers involved terminal alkynes and thus led to mono-substituted 1,3-dithiol-2-ones 7. Accordingly, we did not immediately investigate the method in the context of our molybdopterin research.
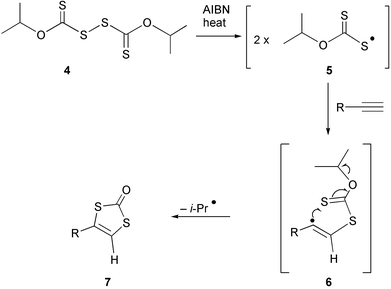 | ||
| Scheme 1 | ||
Later, Gareau reported18 the successful application of his route to 1,3-dithiol-2-ones using disubstituted alkynes, though yields were poorer, especially where the alkynes had carbonyl conjugation and/or bulky substituents.
Based on Gareau's observations, there seemed to be two reasons why the process might not be suitable for our purposes: (1) the considerable size of alkyne substituents that would be required to generate relevant proligands, and (2) the presence of the imine unit of the quinoxaline/pteridine substituent that might act like a conjugated carbonyl group, and discourage reaction. Nevertheless, we have now examined five relevantly disubstituted alkynes, finding in each case a very useful, in some cases, high yielding, synthesis of 4,5-differentially disubstituted-1,3-dithiol-2-ones.
Taylor and co-workers were the first to describe the synthesis of 2-alkynylquinoxalines and 6-alkynylpteridines related to molybdopterin,19 such as would be required for the application of the Gareau method for the assembly of proligands for molybdopterin and its analogues. In our previous studies we have also described the synthesis of various 2-alkynylquinoxalines13,14,20,21 and 6-alkynylpteridines.22,23
Results and discussion
Synthesis of alkynes
The alkynes chosen for study were 8–10,11b,12b. Full details of the synthesis of 8 by palladium(0)-catalysed coupling of 2-chloroquinoxaline with racemic but-3-yn-2-ol have been published.14 We have previously described4 a synthesis of the alkynyl-acetal 9 from 2-(D-arabino-tetrahydroxybutyl)quinoxaline, but for the present work, it was made by coupling of 2-chloroquinoxaline with (4R)-4-ethynyl-2,2-dimethyl-1,3-dioxolane;24 the analogous protected pteridine 10 was made in this way too, coupling to 6-chloro-2-(dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)pteridin-4-one.15 From other, previously unpublished work on an alternative route to such substances, the alkyne-diols 11b and 12b were available to us. Quinoxaline 11b was made by coupling 2-chloroquinoxaline with 4-tert-butyldiphenylsiloxy-3-hydroxybut-1-yne, giving 11a, followed by fluoride deprotection. Diol 12b was prepared by coupling 6-chloro-2-(dimethylaminomethyleneamino-3-(2,2-dimethylpropanoyloxymethyl)pteridin-4-one15 with 3,4-bis(tert-butyldiphenylsiloxy)but-1-yne, giving 12a, followed by fluoride deprotection; in this much more polar pteridine series, the use of the doubly O-silylated C4-alkyne-diol facilitated handling and isolation.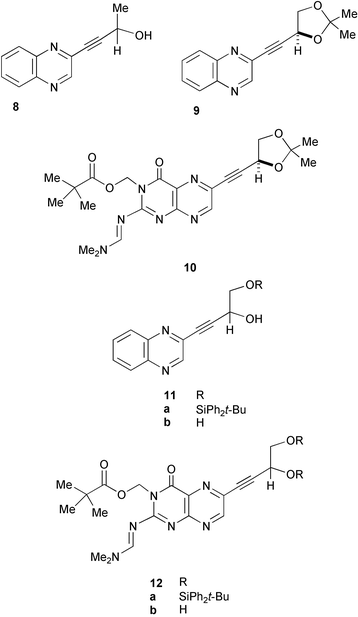
We found that the best route for the synthesis of the alkyne coupling partners started from commercially available (Z)-1,4-dihydroxybut-2-ene which was mono-protected by reaction with tert-butyldiphenylsilyl chloride giving 13, the alkene was then epoxidised using m-chloroperbenzoic acid generating 14, and then this was transformed25 into the alkyne 15a by treatment with n-butyllithium. Finally, the doubly silyl-protected alkyne 15b was prepared by exposure again to tert-butyldiphenylsilyl chloride (Scheme 2).
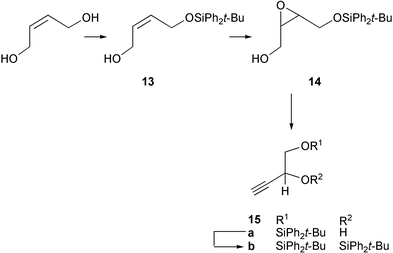 | ||
| Scheme 2 | ||
Synthesis of 1,3-dithiol-2-ones
Notwithstanding the reservations mentioned above, reaction of each of the alkynes 8–10, 11b, and 12b with diisopropyl xanthogen disulfide 4, initiated using AIBN, gave rise to 1,3-dithiol-2-ones, 16–20 respectively. The acetal protected substrates reacted in better yields (17 (77%); 18 (61%)) than the alcohol 8 (41%) or the diols 11b (30%) and 12b (20%). We have previously shown14,15 that it is easy to remove the acetal protection from 17 and 18, revealing diols, 19 and 20, and thus clearly the method of choice for the synthesis of these substances involves the formation of 17 and 18, then acetal hydrolysis.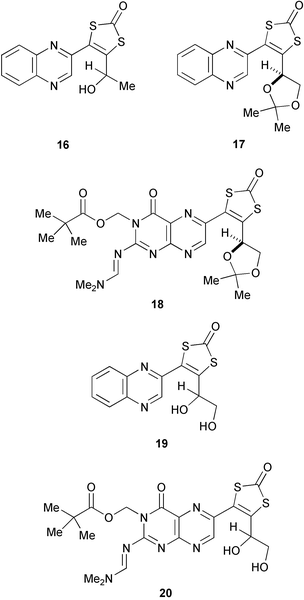
Given the relative efficiency of the alkyne coupling procedure and the directness of this approach (e.g. the synthesis of a 6-iodopterin, as was previously required,15 is not necessary), this route now proves to be the strategy of choice to the key 1,3-dithiol-2-one intermediates 17 and 18 in our route to molybdopterin and pteridine and quinoxaline analogues thereof.
We shall be reporting on the use of this method for the synthesis of suitably modified analogues which will be designed to cast light on the mode of action of the cofactor of the oxomolybdoenzymes.
Experimental
General
Organic solutions were dried over anhydrous MgSO4. Solid products were dried under reduced pressure using P4O10 as a desiccant. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Inova-300 Athos (300 MHz) and Unity 500 (500 MHz) spectrometers. Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on an Inova-300 Athos spectrometer running at 75 MHz. All chemical shifts (δ) are quoted in parts per million (ppm) downfield from tetramethylsilane (TMS). Signal splittings are reported as singlet (s), doublet (d), double doublet (dd), triplet (t), quartet (q), multiplet (m), and broad (br); J values are given in Hertz (Hz). UV spectra were recorded on a Hewlett Packard 8452A diode array spectrophotometer and are given in nm. IR spectra were recorded on an Ati Mattson Genesis Series FTIR spectrometer; only absorptions of importance to structure determination are listed. Mass spectra were recorded on a Fisons VG Trio 2000 (EI/CI{NH3}/ES) (abundance relative to the base peak is given in parentheses as a percentage; only fragment ions of intensity >10% of the base peak are cited), and a Concept IS (MM/FAB) spectrometer for accurate mass determinations. Melting points were recorded on a Reichart heated stage microscope and are uncorrected. Flash column chromatography was carried out using Merck 9385 silica gel 60 (230–400 mesh). Acetonitrile, dichloromethane, and triethylamine were distilled from calcium hydride. Petroleum ether was the fraction bp 40–60 °C.(R)-(+)-4-Ethynyl-2,2-dimethyl-1,3-dioxolane24
To a stirred solution of freshly prepared (+)-2,3-O-isopropylidene-D-glyceraldehyde26 (5.0 g, 38.4 mmol) and the Bestmann–Ohira reagent (7.38 g, 38.4 mmol, prepared according to the modified method of Callant et al.27 using p-acetamidobenzenesulfonyl azide28 as the diazo transfer reagent) in methanol (250 ml) at 0 °C was added K2CO3 (7.1 g, 51.2 mmol) portionwise over 30 min. The resulting mixture was stirred for a further 18 h then a solution of saturated aqueous NH4Cl (250 ml) was added and the aqueous layer was extracted with pentane (3 × 750 ml). The combined organic layers were dried (MgSO4) and the solvent concentrated very carefully at reduced pressure (care!—product is quite volatile). Purification by flash chromatography (silica, 5→10% Et2O in pentane) provided the alkyne (2.66 g, 55%) as a colourless oil with spectroscopic data consistent with the published data.241-(2,2-Dimethyl-1,3-dioxolan-4-yl)-2-(quinoxalin-2-yl)ethyne 9
To a degassed solution of 2-chloroquinoxaline (2.27 g, 13.7 mmol), (R)-(+)-4-ethynyl-2,2-dimethyl-1,3-dioxolane (1.81 g, 14.4 mmol) and Et3N (15 ml) in MeCN (35 ml), was added Pd(OAc)2 (155 mg, 0.69 mmol), Ph3P (362 mg, 1.38 mmol) and CuI (263 mg, 1.38 mmol) and the resulting mixture stirred for 18 h then refluxed for 2 h. On cooling, the mixture was concentrated and the residue partitioned between water (50 ml) and CH2Cl2 (100 ml). The mixture was filtered through Celite to break the emulsion, the layers separated and the aqueous layer re-extracted with CH2Cl2 (2 × 50 ml). The combined organic extracts were dried (MgSO4) and concentrated. Purification by flash chromatography (silica, 10→20% EtOAc in petroleum ether) provided 9 (2.61 g, 75%) as a yellow oil, with physical and spectroscopic data identical to those reported previously.141-[2-(Dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)-4-oxopteridin-6-yl]-2-[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]ethyne 10
To a degassed solution of 6-chloro-2-(dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)pteridin-4-one15 (2.17 g, 5.92 mmol), (4R)-(+)-4-ethynyl-2,2-dimethyl-1,3-dioxolane (1.12 g, 8.88 mmol) and Et3N (15 ml) in MeCN (50 ml), was added Pd(OAc)2 (60 mg, 0.30 mmol), Ph3P (155 mg, 0.59 mmol) and CuI (113 mg, 0.59 mmol) and the resulting mixture stirred at rt for 18 h then refluxed for 2 h. On cooling, the mixture was concentrated and the residue partitioned between water (50 ml) and CH2Cl2 (150 ml). The mixture was filtered through Celite to break the emulsion, the layers separated and the aqueous layer re-extracted with CH2Cl2 (2 × 100 ml). The combined organic extracts were dried (MgSO4) and concentrated. Purification by flash chromatography (silica, 1→2% MeOH in CH2Cl2) provided 10 (1.76 g, 65%) as a yellow solid, mp 186 °C (dec.); νmax(film)/cm−1 2983, 2934, 2875, 1731, 1705, 1634, 1527, 1448, 1423, 1393, 1368, 1330, 1144, 1115, 1061; 1H NMR (300 MHz, CDCl3) δ 8.95 (1H, s, H-7′), 8.78 (1H, s, CHNMe2), 6.38 (2H, s, NCH2OCOt-Bu), 4.98 (1H, t, J = 6.3 Hz, CHO), 4.25 (1H, dd, J = 6.6, 8.1, one of CH2O), 4.08 (1H, dd, J = 6.3, 8.1, one of CH2O), 3.25 (3H, s, NCH3), 3.08 (3H, s, NCH3), 1.55 (3H, s, CH3), 1.41 (3H, s, CH3), 1.08 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 177.7, 161.2, 159.7, 157.8, 153.8, 153.1, 135.0, 130.2, 111.0, 90.8, 82.5, 69.9, 66.1, 66.0, 42.0, 39.0, 36.0, 27.2, 26.4, 26.1; m/z (CI) 457 (MH+, 100%), 391 (40), 279 (20); found C, 57.65; H, 6.01, N, 18.54%; M + H+ 456.2121. C22H28N6O5 requires C, 57.88; H, 6.18, N, 18.41%, M (+ H) 456.2121.(Z)-4-tert-Butyldiphenylsiloxy-1-hydroxybut-2-ene 13
To a stirred solution of (Z)-1,4-dihydroxybut-2-ene (192 g, 2.18 mol), DMAP (1.33 g, 0.012 mol) and Et3N (30 ml) in dry CH2Cl2 (300 ml) at 25 °C, was added tert-butyldiphenylsilyl chloride (30 g, 0.11 mol) over 30 min. After 6 h water (200 ml) was added and the organic layer separated, the aqueous layer was then extracted with CH2Cl2 (2 × 100 ml). The combined organic extracts were washed with 12% citric acid solution (100 ml), brine (100 ml), dried (MgSO4) and the solvent was evaporated. Purification by flash chromatography (silica, 5→10% EtOAc in petroleum ether) provided 13 (35.6 g, 99%) as a colourless oil; λmax(CHCl3)/nm 226, 266; νmax(film)/cm−1 3331, 3070, 3049, 3024, 2957, 2931, 2891, 2857, 1690, 1589, 1471, 1428, 1390, 1361, 1188, 1109, 1079, 823; 1H NMR (300 MHz, CDCl3) δ 7.76 (4H, m, ArH), 7.46 (6H, m, ArH), 5.75 (2H, m, H-2 and H-3), 4.35 (2H, d, J = 5.5 Hz, H-4), 4.08 (2H, d, J = 6.9 Hz, H-1), 2.25 (1H, br s, OH), 1.15 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 135.6, 134.9, 133.4, 130.7, 130.0, 129.8, 127.9, 127.7, 60.2, 58.5, 26.8, 19.1; m/z (CI) 327 (MH+, 100%) 196 (40); found MH+, 327.1784. C20H26O2Si requires M (+ H) 327.1780. The spectroscopic data were exactly comparable with those reported for this compound prepared previously from the diol using n-butyllithium in THF at −78 °C29 or subsequently using sodium hydride in ether at room temperature.303-Hydroxymethyl-2-tert-butyldiphenylsilyloxymethyloxirane 14
To a stirred solution of m-chloroperbenzoic acid (52.5 g, 0.15 mol) in CH2Cl2 (250 ml) was added a solution of alkene 13 (35 g, 0.11 mol) in CH2Cl2 (100 ml) over 0.5 h. After 36 h the solution was cooled to 0 °C, precipitating m-chlorobenzoic acid, which was removed by filtration. Water was added, the organic layer separated and washed with aqueous Na2SO3 (2 × 350 ml), aqueous NaHCO3 (3 × 350 ml), brine (300 ml), dried (MgSO4) and the solvent evaporated. Purification by flash chromatography (silica, 10% EtOAc in petroleum ether) provided epoxide 14 (36.72 g, 100%) as a colourless oil; λmax(CHCl3)/nm 214, 226, 266; νmax(film)/cm−1 3426, 3070, 3048, 3012, 2996, 2954, 2933, 2892, 2859, 1589, 1471, 1428, 1391, 1361, 1260, 1188, 1109, 1091, 1046, 975, 937; 1H NMR (300 MHz, CDCl3) δ 7.72 (4H, m, ArH), 7.45 (6H, m, ArH), 4.0–3.75 (4H, m, H-2,3 and CH2OH), 3.28 (2H, m, CH2OSi), 1.92 (1H, br s, OH), 1.1 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 135.5, 135.4, 132.9, 132.7, 129.9, 127.8, 62.2, 60.7, 56.3, 56.1, 26.7, 19.1; m/z (CI) 360 (MNH+, 100%) 343 (MH+, 50%); found MNH4+ 360.1991. C20H26O3Si + NH4+ requires 360.1995. The spectroscopic data were exactly comparable with those reported previously29 for this compound prepared in homochiral form using Sharpless' method.3-Hydroxy-4-tert-butyldiphenylsiloxybut-1-yne 15a
To a stirred solution of 14 (12 g, 0.033 mol) in THF (150 ml) at −78 °C was added a solution of n-BuLi (62.5 ml, 0.10 mol) over 0.5 h. After 1 h the solution was warmed to room temperature and a solution of sat. aq. NH4Cl (150 ml) was added. The organic layer was separated and the aqueous layer re-extracted with Et2O (2 × 100 ml). The combined organic extracts were washed with brine (100 ml), dried (MgSO4) and the solvent evaporated. Purification by flash chromatography (silica, 5→10% EtOAc in petroleum ether) provided alkyne 15a (8.13 g, 76%) as a colourless oil; λmax(CH2Cl2)/nm 266; νmax(film)/cm−1 3560, 3422, 3291, 3071, 3050, 2957, 2931, 2890, 2859, 1472, 1428, 1132, 1020, 938, 823; 1H NMR (300 MHz, CDCl3) δ 7.62 (4H, m, ArH), 7.34 (6H, m, ArH), 4.4 (1H, m, H-3), 3.7 (2H, m, CH2OSi), 2.58 (1H, d, J = 5.6 Hz, OH), 2.35 (1H, d, J = 2.19 Hz, H-1), 1.1 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 135.7, 135.6, 135.5, 132.8, 132.7, 129.9, 127.8, 82.1 73.6, 67.3, 62.9, 26.9, 26.8, 19.3; m/z (CI) 342 (MNH4+, 100%), 325 (MH+, 5%); found MNH4+ 342.1893. C20H24O2Si (+ NH4) requires M 342.1889. The spectroscopic data were exactly comparable with those reported previously for this compound prepared by mono-silylation of the corresponding diol.313,4-Bis(tert-butyldiphenylsiloxy)but-1-yne 15b
To a stirred solution of alcohol 15a (3 g, 9.26 mmol), DMAP (113 mg, 0.926 mol) and Et3N (5 ml) at 25 °C in dry CH2Cl2 (20 ml), was added tert-butyldiphenylsilyl chloride (3.31 g, 12.0 mmol). After 24 h, water (20 ml) was added and the organic layer separated, the aqueous layer was then extracted with CH2Cl2 (2 × 20 ml). The combined organic extracts were washed with 12% citric acid solution (30 ml), brine (30 ml), dried (MgSO4) and the solvent was evaporated. Purification by flash chromatography (silica, 2.5→5% EtOAc in petroleum ether) provided 15b (5.15 g, 99%) as a white crystalline solid, mp 83–84 °C (previously prepared in 5% yield as a byproduct in a preparation of 3-hydroxy-4-tert-butyldiphenylsiloxybut-1-yne 15a by silylation of the corresponding diol31); λmax(CHCl3)/nm 242, 262; νmax(film)/cm−1 3306, 3071, 3050, 2957, 2931, 2891, 2858, 1472, 1428,1112, 962, 823; 1H NMR (300 MHz, CDCl3) δ 7.74 (8H, m, ArH), 7.4 (12H, m, ArH), 4.56 (1H, m, H-3), 3.8 (2H, m, H-4), 2.28 (1H, d, J = 2.1 Hz, H-1), 1.14 (9H, s, t-Bu), 1.07 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 136.1, 135.8, 135.6, 135.5, 133.4, 133.3, 129.6, 127.6, 127.5, 127.4, 83.0, 73.4, 68.1, 65.0, 26.9, 26.7, 19.3, 19.2; m/z (CI) 580 (MNH4+, 30%), 485 (15), 267 (100); found C, 76.92; H, 7.76%; MNH4+ 580.3077. C36H42O2Si2 requires C, 76.82; H, 7.76%; M (+ NH4) 580.3067.1-(Quinoxalin-2-yl)-3-hydroxy-4-tert-butyldiphenylsiloxybut-1-yne 11a
To a degassed solution of 2-chloroquinoxaline (0.77 g, 4.66 mmol), alkyne 15a (1.78 g, 4.23 mmol) and Et3N (5 ml) in MeCN (10 ml), was added Pd(OAc)2 (47 mg, 0.21 mmol), Ph3P (111 mg, 0.423 mmol) and CuI (80 mg, 0.42 mmol). The resulting mixture was refluxed for 2 h, cooled to 25 °C and concentrated in vacuo. The residue was partitioned between water (50 ml) and EtOAc (50 ml), the layers separated and the aqueous layer extracted with EtOAc (2 × 50 ml). The combined organic extracts were washed with brine (75 ml), dried (MgSO4) and concentrated in vacuo. Purification by flash chromatography (silica, 5→10% EtOAc in petroleum ether) provided 11a (1.40 g, 73%) as a coloured oil; λmax(CHCl3)/nm 256, 334; νmax(film)/cm−1 3329, 2956, 2930, 2888, 2857, 1541, 1487, 1471, 1427, 1364, 1295, 1216, 1114, 824; 1H NMR (300 MHz, CDCl3) δ 8.85 (1H, s, H-3′), 8.08 (2H, m, ArH), 7.74 (6H, m, ArH), 7.36 (6H, m, ArH), 4.90 (1H, t, J = 5.4 Hz, H-3), 4.22 (1H, br s, OH), 4.05 (2H, m, H-4) 1.14 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 146.9, 141.9, 140.8, 138.7, 135.6, 135.5, 132.8, 130.7, 130.6, 129.9, 129.0, 127.8, 92.9, 82.7, 77.6, 77.2, 76.8, 67.3, 63.6, 26.8, 19.3; m/z (CI) 453 (MH+, 70%); found M+ 452.1919. C28H28N2O2Si requires M 452.1919.1-(Quinoxalin-2-yl)-3,4-dihydroxybut-1-yne 11b
To a stirred solution of quinoxaline 11a (0.5 g, 1.11 mmol) in THF (10 ml) at 0 °C was added HF–pyridine (7 : 3, 3 ml). The mixture was then allowed to warm to room temperature. After 2 h the solution was diluted with water (20 ml) and the resulting mixture neutralised by the addition of solid NaHCO3. CH2Cl2 (50 ml) was added, the layers separated, and the aqueous layer re-extracted with CH2Cl2 (2 × 50 ml), the organic layer was washed with brine (30 ml), dried and concentrated. Purification by flash chromatography (silica, 0→2→5→8% MeOH in EtOAc) provided 11b (152 mg, 64%) as a white solid, mp >230 °C; 1H NMR (300 MHz, CD3OD) δ 8.95 (1H, s, H-3′), 8.1 (1H, m, ArH), 8.04 (1H, m, ArH), 7.88 (2H, m, ArH), 4.7 (1H, dd, J = 5.5, 6.3 Hz, H-3), 3.82 (2H, m, H-4); m/z (CI) 215 (MH+, 15%); found MH+ 215.0822. C12H10N2O2 requires M + H 215.0820.1-[2-(Dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)-4-oxopteridin-6-yl]-3,4-bis(tert-butyldiphenylsiloxy)but-1-yne 12a
To a degassed solution of 6-chloro-2-(dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)pteridin-4-one15 (1.0 g, 2.73 mmol), alkyne 15b (2.34 g, 4.10 mol) and Et3N (7 ml) in MeCN (30 ml), was added Pd(OAc)2 (31 mg, 0.14 mmol), Ph3P (72 mg, 0.27 mmol) and CuI (52 mg, 0.27 mmol) and the resulting mixture refluxed for 2 h. On cooling, the mixture was concentrated and the residue partitioned between water (50 ml) and EtOAc (50 ml). The layers were separated and the aqueous layer re-extracted with EtOAc (2 × 25 ml). The combined organic fractions were washed with brine (25 ml), dried (MgSO4) and concentrated. Purification by flash chromatography (silica, 30→50→100% EtOAc in petroleum ether) provided 12a (1.95 g, 80%) as a yellow foam; λmax(CHCl3)/nm 256, 332, 374; νmax(film)/cm−1 2960, 2932, 2859, 1732, 1705, 1634, 1525, 1424, 1365, 1332, 1113, 755; 1H NMR (300 MHz, CDCl3) δ 8.95 (1H, s, H-7′), 8.12 (1H, s, CHNMe2), 7.75 (8H, m, ArH), 7.38 (12H, m, ArH), 7.4 (8H, m, ArH), 6.38 (2H, s, NCH2OCOt-Bu), 4.84 (1H, m, H-3), 3.91 (2H, m, H-4), 3.25 (3H, s, NCH3), 3.17 (3H, s, NCH3), 1.17 (9H, s, t-Bu), 1.13 (9H, s, t-Bu), 1.08 (9H, s, t-Bu); 13C NMR (75 MHz, CDCl3) δ 177.3, 160.8, 159.2, 157.3, 153.1, 152.8, 136.1, 135.8, 135.5, 135.2, 133.5, 133.1, 133.0, 129.7, 129.6, 127.6, 127.4, 92.6, 82.5, 68.0, 65.8, 65.6, 41.6, 38.8, 35.6, 27.0, 26.8, 26.7, 19.1, 19.2, 15.1; m/z (ES+) 892 (M+, 100%).1-[2-(Dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)-4-oxopteridin-6-yl]-3,4-dihydroxybut-1-yne 12b
To a stirred solution of pteridine 12a (1.5 g, 1.68 mmol) in THF (30 ml) at 0 °C was added TBAF (0.97 ml of a 1.0 M solution of 5% H2O in THF). The mixture was then allowed to warm to room temperature. After 5 h the solution was diluted with EtOAc (100 ml) and water (50 ml), the layers separated, the organic layer washed with brine (50 ml), dried and concentrated. Purification by flash chromatography (silica, 0→2→5→10% MeOH in EtOAc) provided 12b (531 mg, 76%) as a yellow solid, mp >230 °C; λmax(CHCl3)/nm 256, 334, 376; νmax(film)/cm−1 3343, 2930, 1731, 1704, 1638, 1518, 1361, 1326, 1145, 1114; 1H NMR (300 MHz, CDCl3) δ 8.88 (1H, s, H-7′), 8.72 (1H, s, CHNMe2), 6.27 (2H, s, NCH2OCOt-Bu), 4.73 (1H, t, J = 5.5 Hz, H-3), 3.83 (2H, m, H-4), 3.19 (3H, s, NCH3), 3.10 (3H, s, NCH3), 1.08 (9H, s, t-Bu); m/z (CI) 417 (MH+, 80%); found MH+ 417.1886. C19H24N6O5 requires M (+ H) 417.1886.4-(1-Hydroxyethyl)-5-(quinoxalin-2-yl)-1,3-dithiol-2-one 16
Alcohol 8 (2.0 g, 0.01 mol), disulfide 4 (3.0 g, 11 mmol), and AIBN (0.75 g, 4.55 mmol) in toluene (1 ml) were heated to reflux for 6 h. The cooled mixture was purified directly by flash chromatography (silica, 0→5→10% EtOAc in CH2Cl2) giving the 1,3-dithiol-2-one 16 (320 mg, 41%) as a yellow crystalline solid, with physical and spectroscopic data identical to those reported previously.144-(2,2-Dimethyl-1,3-dioxolan-4-yl)-5-(quinoxalin-2-yl)-1,3-dithiol-2-one 17
Protected diol 9 (331 mg, 1.30 mmol), disulfide 4 (267 mg, 1.63 mmol), and AIBN (702 mg, 2.6 mmol) in toluene (0.6 ml) were heated at reflux for 1.5 h then further portions of disulfide 4 (267 mg, 1.63 mmol), and AIBN (702 mg, 2.6 mmol) were added and the resulting mixture refluxed for a further 1.5 h. The cooled mixture was purified directly by flash chromatography (silica, 20% EtOAc in petroleum ether) giving 17 (347 mg, 77%) as a yellow crystalline solid, with physical and spectroscopic data identical to those reported previously.4,145-[2-(Dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)-4-oxopteridin-6-yl]-4-[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]-1,3-dithiol-2-one 18
To a solution of protected diol 10 (200 mg, 0.44 mmol) in chlorobenzene (0.25 ml) (NOTE—strong heating was necessary to achieve solution) was added disulfide 4 (474 mg, 1.75 mmol), and AIBN (180 mg, 1.11 mmol). The resulting mixture was heated at reflux for 2 h after which further portions of disulfide 4 (474 mg, 1.75 mmol), and AIBN (180 mg, 1.11 mmol) were added and the refluxing continued for a further 2 h. The cooled mixture was purified directly by flash chromatography (silica, 1→2% MeOH in CH2Cl2) giving 18 (146 mg, 61%) as a yellow solid, with physical and spectroscopic data identical to those reported previously.155-(Quinoxalin-2-yl)-4-(1,2-dihydroxyethyl)-1,3-dithiol-2-one 19
A mixture of diol 11b (2.0 g, 0.01 mol), disulfide 4 (3.0 g, 0.011 mol), and AIBN (0.75 g, 4.55 mmol) in toluene (1 ml) was heated to reflux for 6 h. The cooled mixture was purified directly by flash chromatography (silica, 0→5→10% EtOAc in CH2Cl2) giving 19 (234 mg, 30%) as a yellow solid. All spectroscopic data were identical to those previously reported.145-[2-(Dimethylaminomethyleneamino)-3-(2,2-dimethylpropanoyloxymethyl)-4-oxopteridin-6-yl]-4-(1,2-dihydroxyethyl)-1,3-dithiol-2-one 20
Diol 12b (30 mg, 0.072 mmol), disulfide 4 (21.4 mg, 0.079 mmol), AIBN (5.32 mg, 0.032 mmol) in toluene (0.1 ml) were heated to reflux for 6 h. The cooled mixture was purified directly by flash chromatography (silica, 0→2→5% EtOAc in CH2Cl2) giving 20 (7 mg, 20%) as a yellow crystalline solid with spectroscopic data identical to those reported previously.15Acknowledgements
We gratefully acknowledge support for this work from the EPSRC and latterly from the BBSRC in the form of post-doctoral assistantships (BB).References
- D. J. Rowe, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 1985, 1907 RSC.
- L. Larsen, D. J. Rowe, C. D. Garner and J. A. Joule, Tetrahedron Lett., 1988, 29, 1453 CrossRef CAS.
- L. Larsen, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 1989, 2311 RSC.
- L. Larsen, D. J. Rowe, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 1989, 2317 RSC.
- A. Dinsmore, C. D. Garner and J. A. Joule, Tetrahedron, 1998, 54, 3291 CrossRef CAS.
- A. Dinsmore, C. D. Garner and J. A. Joule, Tetrahedron, 1998, 54, 9559 CrossRef CAS.
- S. Boyde, C. D. Garner, J. A. Joule and D. J. Rowe, J. Chem. Soc., Chem. Commun., 1987, 800 RSC.
- E. M. Armstrong, M. S. Austerberry, J. H. Birks, R. L. Beddoes, M. Helliwell, J. A. Joule and C. D. Garner, Heterocycles, 1993, 35, 563 Search PubMed.
- R. L. Beddoes, A. Dinsmore, C. D. Garner and J. A. Joule, Acta Crystallogr., Sect. C, 1997, 53, 213 CrossRef.
- A. Dinsmore, J. H. Birks, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 1997, 801 RSC.
- E. S. Davies, R. L. Beddoes, D. Collison, A. Dinsmore, A. Docrat, J. A. Joule, C. R. Wilson and C. D. Garner, J. Chem. Soc., Dalton Trans., 1997, 3985 RSC.
- E. S. Davies, G. M. Aston, R. L. Beddoes, D. Collison, A. Dinsmore, A. Docrat, J. A. Joule, C. R. Wilson and C. D. Garner, J. Chem. Soc., Dalton Trans., 1998, 3647 RSC.
- B. Bradshaw, A. Dinsmore, C. D. Garner and J. A. Joule, Chem. Commun., 1998, 417 RSC.
- B. Bradshaw, A. Dinsmore, D. Collison, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 2001, 3232 RSC.
- B. Bradshaw, A. Dinsmore, D. Collison, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 2001, 3239 RSC.
- Y. Gareau, J. Chem. Soc., Chem. Commun., 1995, 1429 RSC.
- Y. Gareau and A. Beauchemin, Phosphorus, Sulfur, Silicon, 1997, 120&121, 393 Search PubMed.
- Y. Gareau and A. Beauchemin, Heterocycles, 1998, 48, 2003 Search PubMed.
- (a) E. C. Taylor, P. S. Roy, I. Darwish, J. L. Johnson and K. V. Rajagopalan, J. Am. Chem. Soc., 1989, 111, 285 CrossRef CAS; (b) R. C. Pilato, K. A. Eriksen, M. A. Greaney, E. I. Stiefel, S. Goswami, L. Kilpatrick, T. G. Spiro, E. C. Taylor and A. L. Rheingold, J. Am. Chem. Soc., 1991, 113, 9372 CrossRef CAS.
- M. Armengol and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 2001, 154 RSC.
- M. Armengol and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 2001, 978 RSC.
- J. R. Russell, C. D. Garner and J. A. Joule, Tetrahedron Lett., 1992, 33, 3371 CrossRef CAS.
- J. R. Russell, C. D. Garner and J. A. Joule, J. Chem. Soc., Perkin Trans. 1, 1992, 1245 RSC.
- J. Pietruszka and A. Witt, J. Chem. Soc., Perkin Trans. 1, 2000, 4293 RSC.
- S. Takano, K. Samizu, T. Sugihara and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1989, 1344 RSC.
- D. Y. Jackson, Synth. Commun., 1988, 18, 337 CAS.
- P. Callant, L. D'Haenens and M. Vandewalle, Synth. Commun., 1984, 14, 155 CAS.
- D. G. Brown, E. J. Velthuisen, J. R. Commerford, R. G. Brisbois and T. R. Hoye, J. Org. Chem., 1996, 61, 2540 CrossRef CAS.
- W. R. Roush, J. A. Straub and M. S. VanNieuwenhze, J. Org. Chem., 1991, 56, 1636 CrossRef CAS.
- D. Crich, M. A. de la Mora and R. Cuz, Tetrahedron, 2002, 58, 35 CrossRef CAS.
- O. W. Gooding, C. C. Beard, D. Y. Jackson, D. L. Wren and G. F. Cooper, J. Org. Chem., 1991, 56, 1083 CrossRef CAS.
Footnote |
| † The IUPAC name for diisopropyl xanthogen disulfide is bis(isopropyloxythiocarbonyl) disulfide. |
| This journal is © The Royal Society of Chemistry 2003 |