Molecular analysis of cis-prenyl chain elongating enzymes
Yugesh Kharel and Tanetoshi Koyama
Institute for Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan
First published on 5th December 2002
Abstract
Covering: the literature up to February 2002
Recent isolation of the gene for an undecaprenyl diphosphate synthase has disclosed the structures of many kinds of cis-prenyl chain elongating enzymes. Not only the primary structure but also the crystal structure of the cis-prenyltransferase is totally different from those of trans-prenyl chain elongating enzymes. This review covers up to February 2002 and contains 72 references.
 Yugesh Kharel | Yugesh Kharel was born in Kavre, Nepal. He obtained a B.E. degree in bio-engineering from Wuxi University of Light Industry, China in 1997, and then moved to Japan where he received an M.S. degree in Chemistry from Tohoku University in 2001. He is now a doctorial student with Professor Tanetoshi Koyama at the same university, where he is currently carrying out research on the molecular mechanism of cis-prenyl chain elongating enzymes. |
 Tanetoshi Koyama | Tanetoshi Koyama was born in Shizuoka, Japan. He obtained a B.S. in chemistry from Tohoku University and a D.Sc. in chemistry from the same university. After postdoctoral work at Stanford University, he joined the Chemical Research Institute of Non-Aqueous Solutions, Tohoku University. He was then promoted to associate professor in the Department of Biochemistry and Engineering, Faculty of Engineering, Tohoku University, and to Professor of the Institute of Chemical Reaction Science (now renamed, Institute of Multidisciplinary Research for Advanced Materials) at the same University. His research interests focus on molecular mechanisms of enzymes, especially those relating to the biosynthesis of isoprenoids. |
1 Introduction
Isoprenoids comprise a very large family of natural products with more than 23 000 structurally diverse compounds of both primary and secondary metabolites. Because of the wide range of physiological roles in all living system, isoprenoids have been the target of extensive studies in the field of organic, biomedical and industrial chemistry.Biological construction of every carbon skeleton of isoprenoid compounds (including derivatives) is catalyzed by the group of enzymes commonly called prenyltransferases (also referred as prenyl diphosphate synthases). Based on the chain length and stereochemistry of the final product, prenyltransferases are classified in two major groups, trans- and cis-prenyltransferases. Enzymes that catalyze the sequential addition of C5-prenyl group in the trans-configuration or the cis-configuration are known as trans-prenyltransferases (TPTs) or cis-prenyltransferases (CPTs), respectively. Further classifications of prenyltransferases are shown in Fig. 1.
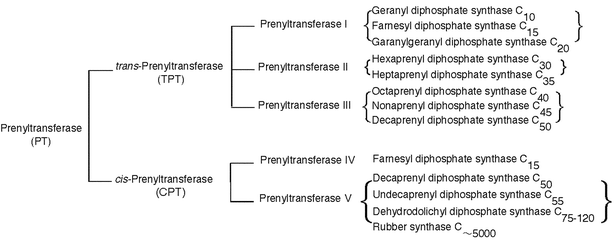 | ||
| Fig. 1 Classification of the enzymes that catalyze prenyl chain elongation. | ||
Prenyltransferases, in a wide sense, catalyze the transfer of prenyl groups to accept molecules of not only isopentenyl diphosphate (IPP) but also aromatic compounds or proteins. In this review, however, prenyltransferase will be restricted to the enzyme whose acceptor molecule is IPP; more specifically, “prenyltransferase” means “prenyl diphosphate synthase”.
Enzymatic studies on the biosynthetic pathway of isoprenoid compounds were developed to an enzymological level after the independent discoveries of IPP as the true “biologically active isoprene unit” by Lynen1 and Bloch,2 whose pioneering works led many researchers to isolate and characterize many enzymes involved in this pathway. IPP was long believed to be synthesized exclusively from the acetate/mevalonate pathway3–5 until Rohmer et al. discovered a novel pathway in which mevalonate is not involved.6,7 Nowadays, the non-mevalonate pathway has been shown to be operative in the biosynthesis of not only particular isoprenoids in bacteria but also of some terpenoids in plants8 and algae.9
In the biosynthetic pathway of isoprenoid compounds, all of the carbon skeleton of the isoprene units is derived from sequential enzymatic condensation of IPP with an allylic diphosphate. The catalytic reaction of prenyltransferase starts with the formation of allylic cation after cleavage of the carbon-oxygen bond in an allylic prenyl diphosphate and then addition of a single isoprene unit, IPP, occurs with the stereospecific removal of a proton at the 2-position, allowing a new C–C bond and a new double bond in the product, which becomes an allylic diphosphate for the next condensation reaction (Fig. 2(A)). In this way, one C5-isoprene unit elongation is carried out and, by repeating this stereospecific condensation of IPP with the allylic diphosphate product, prenyltransferase can synthesize prenyl diphosphate with a certain chain length and stereochemistry according to the product specificity of the individual enzyme.10
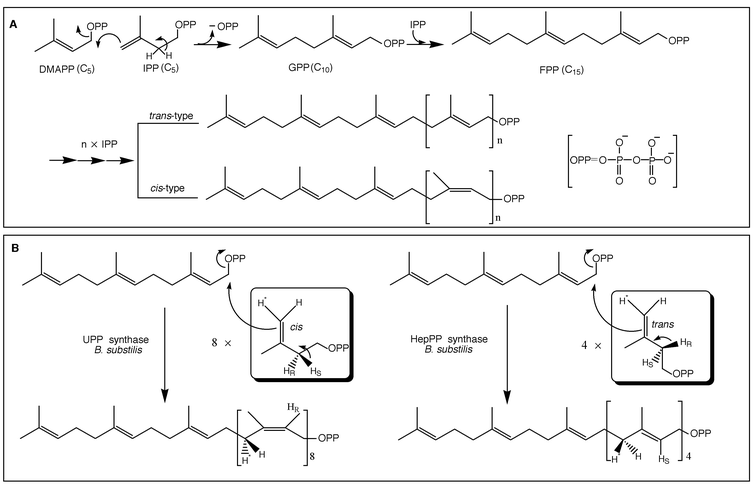 | ||
| Fig. 2 (A) Schematic drawing of the reactions catalyzed by prenyltransferase. (B) Absolute stereochemistry of cis- (left) and trans- (right) prenyltransferase reaction. | ||
The reactions catalyzed by TPTs and CPTs are similar as they both use the same substrates for the enzymatic prenyl chain elongation. The only difference is the prochirality of the proton, which is eliminated from the 2-position of IPP, pro-R for TPT and pro-S for CPT during the condensation reaction, respectively. The absolute stereochemistry involved in the TPT and CPT reactions has been determined, with respect to the reactions catalyzed by (all-E)-heptaprenyl diphosphate (HepPP, C35) synthase or by undecaprenyl diphosphate (UPP, C55) synthase from the same organism, to be that the C–C bond is formed on the same si face of the double bond of IPP as the C–H bond is cleaved during the reaction.11,12 The stereochemistry of the medium-chain prenyl diphosphate synthase, HepPP synthase, has been shown to be exactly similar to that demonstrated for mammalian short-chain prenyl diphosphate synthase, farnesyl diphosphate (FPP, C15) synthase, by the classical work of Cornforth and co-workers.13–15
In both prokaryotes and eukaryotes, TPTs classified in a groups of short chain prenyl diphosphate synthases such as FPP synthase, and geranylgeranyl diphosphate (GGPP, C20) synthase, catalyze the formation of priming allylic substrates which are intrinsic substrates for other groups of prenyltransferases to produce many other longer chain prenyl diphosphates.
CPT catalyzes the sequential cis-condensation of IPP with an allylic diphosphate to synthesize long chain prenyl diphosphates, which occur more or less in association with the membrane fraction of bacterial cell wall and the microsomal fraction of eukaryotic cells. Despite the similarities of the same primer substrates in the prenyl chain elongation reaction, prenyltransferases appear to be two independent families based on the geometric configuration of the products. CPT seems more diverse than TPT in the final product biosynthesis, which varies from cis-farnesyl diphosphate (C15) to natural rubber whose prenyl chain length extends to several millions (Fig. 3).
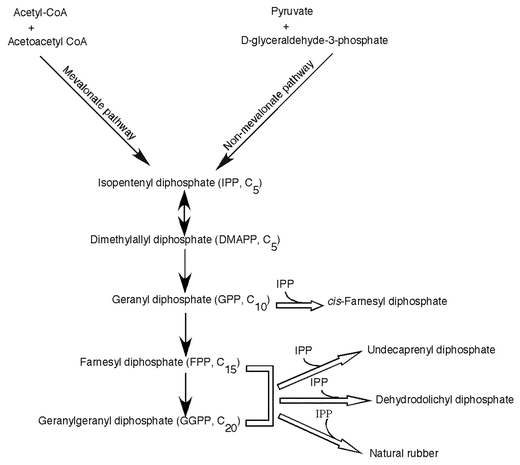 | ||
| Fig. 3 Biosynthetic pathway for cis-prenyl chain elongation. Open arrows indicate the reactions catalyzed by cis- prenyltransferases (CPTs). | ||
In eubacteria and archaea, usually UPP synthase catalyzes the sequential addition of isoprene unit onto FPP to produce UPP, a C55 prenyl diphosphate with mixed E,Z-stereochemistry. As indicated in Table 1, consecutive condensation of all eight molecules of IPP proceeds in the cis-configuration up to the final product UPP. Mycobacteria are exceptional in that they contain two types of CPTs, one catalyzes the short chain C15 product and the other synthesizes a longer prenyl chain of C50.16 Undecaprenyl phosphate (UP, a single dephosphorylated form of UPP), which contains 11 unsaturated isoprene units, is required for carbohydrate carrier lipid in the biosynthesis of bacterial cell wall.17
| Species | Gene name | Enzyme involved | Structurea | Isoprene units | Ref. |
|---|---|---|---|---|---|
| a w: terminal isoprene unit; t: trans-isoprene unit; S: α-saturated isoprene unit; c: cis-isoprene unit. | |||||
| Eubacteria | |||||
| M. luteus | UPS | Undecaprenyl diphosphate synthase | w-t2-c8-PP | C11 | 33 |
| E. coli | UPS | Undecaprenyl diphosphate synthase | w-t2-c8-PP | C11 | 35 |
| H. influenzae | UPS | Undecaprenyl diphosphate synthase | w-t2-c8-PP | C11 | 35 |
| S. pneumoniae | UPS | Undecaprenyl diphosphate synthase | w-t2-c8-PP | C11 | 35 |
| Mycobacteria | |||||
| M. tuberculosis | Rv1086 | Farnesyl diphosphate synthase | w-t-c-PP | C3 | 16 |
| M. tuberculosis | Rv2361c | Decaprenyl diphosphate synthase | w-t-c8-PP | C10 | 16 |
| Archae | |||||
| S. acidocaldarius | CPDS | Undecaprenyl diphosphate synthase | w-t2-c8-PP | C11 | 36 |
| Yeast | |||||
| S. cerevisiae | RER2 | Dehydrodolichyl diphosphate synthase | w-t2-c11-S-PP | C15 | 41 |
| S. cerevisiae | STR1 | Dehydrodolichyl diphosphate synthase | w-t2-c15-S-PP | C19 | 42 |
| Plant | |||||
| A. thaliana | DeDolPS | Dehydrodolichyl diphosphate synthase | w-t2-c15–20-S-PP | C19–24 | 43,44 |
| H. brasiliensis | HRT | Rubber synthase | — | C∼5000 | 46 |
| Human | DeDolPS | Dehydrodolichyl diphosphate synthase | w-t2-c15–16-S-PP | C19–20 | 45 |
In eukaryotes, dehydrodolichyl diphosphate (DedolPP) synthase catalyzes the consecutive cis-condensation of IPP to form DedolPP. Dolichyl phosphate (Dol-P), the α-saturated and a single dephosphorylated form of DedolPP, acts as a direct sugar carrier lipid in the biosynthesis of N-glycosylated proteins and GPI-anchored proteins. The chain length of Dol-P has been shown to be significantly species specific. For example, yeast contains dolichols of 14–17 isoprene units18 whereas plant possesses dolichols with an isoprene unit of C19–2419–22 and in mammalian cells the dolichols of 18–21 isoprene units are predominant.23 The structural difference between the sugar carrier lipids and the bacterial counterpart is the longer prenyl chain and saturation of the α-isoprene unit. In addition to dolichols, many higher plants produce polyprenols with mixed E,Z-stereochemistry, betulaprenols, ficaprenols, and natural rubber.
This review summarizes the recent understanding of prokaryotic and eukaryotic CPTs, including molecular cloning, three-dimensional structure, enzymatic mechanism and regulation of chain length determination.
2 Structure of CPTs
2.1 Primary structure
Little was known about the structure of CPT at the molecular level until the gene cloning of the UPP synthase from M. luteus B–P 26 by Shimizu et al.33 using the colony autoradiography method. This method requires incubation of a replica membrane containing a genomic library in the usual incubation mixture with a radiolabelled substrate for the target enzyme. The replica filters after the incubation were subjected to autoradiography to detect radiolabelled colonies. The principle of this conventional method was developed by Raetz for rapid screening of E. coli colonies for mutants with defective enzymes of phospholipid metabolism.34 Surprisingly the amino acid sequence encoded by the UPP synthase gene is totally different from those of the trans-prenyl chain elongating enzymes. Protein database searches for homologous genes for the UPP synthase yielded more than 25 unknown proteins, which had not yet been characterized at that time. Shortly afterwards, Apfel et al.35 reported the homologous gene for this enzyme from E. coli, Haemophilus influenzae, and Streptococcus pneumoniae. Schulbach et al.16 identified two types of CPTs from Micobacterium tuberculosis, Rv1086, a short (C15) chain Z-prenyltransferase and Rv2361c, a long (C50) chain Z-prenyltransferase. More recently, Hemmi et al.36 cloned and characterized a CPT from an archeon, Sulfolobus acidocaldarius.
 | ||
| Fig. 4 Multialignment of the amino acid sequences of 10 proteins that have been identified as CPTs. In the figure, five conserved regions are indicated and identical amino acid groups are shaded. Sequences: M. tuberculosis Rv1086 (16), M. tuberculosis Rv2361c (16), M. luteus B–P 26 (GenBank accession number AB004319), E. coli (Swiss-Prot Q47675), H. influenzae (Swiss-Prot P444938), S. pneumoniae (33), S. acidocaldarious (GenBank accession number AB048249), Rer2 (Swiss-Prot P35196), Srt1 (Swiss-Prot Q03175), A. thaliana (GenBank accession number AF162441). | ||
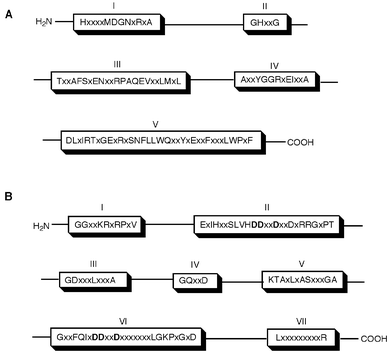 | ||
| Fig. 5 Comparison of conserved regions between cis- (A) and trans- (B) type prenyl chain elongating enzyme. | ||
2.2 Three-dimensional structure
In 1994, Tarshis et al.47 determined the crystal structure of avian FPP synthase, as the first crystal structure of any prenyltransferases, which advanced our understanding of the mechanism of trans-prenyl chain elongating enzymes. This TPT enzyme is a homodimer and possesses a characteristic structure that is composed of many anti-paralleled α-helices forming a large cavity in each subunit. X-Ray crystallographic studies have revealed that many enzymes related to isoprenoid biosynthesis, including pentalenene synthase,48 squalene cyclase,49 5-epi-aristolochene synthase,50 protein farnesyltransferase,51 and FPP synthase,47 have a common structure called isoprenoid synthase fold (or terpenoid synthase fold), which is composed of 11–12 α-helices forming a large central cavity. Since then, it has been believed to be common that isoprenoid synthase fold52 is included in all enzymes related to isoprenoid biosynthesis. Recently, Fujihashi et al.53 determined the crystal structure of M. luteus B–P 26 UPP synthase as the first three-dimensional structure for any cis-prenyl chain elongating enzymes, at 2.2 Å resolution. It is quite interesting that this enzyme shows a novel protein fold possessing a central β-sheet core, which is totally different from the isoprenoid synthase fold. This suggested that the catalytic mechanism of CPTs, including substrate recognition, is also different from those of TPTs. The M. luteus B–P 26 UPP synthase is a homodimeric enzyme of 58 kDa consisting of 20 α-helices and 12 parallel β-sheets (Fig. 6(A)). A large elongated hydrophobic cleft, consisting of highly conserved amino acid residues, surrounded by two α-helices (H2 and H3) and two β-strands (S2 and S4), is found on its molecular surface. This cleft seems to have a suitable volume to keep the large hydrophobic prenyl chain of allylic substrate during the catalytic chain elongation up to C55, the final product of the catalytic reaction. A sulfate ion used for the crystallization as a precipitant is found to bind with a motif in Region I, which is a common motif for phosphate recognition and is called a structural P-loop.54 It is likely that the diphosphate moiety of the allylic substrate FPP is electrostatically recognized by the structural P-loop motif, where the sulfate ion is located. At the entrance of this cleft, a positively charged cluster formed by conserved arginine residues, Arg-197 and Arg-203, is found which is predicted for the recognition of the diphosphate moiety of the homoallylic substrate. On the other hand, the residues from 74S to 85V, which belong to one of the conserved regions, Region III, could not be incorporated in the final crystal structure because of their invisible electron densities. When the two equivalent monomers were superimposed in the dimer structure, it resulted in a good match of the Cα atoms with each other except for in the region from 86N to 95F, which is located adjacent to the disordered loop (74S to 85V). Thus, these unidentified and highly conserved regions seem to be flexible in order to play an important role in the catalytic function. Recently, Ko et al.55 reported the three-dimensional structure of E. coli UPP synthase which is similar to that of M. luteus UPP synthase except for some deviations of some helices, mainly α3, α4 and α6. These differences between the helices might be owing to the different conditions utilized in the protein crystallization. In the E. coli UPP synthase structure the similar region that corresponds to the conserved region III has also been unidentified.55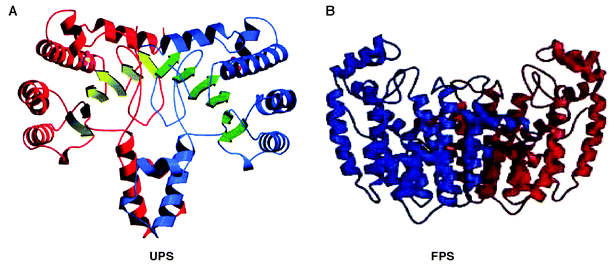 | ||
| Fig. 6 A ribbon representation of (A) M. luteus B–P 26 UPP synthase and (B) avian FPP synthase. (A) Front view of dimeric structure in which α-helices and β-strands of one monomer of UPP synthase are shown in red and yellowish green arrows, and those of the other monomer are shown in blue and green arrows, respectively. A sulfate ion is also shown in each monomer as indicated by small spheres. (B) Front view of avian FPP synthase in which one monomer is shown in blue and the other in red. | ||
3 Catalytic mechanism
In the last 15 years the catalytic mechanism of the TPTs has been elucidated by site-directed mutagenesis56–61 as well as X-ray crystal structural analysis47,62 (with or without substrate bound). The two highly conserved aspartate-rich motifs (DDxxD) characteristic for the trans-prenyl diphosphate synthases are also present in pentalenene synthase, squalene cyclase, 5-epi-aristolochene synthase, and protein farnesyltransferase. The first and second DDxxD motifs in TPTs are very important for binding the diphosphate moieties of the allylic substrate and the homoallylic substrate (IPP) via Mg2+, respectively.On the other hand, the molecular mechanism of CPTs is not yet fully understood. From mutagenesis analysis, Fujikura et al.63 suggested that the Asn-Trp/Phe (NW/F) motif plays an important role in the enzymatic catalysis and substrate binding for cis-prenyl chain elongation. This motif is reminiscent of the Phe-Gln (FQ) motif in Region VI of trans-prenyl chain elongating enzymes, which has been shown to be essential not only for binding of allylic substrate but also for catalytic function.60 From the standpoints of the prenyltransferase reactions, the CPTs and TPTs are similar in that they both catalyze sequential condensations between IPP and allylic diphosphates with the concomitant release of inorganic pyrophosphate in the presence of Mg2+ ion. These NW/F or FQ motif consisting of carbamoyl and aromatic residues may be common and essential parts of the prenyl chain elongating machinery that triggers the consecutive condensation of IPP to produce polyprenyl chains stereospecifically. On the other hand, Pan et al.64 performed site-directed mutagenesis of all conserved acidic residues, Asp and Glu to Ala in the five conserved regions of E. coli UPP synthase. Their results indicated that the highly conserved Asp residue in Region I is critical for catalytic function. Furthermore, the conserved Asp in Region IV and Glu in Region V are essential for IPP binding as well as catalysis. However, these acidic residues seem to be not so important for the allylic substrate binding in CPTs as the DDxxD motifs are in TPTs. It has been shown that TPTs bind with the allylic substrates via a bridge of Mg2+ ion. Although every prenyltransferase requires Mg2+ ions for the catalytic functions, the fact that the optimum concentration of the divalent metal ion for CPTs is relatively lower than that for TPTs, and that there are no similar conserved motifs between CPT and TPT, led us to postulate that a different binding mechanism, i.e. direct binding with the conserved basic residues, may play role for cis-type enzymes. Among the five conserved regions, Region V contains the largest number of amino acid residues, including five highly conserved negatively charged residues and two completely conserved positively charged residues. We have performed site-directed mutagenesis of all the charged residues in Region V and the results indicated that the diphosphate moiety of homoallylic substrate is electrostatically recognized by the RX5RXnE motif in Region V, suggesting that CPTs take a different mode for the binding of IPP from that of trans-prenyl chain elongating enzymes.65 This revealed that positively charged guanidinium groups (R197 and R203) may bind directly with the diphosphate moiety of IPP whereas a carboxyl group (E216) binds, possibly through Mg2+ bridge, similar to that of trans-type enzyme. According to the three-dimensional structure of M. luteus B–P 26 UPP synthase,53 the FS-motif in Region III is located at the position adjacent to the charged triangle formed by R197, R203 and E216. Mutational analysis of the FS-motif revealed their importance in substrate binding as well as catalytic function.65 As predicted by Fujihashi et al. the positively charged amino acid residues in the structural P-loop motif in Region I were found to be critical for binding the diphosphate moiety of FPP as well as for catalysis.66 From the site-directed mutagenesis and X-ray structure analysis, we hypothesized a substrate binding model for M. luteus UPP synthase which is shown in Fig. 7. The model shows that hydrocarbon chain and diphosphate moiety of FPP is recognized by the hydrophobic cleft and structural P-loop motif of the enzyme, respectively. Similarly, the model indicates the importance of five residues, F73, S74, R197, R203, and E216, that bind IPP molecule in the proper direction suitable for stereospecific condensation with FPP. However, the amino acid residues involved in the removal of hydrogen from the 2-position of IPP, pro-R for TPT and pro-S for CPT, have not yet been reported. To enhance the understanding of the entire mechanism of cis-prenyl chain elongation, further X-ray crystallography studies on CPTs with substrate bound are needed.
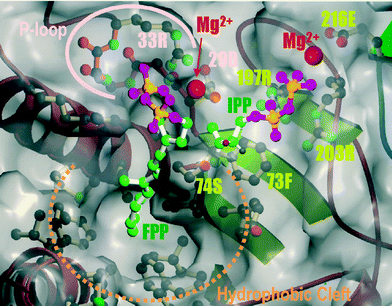 | ||
| Fig. 7 Hypothetical binding model for FPP and IPP for M. luteus B–P 26 UPP synthase. The gray envelope shows the molecular surface of the plausible active site. Carbon, oxygen and phosphorus atoms are shown in green, pink and orange, respectively. The estimated positions of the magnesium ions which are located between the diphosphate moieties of FPP and IPP and carboxyl of 29D and 216E, respectively, are shown as magenta spheres. | ||
4 Chain length determination
The mechanism by which prenyltransferases determine their product chain length has been one of the most interesting subjects for many years. The chain length determination mechanism for some of the TPTs has been investigated precisely. Ohnuma et al. carried out random mutagenesis analysis for B. stearothemophilus FPP synthase67 and S. acidocaldarius GGPP synthase68 and discovered that the bulky amino acid residue located at the fifth position upstream to the first DDxxD motif (FARM) in Region II is the most effective for chain elongation up to the ultimate product. Poulter and his collaborators independently reached a similar conclusion on the product chain length determination mechanism of avian FPP synthase by X-ray crystallographic analysis of the enzyme bound with substrate as well as the mutated enzymes by site-directed mutagenesis.62 Their results have revealed that the Phe-112 and Phe-113, which are situated at the fifth and fourth amino acid upstream to the DDxxD motif in Region II, are important for determining the ultimate length of the hydrocarbon chains. Similarly, Zhang et al.69 and Hirooka et al.70 have shown the important role of the fifth amino acid upstream from the FARM even in the medium chain TPT, HepPP synthase, of B. substilis. In this dissociable heterosubunit enzyme system, both subunits cooperatively participate in product chain length determination.Many reports have shown that CPTs in nature shows distinct product chain length specificities depending on the origin of organisms. For example, bacterial enzyme produces C55 chain length product UPP, whereas yeast enzyme produces C75–C95 products DedolPP, mammalians produce up to C100 chain length dolichols, and plants produce longer dolichols and polyprenyl alcohols up to C120 and natural rubber whose carbon chain length reaches up to several millions. Furthermore, dolichol was found in different chain lengths in various tissues such as liver71 (C90–95) and testis72 (C85–90) in mammals. Some plants show an age dependence of product length, such as young leaves and old leaves of ginko which produce C70–90 and C80–95 compounds, respectively.22 There are many prenyltransferases that show changes in product distribution patterns, shorter or longer than the intrinsic final products under changing some reaction conditions. Pan et al.64 showed that UPP synthase produces longer chains if Triton X-100 is omitted from the reaction mixture. They also indicated that this enzyme produces shorter chain lengths in excess of substrates IPP or FPP. Similar results have also been reported for eukaryotic CPTs. Matsuoka et al.40 have shown that product chain lengths of rat DedolPP synthase are dramatically affected by detergents and phospholipids (such as phosphatidylcholine and deoxycholate) and their concentrations.
Ko et al.55 have reported that mutation of Leu-137 of E. coli UPP synthase to Ala, produces C55 and C60 prenyl chain as a major products in the presence of Triton X-100 and C70 and C75 in the absence of Triton X-100. They suggested that in the hydrophobic tunnel formed by α-helices and β-strands, the “large residue of Leu-137”, which is located on the “floor” of the hydrophobic tunnel, is important for the product chain length determination. This hypothetical mechanism seems to be analogous to that of TPTs in which the residue at the fifth position upstream from the FARM is essential to regulate the product chain length. It is of great interest to understand how CPTs determine the final chain length and by what intrinsic mechanism they can regulate the product chain length. Further investigations to elucidate and confirm the product chain length determination for CTPs might be required for understanding the structure and function of CPTs at the molecular level.
References
- F. Lynen, H. Eggerer, U. Henning and I. Kessel, Angew. Chem., 1958, 70, 738 CAS.
- S. Chaykin, J. Law, A. H. Phillips, T. T. Tchen and K. Bloch, Proc. Natl. Acad. Sci. USA, 1958, 44, 998 CAS.
- S. R. Spurgeon and J. W. Porter, in Biosynthesis of Isoprenoid Compounds, ed. J. W. Porter and S. L. Spurgeon, Wiley, New York, 1981, p. 1 Search PubMed.
- N. Qureshi and J. W. Porter, in Biosynthesis of Isoprenoid Compounds, ed. J. W. Porter and S. L. Spurgeon, Wiley, New York, 1981, p. 47 Search PubMed.
- E. D. Beytia and J. W. Porter, Annu. Rev. Biochem., 1976, 45, 113 CrossRef CAS.
- M. Rohmer, M. Knani, P. Simonun, B. Sutter and H. Sahm, Biochem. J., 1993, 295, 517 CAS.
- M. Rohmer, M. Seemann, S. Horbach, S. Bringer-Mayer and H. Sahm, J. Am. Chem. Soc., 1996, 118, 2564 CrossRef CAS.
- W. Eisenreich, B. Mwnhard, P. J. Hylands, M. H. Zenk and A. Bacher, Proc. Natl. Acad. Sci. USA, 1996, 93, 6431 CrossRef CAS.
- J. Schwender, M. Seemann, H. K. Lichtenthaler and M. Rohmer, Biochem. J., 1996, 316, 73 CAS.
- T. Koyama and K. Ogura, in Comprehensive Natural Products Chemistry, ed. D. Barton and K. Nakanishi, Elsevier Science Ltd., Oxford, 1999, vol. 2, p. 69 Search PubMed.
- M. Kobayashi, M. Ito, T. Koyama and K. Ogura, J. Am. Chem. Soc., 1985, 107, 4588 CrossRef CAS.
- M. Ito, M. Kobayashi, T. Koyama and K. Ogura, Biochemistry, 1987, 26, 4745 CrossRef CAS.
- G. Popjak and J. W. Cornforth, Biochem. J., 1966, 101, 553 CAS.
- J. W. Cornforth, R. H. Cornforth, C. Donninger and G. Popjak, Proc. R.. Soc. (London), Ser. B, 1966, 163, 492 Search PubMed.
- J. W. Cornforth, R. H. Cornforth, G. Popjak and L. Yengoyam, J. Biol. Chem., 1966, 241, 3970 CAS.
- M. C. Schulbach, P. J. Brennan and D. C. Crick, J. Biol. Chem., 2000, 275, 22876 CrossRef CAS.
- W. J. Lannarz and M. G. Scher, Biochim. Biophys. Acta, 1972, 265, 417.
- C. J. Quellhorst, Jr., J. S. Piotrowski, S. E. Steffen and S. S. Krag, Biochem. Biophys. Res. Commun., 1998, 244, 546 CrossRef.
- E. Skoczylas, E. Swiezewska, T. Chojnacki and Y. Tanaka, Plant Mol. Biol., 1994, 32, 825 CAS.
- V. Kulccitsky, J. Hertel, E. Skoczylas, E. Swiezewska and T. Chojnacki, Acta Biochim. Pol., 1996, 43, 707 Search PubMed.
- M. Wanke, T. Chojnacki and E. Swiezewska, Acta Biochim. Pol., 1998, 45, 811 Search PubMed.
- S. Tateyama, R. Wititsuwannakul, D. Wititsuwannakul, H. Sagami and K. Ogura, Phytochemistry, 1999, 51, 11 CrossRef CAS.
- J. W. Rip, C. A. Rupar, N. Chaudhary and K. K. Carroll, Prog. Lipid Res., 1985, 24, 269 CrossRef CAS.
- J. G. Christenson, S. K. Gross and P. W. Robbins, J. Biol. Chem., 1969, 244, 5436 CAS.
- T. Kurokawa, K. Ogura and S. Seto, Biochem. Biophys. Res. Commun., 1971, 45, 251 CrossRef CAS.
- T. Baba, J. Muth and C. M. Allen, Arch. Biochem. Biophys., 1980, 200, 474 CAS.
- I. Takahashi and K. Ogura, J. Biochem. (Tokyo), 1982, 92, 1527 Search PubMed.
- S. Fujisaki, T. Nishino and H. Katsuki, J. Biochem. (Tokyo), 1986, 99, 1327 Search PubMed.
- M. V. Keenan and C. M. Allen, Arch. Biochem. Biophys., 1974, 161, 375 CAS.
- C. M. Allen, M. V. Keenan and J. Sack, Arch. Biochem. Biophys., 1976, 175, 236 CAS.
- T. Baba and C. M. Allen, Biochemistry, 1978, 17, 5598 CrossRef CAS.
- J. D. Muth and C. M. Allen, Arch. Biochem. Biophys., 1984, 230, 49 CrossRef CAS.
- N. Shimizu, T. Koyama and K. Ogura, J. Biol. Chem., 1998, 273, 19476 CrossRef.
- C. R. H. Raetz, Proc. Natl. Acad. Sci. USA, 1975, 72, 2274 CAS.
- C. M. Apfel, B. Takacs, M. Fountoulakis, M. Stieger and W. Keck, J. Bacteriol., 1999, 181, 483 CAS.
- H. Hemmi, S. Yamashita, T. Shimoyama, T. Nakayama and T. Nishino, J. Bacteriol., 2001, 183, 401 CrossRef CAS.
- W. L. Adair, N. Cafmeyer and R. K. Keller, J. Biol. Chem., 1984, 259, 4441 CAS.
- Z. Chen, C. Morris and C. M. Allen, Arch. Biochem. Biophys., 1988, 266, 98 CrossRef CAS.
- Y. E. Bukhtiyarov, Y. A. Shabalin and I. S. Kulaev, J. Biochem. (Tokyo), 1993, 113, 721 Search PubMed.
- S. Matsuoka, H. Sagami, A. Kurisaki and K. Ogura, J. Biol. Chem., 1991, 266, 3464 CAS.
- M. Sato, K. Sato, K. Nishikawa, A. Hirata, J. Kato and A. Nakano, Mol. Cell. Biol., 1999, 19, 471 CAS.
- J. Kato, S. Fujisaki, K. Nakajima, Y. Nishimura, M. Sato and A. Nakano, J. Bacteriol., 1999, 181, 2733 CAS.
- S. K. Oh, K. H. Han, S. B. Ryu and H. Kang, J. Biol. Chem., 2000, 275, 18482 CrossRef CAS.
- N. Cunillera, M. Arro, O. Fores, D. Manzano and A. Ferrer, FEBS Lett., 2000, 477, 170 CrossRef CAS.
- S. Endo, Y.-W. Zhang, S. Takahashi and T. Koyama, submitted.
- K. Asawatraratanakul, Y.-W. Zhang, D. Wititsuwannakul, R. Wititsuwannakul, S. Takahashi and T. Koyama, submitted.
- L. C. Tarshis, M. Yan, C. D. Poulter and J. C. Sacchettini, Biochemistry, 1994, 33, 10871 CrossRef CAS.
- C. A. Lesburg, G. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820 CrossRef CAS.
- K. U. Wendt, K. Poralla and G. E. Schulz, Science, 1997, 277, 1811 CrossRef CAS.
- C. M. Starks, K. Back, J. Chappell and J. P. Noel, Science, 1997, 277, 1815 CrossRef CAS.
- H. W. Park, S. R. Boduluri, J. F. Moomaw, P. J. Casey and L. S. Beese, Science, 1997, 277, 1800 CrossRef CAS.
- J. C. Sacchettni and C. D. Poulter, Science, 1997, 277, 1788 CrossRef.
- M. Fujihashi, Y.-W. Zhang, Y. Higuchi, X.-Y. Li, T. Koyama and K. Miki, Proc. Natl. Acad. Sci. USA, 2001, 98, 4337 CrossRef CAS.
- K. Kinoshita, K. Sadanami, A. Kidera and N. Go, Protein Eng., 1999, 12, 11 CrossRef CAS.
- T. P. Ko, Y. K. Chen, H. Robinson, P. C. Tsai, Y.-G. Gao, A. P.-C. Chen, A. H.-J. Wang and P. H. Liang, J. Biol. Chem., 2001, 276, 47474 CrossRef CAS.
- A. Joly and P. A. Edwards, J. Biol. Chem., 1993, 268, 26983 CAS.
- L. Song and C. D. Poulter, Proc. Natl. Acad. Sci. USA, 1994, 91, 3044 CAS.
- P. F. Marrero, C. D. Poulter and P. A. Edwards, J. Biol. Chem., 1993, 267, 21873.
- T. Koyama, M. Tajima, H. Sano, T. Doi, A. Koike-Takeshita, S. Obata, T. Nishino and K. Ogura, Biochemistry, 1996, 35, 9533 CrossRef CAS.
- T. Koyama, M. Tajima, T. Nishino and K. Ogura, Biochem. Biophys. Res. Commun., 1995, 212, 681 CrossRef CAS.
- T. Koyama, K. Saito, K. Ogura, S. Obata and A. Takeshita, Can. J. Chem., 1994, 72, 75 CAS.
- L. C. Tarshis, P. J. Proteau, B. A. Kellogg, J. C. Sacchetinni and C. D. Poulter, Proc. Natl. Acad. Sci. USA, 1996, 93, 15018 CrossRef CAS.
- K. Fujikura, Y.-W. Zhang, H. Yoshizaki, T. Nishino and T. Koyama, J. Biochem. (Tokyo), 2000, 128, 917 Search PubMed.
- J. J. Pan, L. W. Yang and P. H. Liang, Biochemistry, 2000, 39, 13856 CrossRef CAS.
- Y. Kharel, Y.-W. Zhang, M. Fijihashi, K. Miki and T. Koyama, J. Biol. Chem., 2001, 276, 28459 CrossRef CAS.
- K. Fujikura, Y.-W. Zhang, M. Fijihashi, K. Miki and T. Koyama, submitted.
- S.-i. Ohnuma, T. Nakazawa, H. Hemmi, A.-M. Hallberg, T. Koyama, K. Ogura and T. Nishino, J. Biol. Chem., 1996, 271, 10087 CrossRef CAS.
- S.-i. Ohnuma, K. Narita, T. Nakazawa, C. Ishida, Y. Takeuchi, C. Ohto and T. Nishino, J. Biol. Chem., 1996, 271, 30748 CrossRef CAS.
- Y.-W. Zhang, X.-Y. Li and T. Koyama, Biochemistry, 2000, 39, 12717 CrossRef CAS.
- K. Hirooka, S. Ohnuma, A. Koike-Takeshita, T. Koyama and T. Nisino, Eur. J. Biochem., 2000, 267, 4520 CrossRef CAS.
- K. Yamada, H. Yokohama, S. Abe, K. Katayama and T. Sato, Anal Biochem., 1985, 150, 26 CrossRef CAS.
- K. Yamada, S. Abe, T. Suzuki, K. Katayama and T. Sato, Anal. Biochem., 1986, 156, 380 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |