The chalcone synthase superfamily of type III polyketide synthases
Michael B. Austin and Joseph P. Noel
Structural Biology Laboratory, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, CA 92037, USA
First published on 13th December 2002
Abstract
Covering 1970–2001
This review covers the functionally diverse type III polyketide synthase (PKS) superfamily of plant and bacterial biosynthetic enzymes, from the discovery of chalcone synthase (CHS) in the 1970s through the end of 2001. A broader perspective is achieved by a comparison of these CHS-like enzymes to mechanistically and evolutionarily related families of enzymes, including the type I and type II PKSs, as well as the thiolases and β-ketoacyl synthases of fatty acid metabolism. As CHS is both the most frequently occurring and best studied type III PKS, this enzyme's structure and mechanism is examined in detail. The in vivo functions and biological activities of several classes of plant natural products derived from chalcones are also discussed. Evolutionary mechanisms of type III PKS divergence are considered, as are the biological functions and activities of each of the known and functionally divergent type III PKS enzyme families (currently twelve in plants and three in bacteria). A major focus of this review is the integration of information from genetic and biochemical studies with the unique insights gained from protein X-ray crystallography and homology modeling. This structural approach has generated a number of new predictions regarding both the importance and mechanistic role of various amino acid substitutions observed among functionally diverse type III PKS enzymes.
 Michael B. Austin | Mike Austin was born and reared in Springfield, Missouri. He studied philosophy briefly before engaging science, and received a B.S. in Chemistry from Southwest Missouri State University (SMSU) in 1998. Entering the Chemistry and Biochemistry Ph.D. program at the University of California at San Diego (UCSD), Mike joined Joe Noel's group in the Structural Biology Laboratory at the Salk Institute for Biological Studies. Mike's current interests include the use of such tools as X-ray crystallography, site-directed mutagenesis, and biochemical characterization to investigate the structural basis for the mechanistic functional divergence of evolutionarily related natural product biosynthetic enzymes, accompanied by the application of this knowledge to the engineering of both known and novel enzymatic activities of interest. |
 Joseph P. Noel | Joe Noel was born and raised in rural western Pennsylvania. Upon obtaining a B.S. degree in Natural Sciences with a concentration in Chemistry from the University of Pittsburgh at Johnstown, Joe entered the graduate program in Chemistry at the Ohio State University in the summer of 1985, and graduated from the Chemistry Department in 1990 with a PhD centered on the examination of the mechanistic enzymology of phospholipases with Professor Ming-Daw Tsai. Joe then completed his postdoctoral training with the late Paul B. Sigler in the Department of Molecular Biophysics and Biochemistry at Yale. While at Yale University, he undertook the X-ray crystallographic examination of heterotrimeric G-proteins. Joe and his group are now utilizing a combination of traditional mechanistic enzymology, molecular biology, plant biology, and tools in structural biology including protein X-ray crystallography and NMR to decipher the structure, function, and evolutionary lineage of a large number of enzymes that act in plant cells and many microorganisms to produce biologically active natural products including terpenes, polyketides, and alkaloids. Armed with the three-dimensional structure of the enzymes in plant cells responsible for the creation of this diverse array of bioactive compounds, his group is also working to engineer new specificities into these pathways to create novel products using a structurally-guided approach. |
1 Introduction and background
Covering type III polyketide synthase (PKS) research between the discovery of chalcone synthase (CHS) in the1970s through the end of 2001, this review additionally touches on related and ancestral enzymes, and discusses the biological and medicinal applications of the natural products of type III PKS pathways. A recurring theme of this review integrates information from genetic and biochemical studies with the unique insights gained from both protein X-ray crystallography and homology modeling to formulate the structural basis for the functional diversity observed within the type III PKS superfamily of enzymes.1.1 Metabolism and natural product biosynthesis
Natural products include a diverse collection of small molecules, which confer upon their hosts a multitude of attractive, defensive, communicative, or other biological advantages. An organism's collection of natural products often determines both intra- and inter-species interactions, thus contributing to both the identity and individuality of a species. Remarkably, most natural products are built up from a handful of simple building blocks, usually derived from one or more primary metabolic pathways. For example, each of the three types of PKSs catalyzes the sequential condensation of two-carbon acetate units derived from a malonate thioester into a growing polyketide chain, in a reaction sequence that mirrors the biosynthetic pathway of the fatty acid synthase (FAS) enzymes of primary metabolism. This review focuses on the third class of PKS (also known as CHS-like PKSs), traditionally associated with plants but recently discovered in a number of bacteria.1.2 The chalcone synthase/stilbene synthase (CHS/STS) superfamily of plant and bacterial type III polyketide synthases
As a ubiquitous enzyme in higher plants, CHS provides the first committed step in flavonoid biosynthesis by catalyzing the sequential decarboxylative addition of three acetate units from malonyl-CoA to a p-coumaroyl-CoA starter molecule derived from phenylalanine via the general phenylpropanoid pathway. In the same active site, CHS then forms chalcone via the intramolecular cyclization and aromatization of the resulting linear phenylpropanoid tetraketide (Fig. 1). Downstream enzymes in branching pathways variously modify this chalcone scaffold to produce a number of biologically important compounds. These plant natural products are utilized for such diverse purposes as antimicrobial defense, anthocyanin flower pigmentation, UV photoprotection, pollen fertility, and induction of symbiotic root nodulation by Rhizobium bacteria leading to nitrogen fixation.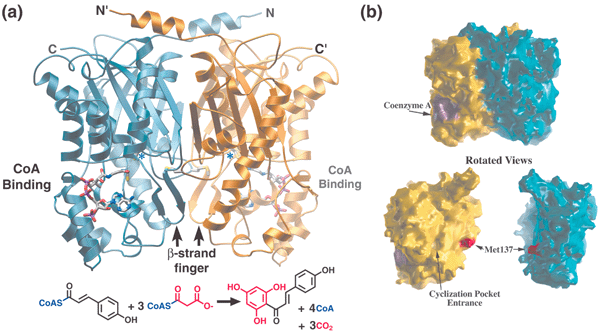 | ||
| Fig. 1 Structure, CoA binding, and overall reaction of chalcone synthase (CHS). (a) Each monomer is colored in gold and blue, respectively, in this ribbon diagram. The N- and C-termini for each monomer are indicated. CoA is depicted as a stick diagram while the position of the active site cysteine is highlighted by *. The bottom panel shows the overall reaction catalyzed by CHS with the malonyl derived portions of chalcone shown in red. (b) Molecular surface representation of the CHS-CoA complex oriented as shown in (a). In the bottom panel, the two CHS monomers are separated and rotated slightly to highlight the flat dimerization interface along with the methionine side-chain and dyad related hole in the backside of the CHS active site. | ||
Aside from the biosynthetic diversity provided by downstream tailoring enzymes, duplication and functional divergence of the CHS gene in plants has given rise to an expanding superfamily of homologous enzymes, many of which have only recently been cloned or functionally characterized. These type III PKS enzymes can differ from CHS in their preference for starter molecules, the number of acetyl additions they catalyze, and their mechanism of chain termination including alternative patterns of intramolecular cyclization, as is the case with the stilbene synthases (STSs). Because this important class of divergent CHS-like enzymes was discovered not long after CHS, known type III PKS enzymes are often referred to as the CHS/STS superfamily. Only a few years ago, less than five bacterial gene sequences in sequence databases were annotated as CHS-like. None of the enzymes encoded by these original depositions had been functionally characterized. Their scarcity, lower sequence homology, and the uncertain evolutionary history of type III PKS enzymes prompted speculation that these few bacteria had only recently acquired CHS-like genes via horizontal transfer from plants. However, the recent explosion of genome sequences has revealed many more bacterial CHS-like sequences, most sharing about 25% amino acid sequence identity with CHS and with each other. Only a small number of these bacterial enzymes have been functionally characterized to date, but this work has uncovered three new type III biosynthetic activities, and demonstrated the involvement of bacterial CHS-like enzymes in important secondary metabolic pathways. Given the rapid expansion in the number of deposited bacterial and plant CHS-like sequences, the initial elucidation of their biosynthetic capabilities, and the availability of high resolution structural information for some members of this family, increased attention has focused on these enzymes, resulting in a deeper appreciation for the CHS/STS superfamily and the general acceptance of the type III PKS designation.
While this review covers the essential features of several decades of type III PKS superfamily research, the reader is in many places referred to older or more focused reviews for more detailed information and additional primary references. This approach allows us to integrate recent insights into the structural basis of functional diversity within the type III PKS superfamily with a wealth of phytochemical and biological information. Clues obtained from studies of several functionally divergent type III enzymes, combined with mechanistic knowledge gained from recent crystal structures of CHS and related condensing enzymes, provide a fascinating example of the evolutionary process of metabolic divergence, an important contributor to the remarkable diversity of life on earth. Additionally, the comprehensively annotated primary sequence alignments of known divergent type III PKS enzymes presented here, viewed in light of this structural and functional information, should facilitate more reliable annotations and more appropriate selections of existing and newly discovered type III PKS gene sequences for biochemical analysis.
The remainder of Section 1 provides an introduction to related enzymes of both primary and secondary metabolism, and summarizes insights derived from various crystallographic and mechanistic studies of these PKS relatives. Section 2 covers CHS and summarizes the branching metabolic pathways associated with CHS products, while Sections 3 and 4 address other plant and bacterial type III PKS enzymes, respectively. A summary and concluding remarks are provided in Section 5.
1.3 PKS ancestors and related enzymes
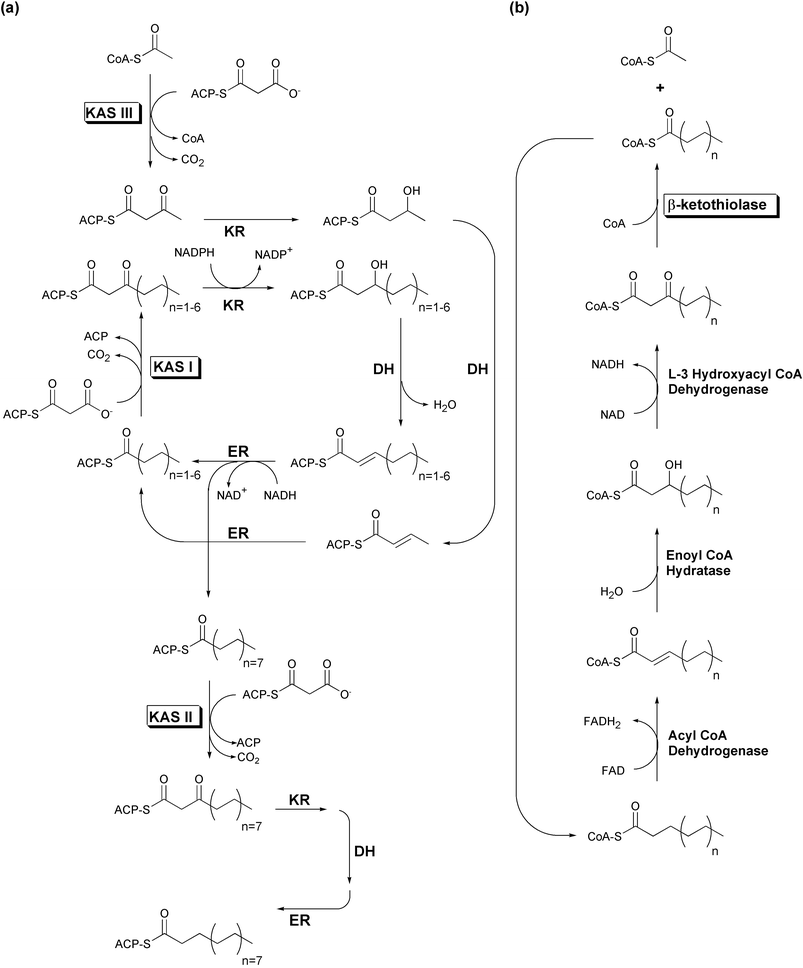 | ||
| Fig. 2 Simplified E. coli fatty acid metabolism. Boxed labels denote enzymes possessing the αβαβα-fold. (a) Fatty acid biosynthesis. (b) Fatty acid degradation. | ||
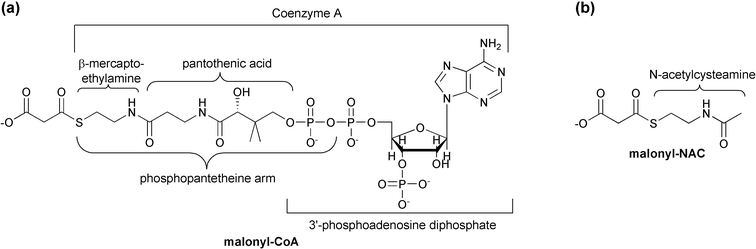 | ||
| Fig. 3 (a) Structure and composition of malonyl-CoA. Note the phosphopantetheine arm also utilized by ACP (acyl carrier protein). (b) A NAC (N-acetylcysteamine) synthetic thioester. | ||
While short chain acyl-CoAs such as acetyl-CoA are common end products of various degradative pathways, CoA is not the preferred carrier for most FAS biosynthetic enzymes. Substrates must first be activated through transfer to an ACP by an acyltransferase (AT) activity. This activity is sometimes labeled as a malonyl acyltransferase (MAT) to reflect its additional role in the transfer of the malonyl unit to ACP whereupon it is used for polyketide chain extension. Following the transfer of the substrate to the ketoacyl synthase (KAS or KS) domain's catalytic cysteine, this condensing enzyme catalyzes the addition of a two-carbon acetate unit to the enzyme bound thioester end of the fatty acid, via a decarboxylative condensation with malonyl-ACP. The resulting ACP-bound β-ketoacyl thioester is presented to an NADPH-dependent β-ketoacyl-ACP reductase (KR), which reduces the original substrate carbonyl (now the β-keto carbonyl) to an alcohol. A β-hydroxyacyl dehydratase (DH) catalyzes loss of water, leaving a carbon–carbon double bond. An NADH-dependent enoyl-ACP reductase (ER) module completes the reduction of the β-carbon, resulting in an acyl-ACP that resembles the original substrate, but with two additional methylene moieties. Most FAS complexes perform several cycles of elongation before a thioesterase (TE) activity releases the product as a free fatty acid. In vivo, it can be difficult to assess whether the final product length specificity of a FAS system depends more upon its thioesterase or its KAS activity.
Fatty acid biosynthetic systems are divided into two classes based upon genetic organization. Type I FAS systems, found in animals and fungi, encode the above activities in distinct domains on one or two multifunctional, multidomain protein chains. For example, while mammalian FAS activities are encoded in a single polypeptide that functions as a homodimer,5 the yeast FAS activities are distributed across two polypeptide chains (α and β), functioning as a multimeric complex incorporating six copies of each chain.1 Conversely, plants and most bacteria instead have type II fatty acid biosynthetic systems. These pathways use similar enzymes to carry out the same functions as type I FAS systems. However, each of the type II FAS activities is catalyzed by a distinct and dissociable enzyme.3 Although some evidence from plant plastids suggests that type II FAS enzymes do associate in vivo, unequivocal stoichiometric information is not yet available.6
The FAS system of E. coli has been intensively studied, yielding many insights into type II fatty acid biosynthesis.3 Three different homodimeric KAS enzymes, varying in specificity for both substrate and product length, are found in E. coli (Fig. 2(a)). KAS I (FabB) elongates C4 fatty acids to C16 acyl chains, while KAS II (FabF) expands these C16 fatty acids to C18 acyl chains through the addition a single acetate unit. KAS III (FabH) initiates this process by elongating acetyl-CoA to a C4 intermediate, thus activating C2 fatty acids for entry into the iterative KAS I fatty acid biosynthetic cycle. All three E. coli KAS enzymes use decarboxylative condensations of malonyl-ACP to extend acyl substrates, but while KAS I and II utilize acyl-ACP starters as well, KAS III is unique in that it acts directly upon an acyl-CoA starter, but releases its product as an ACP thioester.
Interestingly, pathogenic Mycobacterium tuberculosis bacteria possess both type I and type II FAS systems. A conventional type I FAS produces C16–C24 long chain fatty acids, some of which are further extended to C50–C56 acyl products by a type II FAS system. The products of both FAS systems are combined in a high molecular weight α-alkyl-β-hydroxy fatty acid called mycolic acid (Fig. 4), which serves as a unique and critical component of the mycobacterium cell wall. As in E. coli, the initial biosynthetic step catalyzed by the type II FAS is carried out using a KAS III enzyme that utilizes malonyl-ACP. However, the mycobacterial KAS III is inactive against acetyl-CoA, and instead elongates a C12 acyl-CoA product of the type I FAS.7
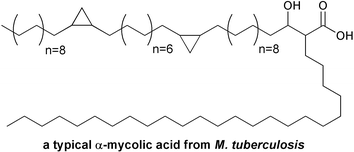 | ||
| Fig. 4 Branched structure of a typical mycolic acid from Mycobacterium tuberculosis. | ||
The iterative fatty acid β-oxidation degradative (FAD) pathway, although it uses CoA-instead of ACP-tethered substrates, is essentially the reverse of the FAS biosynthetic process, generating the same intermediate forms as each β-carbon is successively oxidized from a methylene to a ketone (Fig. 2(b)). However, rather than a KAS condensing enzyme, β-oxidation pathways utilize the distantly related 3-ketoacyl-CoA thiolase to eliminate the original carboxyl group and adjacent methylene of the oxidized β-ketoacyl-CoA, as an acetyl-CoA molecule. While 3-ketoacyl-CoA thiolase catalyzes both the forward and reverse reactions, thermodynamic equilibrium favors the degradative process.10
Biosynthetic thiolases maintain a much narrower substrate tolerance than the iterative FAD thiolases, converting two acetyl-CoA molecules into acetoacetyl-CoA, or vice versa. The condensing reaction of thiolase-II is utilized by eukaryotes for the biosynthesis of ketone bodies, cholesterol and steroid hormones, and by some prokaryotes for the production of their primary energy storage molecule, poly-3-D-hydroxybutyrate.11,12 Biosynthetic thiolases differ from FAS and PKS condensing enzymes most notably in their method of generating the acetyl carbanion moiety used in the Claisen condensation reaction catalyzed by all three families of enzymes. While KAS and PKS enzymes achieve this via the decarboxylation of malonyl-CoA or malonyl-ACP, thiolases use a second active site cysteine in an anionic thiolate form to abstract a proton directly from acetyl-CoA.13
1.4 Three types of polyketide synthase
All PKSs, like their FAS ancestors, possess a β-keto synthase (KS) activity that catalyzes the sequential head-to-tail incorporation of two-carbon acetate units into a growing polyketide chain. However, whereas FAS systems perform reduction and dehydration reactions on each resulting β-keto carbon to produce an inert hydrocarbon, PKS systems omit or modify some of these latter reactions, thus preserving varying degrees of polar chemical reactivity along portions of the growing linear polyketide chain. Various PKS enzymes selectively exploit the reactivity of polyketide intermediates to promote intramolecular cyclization and π-bond rearrangements, generating an amazingly diverse collection of substituted monocyclic and polycyclic products from a simple acetyl building block. The major classes of PKSs are briefly described here, but the reader is referred to a number of outstanding and current reviews14–20 for additional information regarding the extensive literature on polyketide biosynthesis and its comparison to fatty acid metabolism.21PKSs have traditionally been divided into two classes by the same genetic organization criteria used to classify FAS systems. The enzymology and genetic organization of PKS types I and II, found in the biosynthetically prolific fungi and also in some bacteria, suggest that they evolved via FAS pathway gene duplication and partial loss of function, or perhaps, in some cases, more directly through partial gene duplication.
Type I PKSs resemble the yeast and animal FASs, consisting of multi-domain polyproteins that form large multi-functional biosynthetic complexes and act in either a modular or iterative fashion. Erythromycin biosynthesis in the bacterium Saccharopolyspora erythraea is the best-characterized example of a modular type I PKS enzymatic process, with two sets of most or all of the four core FAS domains (described in Section 1.3) encoded on three ∼350 kDa polypeptides, each linearly arranged set catalyzing a single round of elongation and full or partial reduction (reviewed in Refs. 14,17,19). The first module is preceded by AT and ACP domains, and the last module is followed by a TE domain for product off loading. Conversely, the biosynthesis of 6-methylsalicylic acid (6-MSA) in the fungus Penicillium patulum is carried out by an iterative, homotetrameric type I PKS, which uses a single copy each of KS, MAT, DH, KR and ACP to link four acetate units that cyclize via an intramolecular aldol condensation and aromatization (reviewed in Refs. 14,18,20). Interestingly, the KR and DH domains act upon only one of the four ketone moieties on the elongated polyketide.
In contrast, carbon flow through type II PKSs is directed by putative multienzyme complexes consisting of discrete, separable proteins, like the FAS type II systems found in plants and bacteria. Although enzymes of this second class are dissociable, and not always encoded in a linear fashion within the genome, they are believed to form in vivo complexes similar to those of the type I PKS systems. Type II PKS complexes lack the AT domains seen in FAS and type I PKS enzymes, but nonetheless possess acyltransferase activity. Actinorhodin biosynthesis in Streptomyces coelicolor (reviewed in Refs. 14,15) is catalyzed by a typical type II PKS, which uses six gene products to fashion eight acetate units into three fused, substituted six-membered rings (two aromatic carbon rings and a lactone). Three of these subunits, a KR, an aromatase (ARO), and a cyclase (CYC), are reaction-specific, while the remaining three subunits, an ACP, a KS, and a third domain with KS homology, comprise the iterative “minimal PKS” common to all type II PKS systems. This mysterious third domain was originally labeled a “chain length factor” (CLF), as swapping this domain between various type II minimal PKS complexes alters polyketide chain length. However, subsequent analysis noted that the missing catalytic cysteine is replaced by a glutamine in all known CLF subunits, a substitution that converts KS domains into efficient decarboxylases. This decarboxylation activity of the so-called CLF was subsequently proposed to provide the KS module with an acetyl starter, and the CLF was dubbed a “chain initiation factor” (CIF). A more recent and neutral label for this domain is KS-β, with the actual ketosynthase designated KS-α. KS-α and KS-β are translationally coupled and form a heterodimer. This is unusual among ketosynthase domains, as even the multidomain type I FAS and PKS polypeptides appear to associate in such a way as to allow a homodimeric arrangement of their KS domains.
NMR structures of ACP domains from E. coli and B. subtilis FAS22–24 and Streptomyces PKS25 have been solved, revealing subtle structural differences consistent with the observed inability of ACPs to substitute for one other.25 Additionally, crystallographic efforts with individual PKS domains such as the macrocycle-forming thioesterase from erythromycin biosynthesis are beginning to bear fruit.26 However, the lack of detailed information about the three-dimensional organization of the biosynthetic complexes of PKS types I and II limits our understanding of the structural determinants of substrate preference, polyketide chain length, and cyclization specificity in these large multi-enzyme complexes. Questions of PKS quaternary structure become even more complicated when enzymes are recruited from other biosynthetic pathways to act on polyketide intermediates, thus tailoring the chemical and biological properties of the final products.
Conversely, type III or CHS-like PKS enzymes maintain a simple architecture (a homodimer of identical KS monomeric domains—see Fig. 1), making them much more amenable to in vitro examination and manipulation as well as detailed structural analyses. Recent structural evidence,27 summarized in Figs. 5 and 6, and discussed in Section 1.5, suggests that type III PKS enzymes emerged by gain of function from the structurally similar homodimeric KAS III. As was described in Section 1.3.2, KAS III initiates type II FAS biosynthesis by adding a single acetate unit to a small starter molecule, via a decarboxylative condensation with malonyl-ACP. In contrast, type III PKS enzymes have acquired multiple activities to catalyze varying numbers of iterative condensations utilizing a diverse set of much larger starter molecules. Moreover, these mechanistically complex type III PKSs have further developed three distinct polyketide cyclization mechanisms, all the while maintaining a simple homodimeric organization of KS monomers.
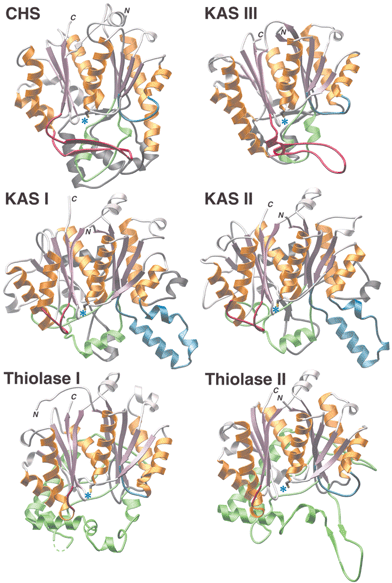 | ||
| Fig. 5 Structural similarities and differences among αβαβα-fold enzyme families. The conserved αβαβα-folds are shown in rose and gold. Use of blue, green, red, or charcoal for additional highlighting facilitates comparisons by denoting specific insertion points into the primary sequence of the conserved fold. Blue asterisks mark the locations of conserved catalytic cysteines. | ||
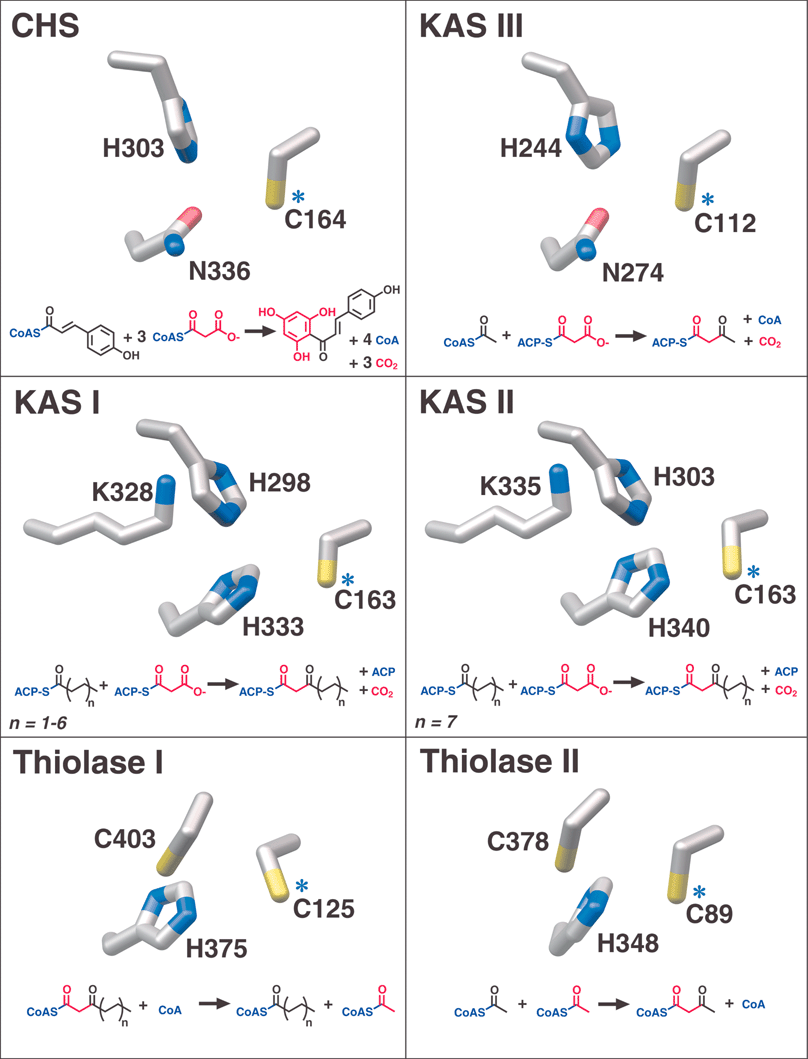 | ||
| Fig. 6 Catalytic active site residues and overall reactions of αβαβα-fold enzyme families. Blue asterisks identify the conserved catalytic cysteines also denoted in Fig. 5. Red is used to track the incorporation or removal of acetate units in the various reactions. | ||
In at least one case, a type III PKS enzyme (CHS) has recruited an NADPH-dependent monomeric ketoreductase28 to specifically reduce to an alcohol one carbonyl position in either a linear polyketide intermediate or the cyclized trione. The resulting alcohol is eliminated as water in the subsequent aromatization that produces chalcone. This ketoreductase was called chalcone reductase (CHR), but the name polyketide reductase (PKR)29 has been championed due to the enzyme's inactivity towards chalcone itself. While “chalcone reductase” is a somewhat misleading label, ketoreduction is also invoked in the proposed type III PKS catalyzed biosyntheses of several other plant natural products (see Section 3.3), suggesting that PKR is an overly general name for the CHS-specific ketoreductase. Although the synthesis of 6′-deoxychalcone by the combined action of CHS and CHR has been amply demonstrated in vitro, evidence supporting the formation of a CHS-CHR complex remains elusive, and CHR access to the buried CHS active site is improbable. Most likely, CHR acts on a diffusible linear CoA-thioester intermediate or an exchangeable and cyclic trione product (discussed in Section 2.2.2). Interestingly, CHR is more closely related to carbohydrate reductases than to the analogous KR domains of FAS and PKS types I and II.30
1.5 Structural comparison of thiolase-fold enzymes illuminates type III PKS evolution and function
Pre-structural bioinformatic analysis successfully predicted the roles and identities of some catalytic residues and hinted as to the evolutionary relationships between condensing enzymes.31 The αβαβα fold, apparently common to all β-ketoacyl synthases and thiolases, was first observed in the 1994 structure of a homodimeric yeast 3-ketoacyl-CoA thiolase.9 Subsequently, in 1998, the crystal structure of E. coli KAS II (FabF) confirmed the conservation of the αβαβα fold in this condensing enzyme.32Along with a higher resolution structure of yeast thiolase,8 structures of a tetrameric biosynthetic thiolase from the prokaryote Zoogloea ramigera were elucidated, in various unliganded, CoA-bound, and cysteine-acetylated forms, thus facilitating improved modeling of thiolase reaction intermediates.11,13 Multiple crystal structures of all three condensing enzymes from the model E. coli type II FAS system, namely KAS I (FabB),33–35 KAS II (FabF),32,36 and KAS III (FabH),27,37,38 are also now available, as are the structures of Synechocystis KAS II39 and the divergent Mycobacterium tuberculosis KAS III enzyme described in Section 1.3.2.40 Both apo and complexed forms of most of these later enzymes are known, and some of the functional hypotheses suggested by individual structures have been tested by mutagenesis.41–43
In 1999, our laboratory published the first crystal structure of a type III PKS, a CHS from alfalfa, in both apo and complexed forms.44 Additional structures of various CHS mutants divulged further details of the condensation mechanism.45 Subsequently, the structure of a 2-pyrone synthase (2-PS) from daisy sharing 74% amino acid identity with CHS confirmed anticipated alterations in the active site volume available to polyketide intermediates.46 Most recently, the as-yet unpublished structures of two stilbene synthases (STSs) elucidated in our laboratory revealed the structural basis for these enzymes' divergent aldol-based cyclization mechanisms.
Comprehensive comparisons of thiolase-fold enzymes accompany two of the most recent structural studies.34,39 Comparison of structural and mechanistic traits allows the grouping of structurally characterized thiolase-fold enzymes into three families (Figs. 5 and 6). One group encompasses KAS I and II, another group consists of KAS III and the CHS-like type III PKS enzymes, and the final group contains the biosynthetic and degradative thiolases. The features shared by all of these enzymes include conservation of the core αβαβα structural architecture, the location of the extensive dimerization interface, placement of the active site, and use of the same catalytic cysteine residue for covalent attachment of substrates and intermediates (Fig. 6). Differences include the extent and structure of the loops located on the opposite side of the active site cavity from the conserved αβαβα core, the number of active site residues contributed by the dyad-related monomer, the position and identity of key catalytic residues (excluding the universally conserved cysteine), and substrate preference for either CoA-linked or ACP-linked thioesters. The observed differences in the catalytic machinery of thiolase-fold enzyme families account for their varied susceptibility to inhibition by the antibiotic cerulenin, a feature historically exploited to help classify these related enzymes.35
The homology-based structure/function analysis of divergent type III PKS enzymes presented in this review relies heavily on comparisons of both conserved and varied active site residues, both within the type III PKS enzyme family and between CHS-like enzymes and their closest structural and functional relatives, the ancestral KAS III enzymes of primary metabolism. For consistency, references to type III PKS residues throughout this review will utilize the numbering of the structurally characterized Medicago sativa CHS2 enzyme,44 unless otherwise specified.
2 Chalcone synthase (CHS) and its biosynthetic pathways
2.1 CHS introduction
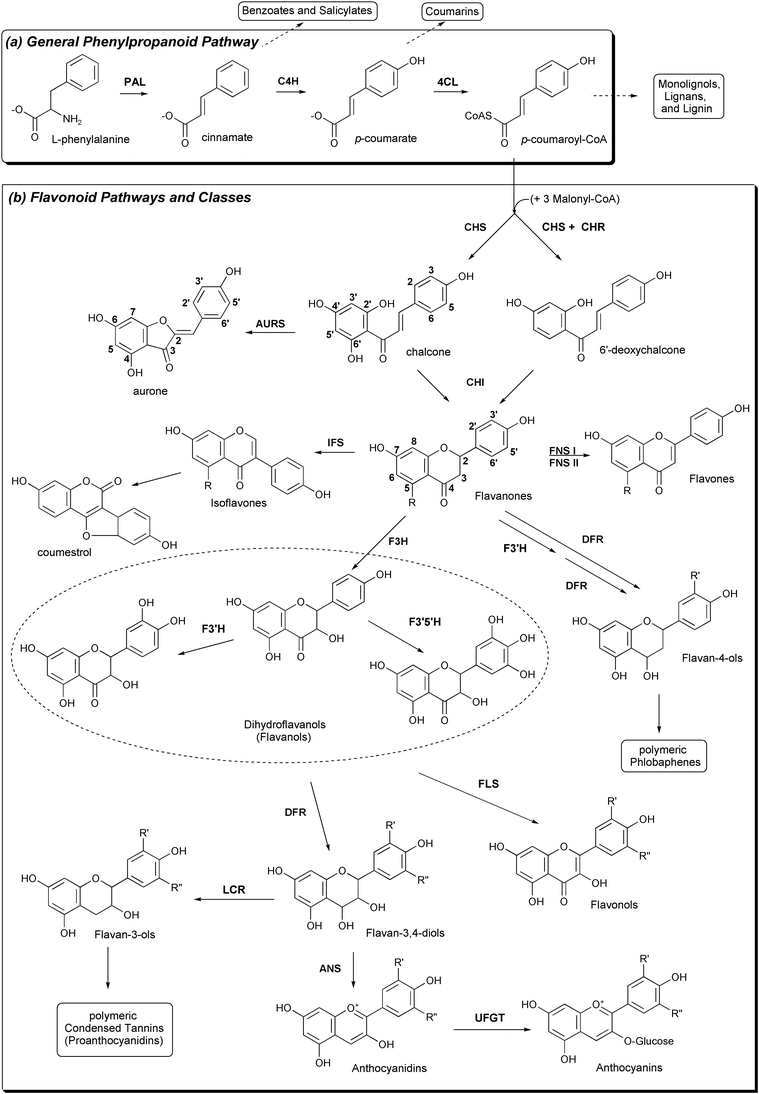 | ||
| Fig. 7 Plant pathways relevant to CHS and resulting classes of natural products. (a) The general phenylpropanoid pathway. (b) CHS and its importance in flavonoid biosynthesis. Note the different numbering conventions for chalcones and flavanones. | ||
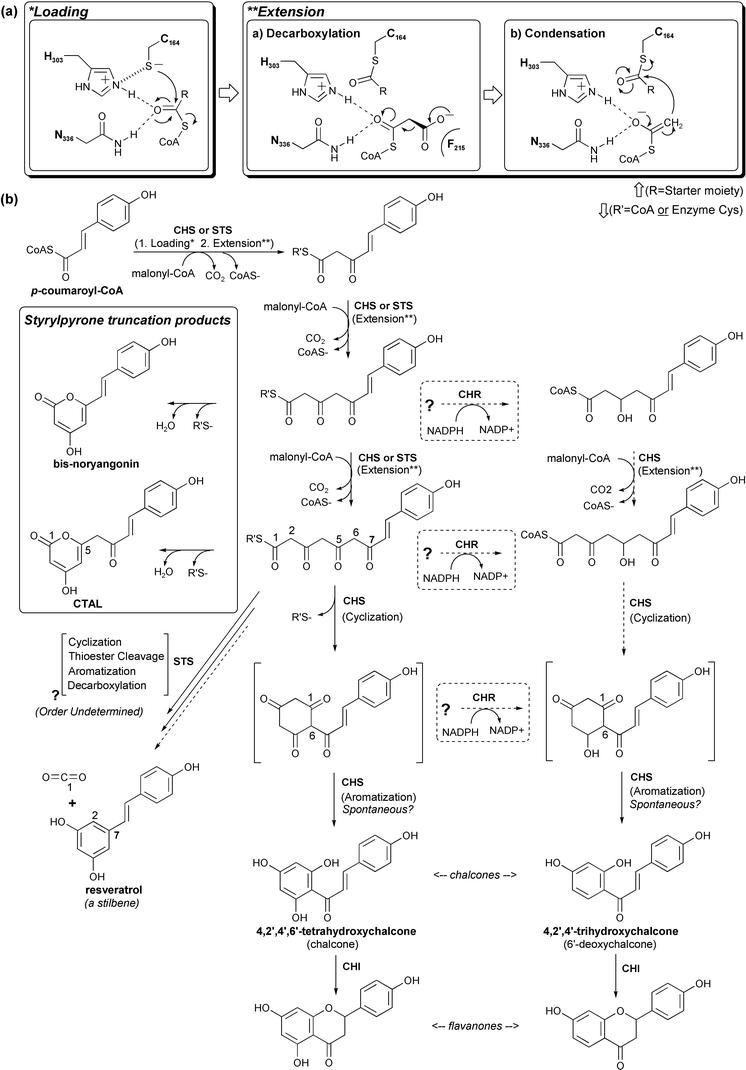 | ||
| Fig. 8 CHS chemistry, reaction intermediates, and related products. (a) Our current understanding of CHS substrate loading, malonyl decarboxylation, and polyketide extension. (b) CHS reaction intermediates, truncation products, and possible routes to reduced chalcones are shown in the bottom panel. The related STS reaction (see text) is included for comparison. | ||
While CHS is present in all gymnosperms and angiosperms examined thus far, the distribution and specificity of downstream tailoring enzymes varies dramatically across different species, within different tissues, and at different times during the life cycle of an individual plant. Due to this biochemical diversity, most plants maintain multiple copies of chs genes,52 which are expressed in various tissues at different developmental stages. Some of these isoenzyme genes are constitutively expressed, while others are transcriptionally induced by environmental stress including UV light, wounding, and pathogen infestation.53–55 These diverse expression patterns reflect the wide range of biological roles fulfilled in plants by products of the downstream flavonoid metabolic grid.56
Although chalcone synthase catalyzes the first committed step of the multi-branched flavonoid pathway, CHS is one of several enzymes using products of the upstream general phenylpropanoid pathway, which synthesizes p-coumaroyl-CoA from L-phenylalanine in three enzymatic steps (Fig. 7). Aside from flavonoid biosynthesis, intermediates and products of the general phenylpropanoid pathway are diverted to synthesize benzoic acid, salicylic acid, coumarins, and the monolignol precursors of lignin, an important structural and defensive polymer.55,58 Evolution of lignin biosynthesis appears to have coincided with the movement of plants onto land; rudimentary flavonoid biosynthesis may have emerged soon after, co-opting phenylpropanoid substrates to protect the new land plants from harmful UV rays. Alternatively, early flavonoids may have first been used as signaling molecules or anti-microbial phytoalexins.63
Plants have recruited a number of tailoring enzymes to modify the flavonoid scaffold (summarized in Fig. 7 and discussed below). Further diversity is achieved by various combinations of hydroxylation, O-methylation, glycosylation, acylation, prenylation, and conjugation at multiple positions, resulting in the identification so far of over 6000 naturally occurring flavonoids.64 Flavonoid nomenclature can be confusing, as chalcones use a different numbering convention than other flavonoids. The carbons of chalcones' phenylpropanoid-derived rings are numbered 1–6 and the acetate-derived ring is numbered 1′–6′. Conversely, in the flavanone naringenin (and downstream flavonoids), the acetate-derived and middle rings are numbered 1–10, and the phenylpropanoid-derived ring is renumbered 1′–6′ (Fig. 7).
The chalcone product of CHS is stereospecifically isomerized by chalcone isomerase (CHI), forming the central heterocyclic ring of the flavanone (2S)-naringenin. The sequential oxidation, reduction, and dehydration of this central ring by flavanone 3-hydroxylase (F3H), dihydroflavonol reductase (DFR), and anthocyanidin synthase (ANS) [also known as leucoanthocyanidin dioxygenase (LDOX)], produces anthocyanidin (Fig. 7). Glycosylation of this aglycone yields the red pigment anthocyanin (reviewed in Ref. 59). Some species utilize additional B-ring hydroxylation of upstream intermediates, by flavonoid 3′ (or 3′5′) hydroxylase (F3′H and F3′5′H) P450s, to alter the color patterns of resultant anthocyanin pigments (Fig. 7). Anthocyanin pigments are transported to and accumulate in vacuoles, where pH and conjugation or complexation (with metals, malonic acid, or other flavones) enhances or modifies their colors to yield orange, pink, red, purple, or blue pigmentation. Flower color is an important determinant of a number of interspecies interactions: bees, for example, are attracted to blue flowers.56
Other tailoring enzymes utilize intermediates in the anthocyanidin pathway to create additional classes of flavonoids (reviewed in Refs. 56,57 and summarized in Fig. 7). The CHI-catalyzed flavanone formation can be blocked by methylation of 6′-deoxychalcone's 2′-hydroxyl moiety. Alternatively, chalcone can be modified to produce aurones. Flavones are produced from flavanones by flavone synthase (FS or FNS) I and II, and in legumes isoflavones result from oxidation and a dramatic rearrangement of the flavanone carbon skeleton through the action of isoflavone synthase (IFS), which shifts the B ring from C-2 to C-3 on the lactone ring. Downstream modification of this isoflavonoid scaffold produces coumestrol and dihydropterocarpan natural products.
Flavonol synthase (FLS) converts dihydroflavanols into flavonols, and leucoanthocyanidin reductase (LCR) converts flavan-3,4-diols into flavan-3-ols. Both of these products are conjugated to each other forming condensed tannins (proanthocyanidins). It is important to note that the seemingly trivial introduction of a C2–C3 double bond by many of these enzymes transforms the non-planar flavanone and dihydroflavanol anthocyanidin pathway intermediates into planar molecules, and also completes the conjugation of the previously separate A- and B-ring π-systems.
In legumes, a branched pathway results from the regiospecific reduction of a CHS polyketide intermediate to an alcohol by the so-called chalcone reductase (CHR) enzyme introduced in Section 1.4. The resultant hydroxyl group is eliminated during the final aromatization step of the CHS reaction, producing 4,2′4′-trihydroxychalcone (commonly referred to as 6′-deoxychalcone). This product is also a candidate for methylation as well as the CHI and IFS reactions discussed above. (CHR is further discussed in the context of the CHS mechanism, in Section 2.2.2)
While CHI appears to possess a unique fold unrelated to any other enzyme of known function,65 most of the other flavonoid tailoring enzymes belong to one of three common enzyme families: NADPH-dependent reductases, oxoglutarate-dependent dioxygenases, or cytochrome P450 hydroxylases.57 Several lines of evidence implicate the co-expression and co-localization of most of these enzymes to the endoplasmic reticulum, as well as the in vivo channeling of intermediates ( i.e. flavonoid pathways are not dependent on a cytoplasmic diffusion of reactive or unstable intermediates). Further evidence (reviewed in Ref. 57) suggests the temporally regulated occurrence of loosely associated multi-enzyme complexes consisting of these predominantly cytoplasmic (soluble) proteins, perhaps on or near the membrane-associated P450 enzymes. However, no such complexes have yet been observed in vitro, even in the case of CHS and CHR, where the apparent exchange of reactive intermediates would suggest a particularly intimate interface.
The distribution and substrate specificity of many of these enzymes vary in different tissues and in different species. As a result, multi-branching pathways sometimes form metabolic grids that compete with each other for substrates, resulting in a diverse mixture of flavonoid products. For example, while DFR, FLS, F3′H, and F3′5′H might all potentially compete for the dihydroflavonol product of F3H, FLS is also able to accept the hydroxylated products of F3′H and F3′5′H as substrates. In some cases, mixtures of flavonoids are known to exhibit synergistically enhanced activities. On the other hand, some branch point enzymes are developmentally or constitutively expressed, while others are only transiently upregulated in response to a specific environmental cue.58,66
Most flavonoids absorb harmful UV-B radiation (280–315 nm). Almost all green leaves contain epidermal flavonoids, especially flavonols and anthocyanin pigments (chlorophyll masks their color) (reviewed in Ref. 56), and mutant plants lacking these epidermal flavonoids are particularly sensitive to UV-B damage.67 Besides acting as a sunscreen, various 3′4′-dihydroxyflavonoids (downstream of F3′H) are also effective free radical scavengers. Plants with UV-resistant genotypes or with sustained exposure to UV radiation generally maintain higher ratios of 3′,4′-hydroxylated to 4′-hydroxylated flavonoids.56
Although a few biological or medicinal properties are confined to (or enhanced in) specific flavonoid subclasses, more often a particular activity or a set of related physiological activities will be represented by a distribution of variously modified natural products from different flavonoid subclasses. In other words, different species often arrive at different phytochemical solutions to the same or similar biological problems, in the form of varied flavonoid products exhibiting different combinations of chemical constituents. This kind of biological diversity also occurs within individual species, and is expected to confer an adaptive advantage to plants. Rapid elaboration of a plant's chemical repertoire makes intuitive sense, as many of the uses of flavonoids involve species–species interactions, often of a defensive nature.54,56 Chemical defense strategies must continue to evolve, as pathogens or predators also continue to evolve countermeasures to battle with plant chemical defenses.
Many diverse flavonoids, including aurones, flavanols, isoflavans, 3-deoxyanthocyanidins, prenylated isoflavonoids, and pterocarpans, have significant antibacterial, antifungal, or antiviral activity against various pathogens (reviewed in Refs. 56,58). Some are inhibitors of the multidrug resistance (MDR) efflux pump implicated in the emergence of bacteria resistant to a wide range of antibiotics.68,69 Conversely, several plant flavonoids, including chalcones, flavanones, flavones, and isoflavones, are known to promote symbiosis with nitrogen-fixing Rhizobia bacteria. Secreted from plant roots, these compounds act by inducing expression of the bacterial nod genes that allow Rhizobia to form symbiotic nodules on the plant root hairs, a relationship that provides the plant with usable nitrogen (reviewed in Refs. 61,62).
Plants also use various flavonoids, including flavones, flavonols, isoflavones, and condensed tannins, to control herbivory by both insects and animals, often with species–specific consequences (reviewed in Ref. 56). For example, the diets of primate species reflect their differing ability to tolerate condensed tannins.70 In herbivores adapted to certain diets, flavonoids often play other roles. Some butterflies lay eggs only on leaves containing a particular combination of flavonol glycosides.71 Other butterflies sequester UV-absorbing flavonoids they ingest from their host plant in their wings; the amount and type of flavonoids sequestered influences mate selection.71
It is unknown to what extent flavonoids affect overall human health, as they are almost ubiquitously present in human diets. Nevertheless, a number of significant medicinal properties have been attributed to the flavonoids present in plant-rich diets. Given the functions of flavonoids in plants, their utility as free radical-scavenging antioxidants72 and antimicrobial agents ( e.g. antimalarials)73 in humans is not surprising. However, flavonoids have also been shown to exhibit vasodilatory,74 anti-cancer,75 anti-mitotic,76 anti-inflammatory,77 anti-asthmatic,78,79 and estrogenic80 activities. Some flavonoids, particularly isoflavones, are called phytoestrogens due to their ability to mimic steroid hormones by binding to estrogen receptors and to type II receptors (which also bind estrogen).80 Actually, the NodD receptor itself (the rhizobial target of flavonoids that some plants secrete to induce root nodulation leading to nitrogen fixation) may bear some structural as well as functional similarity to the ER-α estrogen receptor.81 Several flavonoids non-specifically inhibit tyrosine kinases,82 while others specifically inhibit83 the Iκ-B kinases that activate the transcription factor NF-κB.84 And finally, chalcone derivatives interfere with MDM2-mediated inhibition of the p53 tumor suppressor protein implicated in some types of tumors.85 These important physiological interactions in humans are consistent with the observed health benefits of flavonoids. The differential effects of flavonoids on multiple vital signaling pathways, combined with their diversity and number, suggest that we have just begun to sample the medicinal potential of flavonoid natural products.
2.2 CHS structure and mechanism
The CHS homodimer contains two distinct bi-lobed active site cavities, situated at the bottom edge of each monomer's conserved αβαβα core (if the N-terminal helices are considered to be on the top). Identical six-residue loops from each monomer, which meet at the dimer interface, separate the active sites of the two monomers from each other. Each of these loops begins with residue Thr132 in the active site cavity and ends with a cis-peptide bond to Pro138. The adjacent side-chain of Met137 forms the only contribution of either polypeptide chain to the opposing monomer's active site cavity, and this thioether side-chain neatly plugs a hole in the other monomer (Fig. 1). A second series of larger loops specific to KAS III and type III PKSs surround the bottom half of the active site, and form an additional domain. Thus, the active site cavity is buried, except for a 16 Å CoA-binding tunnel opposite the dimer interface that connects the catalytic surface to the outside aqueous milieu. Interestingly, the width of this tunnel is slightly too narrow for the aromatic substrates and products which must pass through it, indicating that at least some dynamic mobility within and around this tunnel must occur in solution. Furthermore, in a single apo E. coli KAS III crystal structure, the lower domain that forms the floor of the CoA-binding tunnel is extensively disordered.27 While this may be an artifact of crystallization, it might also indicate the presence of sufficient conformational flexibility to accommodate bulky substrate thioesters. However, no significant disorder in this lower domain has been observed in any of the other dozen or so available apo or complexed KAS III or type III PKS crystal structures.
The CHS reaction includes an acyltransferase activity that loads the p-coumaroyl starter moiety onto the catalytic cysteine, a decarboxylase activity that activates malonyl-CoA, an iterative condensing activity that couples the resulting acetyl anion to the growing ketide chain, a cyclase activity that forms the cyclized polyketide precursor of chalcone via an intramolecular Claisen condensation of the linear tetraketide intermediate, and finally an aromatase-like activity (Fig. 8). In CHS, the cyclization activity also performs the function of a thioesterase, by severing the polyketide's covalent attachment to the active site cysteine. The alfalfa CHS crystal structure44 revealed the configuration of residues in the active site responsible for many of these activities, which led to a number of additional experiments to probe the CHS mechanism.
Examination of the CHS crystal structure led to the identification of three conserved residues including the catalytic cysteine. This catalytic triad is likely to participate in the multiple decarboxylation and condensation reactions necessary during the course of the CHS-catalyzed mechanism.44 Subsequent mutational and kinetic analysis of these residues,45 along with pH-dependent inactivation studies,93,94 provided further clarification of the roles of these residues in the CHS mechanism.
The core chemical machinery of the type III PKS active site is a catalytic trio of residues consisting of Cys164, His303, and Asn336 (Figs. 6 and 8). Positioned at the top of the active site cavity, these residues act on substrates inserted into the active site by the pantetheine arm of CoA-linked molecules. Interestingly, the catalytic cysteine is conserved in all thiolase-fold enzymes, and is located at the N-terminal cap of one of a pair of buried amphipathic α-helices that make up the buried central α-layer of the thiolase-fold architecture (Fig. 5). A mechanistic role for these helices was proposed for thiolase8 and has since been invoked for other thiolase-fold enzymes. In model peptides, the placement of a cysteine near the N-terminus of an α-helix lowers its pKa from 8.8 to 7.2.95 The central thiolase-fold α-helices are presumed to exert the same effect, but this mechanistic inference has not been directly tested. The superimposability of the cysteine with the catalytic serine residue at the N-terminal cap of a similar helix in the unrelated serine protease family has also been cited as circumstantial evidence for this mechanistic proposal.34
An “oxyanion hole” formed by protonated nitrogens on His303 and Asn336's adjacent side-chains positions the thioester carbonyl oxygen of CoA-bound substrates, and provides an electron sink to stabilize the tetrahedral transition state formed during both the transfer of starters to the catalytic cysteine and upon attack of the acetyl carbanion (derived from malonyl-CoA decarboxylation) on thioester-linked substrates. Furthermore, the activation of acetyl groups via the decarboxylation of malonyl-CoA is promoted through the stabilization of the enol tautomer by this oxyanion hole (Fig. 8(a)). This latter activity does not require the participation of the Cys164 nucleophile, which is within hydrogen bonding distance of His303. Both mutational and pH-dependent inactivation studies preclude a role as a general base for CHS's His303 Nε, which instead forms a stable imidazolium-thiolate ion pair with Cys164.45,93,94 Mutations of CHS His303 to glutamine and alanine shift the cysteine's pKa from 5.5 to 6.6 and 7.6, respectively.93 The still somewhat acidic pKa of the H303A mutant's cysteine most likely results from the helical dipole effect described above, but may also be influenced by other adjacent active site residues. Conversely, the Cys164 thiolate also modulates His303's decarboxylation activity. Loading of a starter molecule neutralizes the thiolate, which stimulates decarboxylation activity. Conversely, in the absence of a starter, the thiolate-imidazolium pair is less likely to catalyze the unproductive decarboxylation of the malonyl extender unit, in a rather elegant example of enzymatic self-regulation.94 Notably, the H303Q mutant retains near wild-type catalytic activity. The amide nitrogen of the mutant's Gln303 side-chain superimposes perfectly with the Nε of the wild-type enzyme's His303 side-chain, and thus serves as a reasonable mimic of the hydrogen-bonding arrangement of His303 to Cys164.45
The hydrophobic side-chain of another conserved active site residue, Phe215, is also thought to contribute to decarboxylation by encouraging the formation of neutral CO2 from the charged malonyl-CoA terminal carboxylate.45 In addition, it likely serves to reposition the sigma bond of malonyl-CoA that undergoes cleavage (Fig. 8(a)). The predicted perpendicular arrangement of this sigma bond is such that the extended π-systems of the keto-enol tautomers are parallel with the newly released electrons. Interestingly, Phe215 is one of two “gatekeeper” phenylalanines that block the lower portion of the opening between the CoA-binding tunnel and the active site cavity,44 not unlike the swinging saloon doors in a cowboy movie (Fig. 9). The conformational flexibility of these residues, inferred from comparisons of several crystal structures, may confer upon CHS the ability to limit the access of water to the active site, while accommodating substrates and intermediates of varying size.
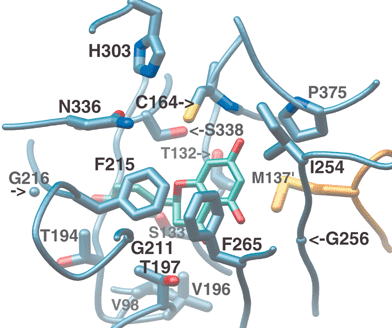 | ||
| Fig. 9 Buried active site cavity of CHS with naringenin bound. View is from the outside, looking through the CoA-binding tunnel. The interface of the active site cavity and the CoA-binding tunnel is mediated by the “gatekeeper” phenylalanines at positions 215 and 265. Other active site residues discussed in the text are also shown. | ||
Following transfer of the starter moiety to the catalytic cysteine and the subsequent decarboxylation of malonyl-CoA with loss of CO2, the resulting acetyl-CoA carbanion, stabilized by the His-Asn oxyanion hole, is poised to attack the cysteine-bound starter's thioester carbonyl carbon using a classical Claisen condensation reaction. This results in the transfer of the starter to the CoA-linked acetyl group, and its simultaneous elongation by two carbons (Fig. 8). The negative charge found on the malonyl's carboxylate moiety is thus retained by acetyl-CoA upon decarboxylation, and then used to restore the negative charge of the thiolate leaving group. The resultant CoA-linked diketide group is once again poised for loading onto Cys164, and another round of decarboxylation and condensation. The Cys164 thiol thus cycles between an anionic thiolate form and a neutral acylated form, with no additional protonation or deprotonation required.
This cycle is repeated until the elongated polyketide completely fills the active site, which in CHS occurs after three acetyl additions. Active site mutants such as T197L, which impair CHS's Claisen cyclization activity but not its condensing activity, still make tetraketide products, indicating that termination of elongation is independent of the cyclization activity of CHS.46 The location of active site mutations which impair cyclization, the scarcity of reactive residues in the CHS active site, and the relatively acidic nature of the tetraketide intermediate's methylene β-carbons make it likely that intramolecular Claisen cyclization of the linear intermediate occurs spontaneously, possibly through an internal proton transfer upon proper elongation, folding, and exclusion of bulk water within the CHS active site.44 A number of inert active site residues, including Thr132, Ser133, Thr194, Thr197, Gly256, Phe265, and Ser338 (Fig. 9), are conserved in CHSs but vary in other type III PKSs, and are likely critical for providing the necessary steric guidance to the linear polyketide.44 The contribution of polarity to these interactions is currently under investigation in our laboratory. Moreover, it is not established whether the proper cyclization conformation is achieved while the linear tetraketide's C1 carbon is attached to the active site cysteine or to the sulfhydryl group of enzyme-bound CoA. Nevertheless, the position of these C1-derived carbons observed in naringenin and resveratrol crystallographically determined complexes with CHS implicates the active site cysteine as the likely choice for final attachment prior to cyclization.44
Except for thiolases themselves, which instead of decarboxylating a malonyl thioester employ a second catalytic cysteine to deprotonate acetyl-CoA, thiolase-fold enzymes use similar or analogous collections of lysines, histidines, or asparagines to interact with substrates and modify the conserved catalytic cysteine.34 However, only KAS III enzymes, which catalyze a single addition of malonyl-ACP to a CoA-thioester starter, share the Cys-His-Asn catalytic triad used by CHS and other type III PKS enzymes. Despite this similarity, CHS and KAS III differ in their use of this catalytic machinery. While both enzymes use the His303-Asn336 oxyanion hole to bind and decarboxylate the malonyl moiety for polyketide elongation, KAS III utilizes an additional oxyanion hole, formed by the backbone amides of the catalytic cysteine and a nearby glycine residue, to orient and stabilize the carbonyl oxygen of the bound starter molecule. This additional backbone amide oxyanion hole is abolished in type III PKSs by an absolutely conserved proline (the only residue with a substituted backbone amide) at position 375 (Fig. 9).34 Lacking this auxiliary oxyanion hole, CHS uses only the His303-Asn336 oxyanion hole for both loading starter molecules on Cys164 and for malonyl-CoA decarboxylation.93 Looking down the CoA-binding tunnel from the surrounding solvent, KAS III orients starters to the right and towards the dimer interface, while in CHS starters are positioned to the left. Interestingly, even the divergent KAS III enzyme from M. tuberculosis that has evolved to utilize a bulky starter does so by creating a new binding pocket, downward and to the right, thus preserving its ability to use this second oxyanion hole.40 The abolition of this second oxyanion hole by the presence of Pro375, rather than specificity for a particular carrier moiety or starter molecule, may well have been the watershed event in the evolutionary divergence of type III PKS enzymes from their KAS III ancestors.
Aside from a number of van der Waals contacts, CHS crystal structures determined in the presence of CoA reveal four residues that form hydrogen bonds with CoA.44 Helix-2 forms the lower left side of the outer rim of the CoA-binding tunnel, and positions three solvent-exposed residues, namely Lys55, Arg58 and Lys62, for electrostatic interactions with two CoA phosphates. These basic residues are each highly conserved in plant CHS-like enzymes, but conservative substitution (or transfer of the basic side-chain to an adjacent residue) occurs in a small number of plant type III PKSs. A fourth highly conserved interaction with CoA forms between the backbone amide nitrogen of Ala308, found on the N-terminal end of helix-9, and the pantetheine moiety's outer carbonyl oxygen. In some type III PKS enzymes, the adjacent Pro307 is replaced with a lysine or an arginine, which can also contribute to CoA-binding. The subsequently determined crystal structure of 2-PS (see Section 3.2.1) demonstrates such an interaction, and also reveals a few other contacts with CoA not observed in CHS,46 confirming that slight variations in CoA-binding are permitted within the type III PKS superfamily. The deletion or addition of CoA interactions may alter the kinetics of association and dissociation of CoA thioesters and thus influence the ultimate fate of type III PKS reaction intermediates.
While CoA binds to KAS III enzymes in a similar position, the E. coli FabH crystal structure reveals an alternate set of conserved residues used by this enzyme family to stabilize the bound cofactor. Additionally, the pantethiene arm of KAS III-bound CoA penetrates that enzyme's active site more deeply (by 1.8 Å) than does type III PKS-bound CoA. While these observed differences in CoA binding might reflect KAS III's need to maintain specificity for both CoA and ACP, the functional relevance of these changes has not been tested.
The intermediate CoA-linked polyketide products are believed to diffuse in and out of the CHS active site, based upon the following evidence. First, as the concentration of 2-mercaptoethanol or other thiol-bearing reducing agents is increased in in vitro assays, less chalcone and more CHS truncation products (discussed in Section 3.1.3) are produced.88 Given the buried nature of the CHS active site, these products most likely form as a result of the diffusion of CoA-linked intermediates into the surrounding solution, where reducing agents can access and sever their CoA-thioester bonds. Likewise, linear polyketides form heterocyclic lactones when acidified in aqueous solution, and acidification of cyclization-impaired CHS mutant reaction mixtures results in a correspondingly increased yield of lactone products.96 Additional supporting evidence for the exchangeability of reaction intermediates with the bulk solution comes from studies of the biosynthesis of reduced chalcone products, resulting from the combined action of CHS and CHR (introduced in Section 2.1.2). CHR is believed to reduce its target oxygen when presented with a CoA linked linear polyketide intermediate, prior to the CHS-catalyzed cyclization and aromatization that eliminates the resulting alcohol.28 Alternatively, a CHR-mediated reduction of the unstable cyclized trione intermediate prior to aromatization would produce the same result (Fig. 8). The buried nature of the CHS active site precludes CHR from accessing the bound linear intermediate polyketide, which again suggests that CHS reaction intermediates can readily exchange with the bulk solvent. And finally, CHR is able to reduce only a portion of the chalcone produced in mixed assays with CHS,28,30 as would be expected if further elongation, cyclization, and/or aromatization of polyketide intermediates competes kinetically with CHR-mediated reduction.
Another clouded issue concerns the movement of intermediate CoA-thioesters into and out of the active site. Evidence outlined in Section 2.2.2 suggests such movement occurs. However, the kinetics of these events, and the relationship of such on and off rates to additional chemical steps during the mechanistically complex series of biosynthetic steps catalyzed by CHS, remain elusive. Several experimental problems, in addition to the microscopic kinetic concerns discussed above, combine to obscure the issue of polyketide intermediate diffusion. The difficulty of stabilizing and separating these linear intermediates from the pool of available substrates in vitro contributes to mechanistic uncertainty. Acidification of reaction mixtures facilitates extraction of tri- and tetraketides via lactone formation, but whether such lactonization occurs in bulk solution or when bound to the enzyme has not been studied. For instance, what percentage, if any, of the lactonization of CHS's intermediate linear polyketides leading to truncation products such as bis-noryangonin and CTAL (see 3.1.3), or lactone products of divergent enzymes such as 2-PS (see Section 3.2.1) occur while linear intermediate-CoAs or Cys164-linked thioesters are bound in the active site? Furthermore, an understanding of the diffusion of intermediates based on the product distribution of in vitro reactions carried out in the presence of CHS and CHR is complicated by an incomplete appreciation of substrate channeling. While channeling probably occurs in vivo (see Section 2.1.2), CHS-CHR complexes have not been observed crystallographically or in solution, even at high concentrations, and therefore might require the presence of a particular enzyme isoform, additional scaffolding proteins, or other enzymes in the pathway.
Many of these and other fundamental mechanistic questions may be addressed using creative approaches that employ some combination of substrate, polyketide intermediate, or carrier moiety mimics amenable to spectroscopic analysis, the implementation of stop-flow and quenched-flow technology, or time-resolved structural techniques including NMR spectroscopy and X-ray crystallography.
3 Evolutionary divergence of plant type III PKSs
3.1 Introduction to type III PKS mechanistic divergence
However, CHS is by far the most common and widely distributed type III PKS in higher plants, and most or all of the divergent plant CHS-like enzymes characterized to date have arisen via extensive duplication and subsequent genetic variation of the chs gene.99–101 This duplication provides CHS activity with functional redundancy, allowing the chs gene to mutate without endangering flavonoid biosynthesis. However, while bioinformatic analysis shows that grasses, for example, duplicate the chs gene approximately every 15–25 million years, these redundant chs genes have only a few million years to develop novel advantageous type III PKS phenotypes before random genetic drift introduces stop codons, dismantles the catalytic machinery, or interferes with proper protein folding.102 Bacterial CHS-like enzymes (Section 4), in contrast to their plant counterparts, exhibit so much sequence divergence that they may have evolved independently of CHS, either from more ancient type III PKS enzymes, or directly from KAS III ancestors. For the sake of clarity, however, all structure/function analyses of divergent enzymes presented here will be discussed in comparison to CHS.
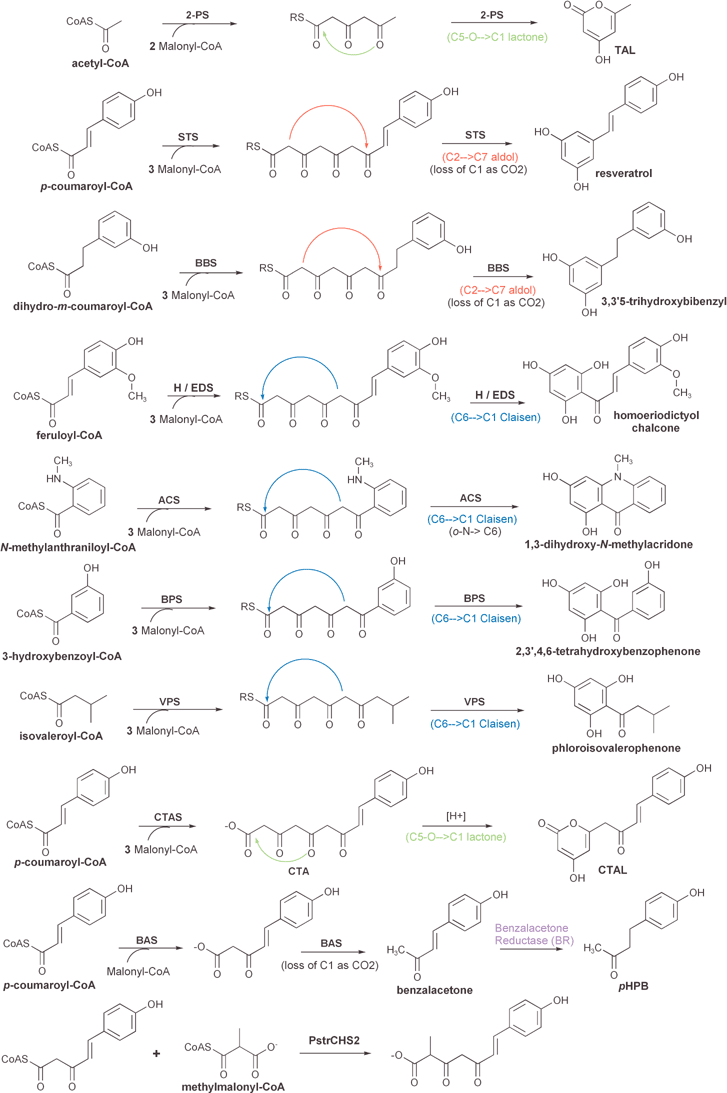 | ||
| Fig. 10 Comparison of the reactions and products of known divergent plant type III polyketide synthases (see text for details). The position and type of cyclization reaction (Claisen, aldol, or lactone) of each presumed linear polyketide intermediate is depicted. One reaction that is not catalyzed by a type III PKS, namely the reduction of benzalacetone to pHPB by benzalacetone reductase (BR), is also included. Other post-PKS modifications are shown in Fig. 13. | ||
Derailed triketide and tetraketide intermediates are isolated as the heterocyclic lactones bis-noryangonin and coumaroyl triacetic acid lactone (CTAL), respectively (Fig. 8). A divergent type III PKS enzyme, coumaroyl triacetic acid synthase (CTAS), that produces coumaroyl triacetic acid as its major product in in vitro assays with standard substrates has been characterized,96 and is discussed in Section 3.2.8. Acidification of CTAS reaction mixtures prior to organic extraction greatly increases the yield of the extracted CTAL lactone product. Unfortunately, this result is consistent with each of two mechanistic alternatives: either an acid-catalyzed lactonization of a linear product in solution, or the acid stabilization of a lactone originally formed within the PKS active site.
While the promiscuity of CHS-like enzymes complicates the assessment of in vivo biological function by making it difficult to categorize the significance of in vitro starter usage, overall trends (preference for larger or smaller, aromatic or aliphatic, straight-chain or branched starters) provide clues about the likely physiological substrate. Relative activities with a range of substrates must be weighed against both the in vivo availability of CoA thioesters and the cellular distribution of products. In fact, the promiscuity of CHS-like enzymes suggests that changes in the in vivo availability of particular substrates may initially play a more significant role than mutational variation in the emergence of new type III PKS activities, with subsequent mutational reinforcement of desirable activities. If true, this hypothesis implies that CHS's loose substrate specificity may be an evolutionary strategy for the rapid emergence of new activities. This is an attractive hypothesis, as many of the natural products synthesized by divergent plant type III PKSs provide resistance to fast-evolving microbial pathogens. Nonetheless, all divergent type III enzymes characterized to date exhibit one or more altered in vitro characteristics relative to CHS. These changes can include increased affinities for alternate starters, variable numbers of extension reactions, loss or gain of intramolecular aldol or Claisen cyclization activity, or a combination of these factors.
Interestingly, the ancestral KAS III enzymes are also quite promiscuous, accepting a broad range of linear and branched short chain fatty acyl substrates.108 A similar change in in vivo substrate availability, perhaps due to the emergence of the general phenylpropanoid pathway or relocalization of the enzyme to the ER, might also have lead to the initial divergence of type III PKS enzymes from KAS III enzymes, prior to the alteration of starter positioning due to the aforementioned Pro375 mutation.
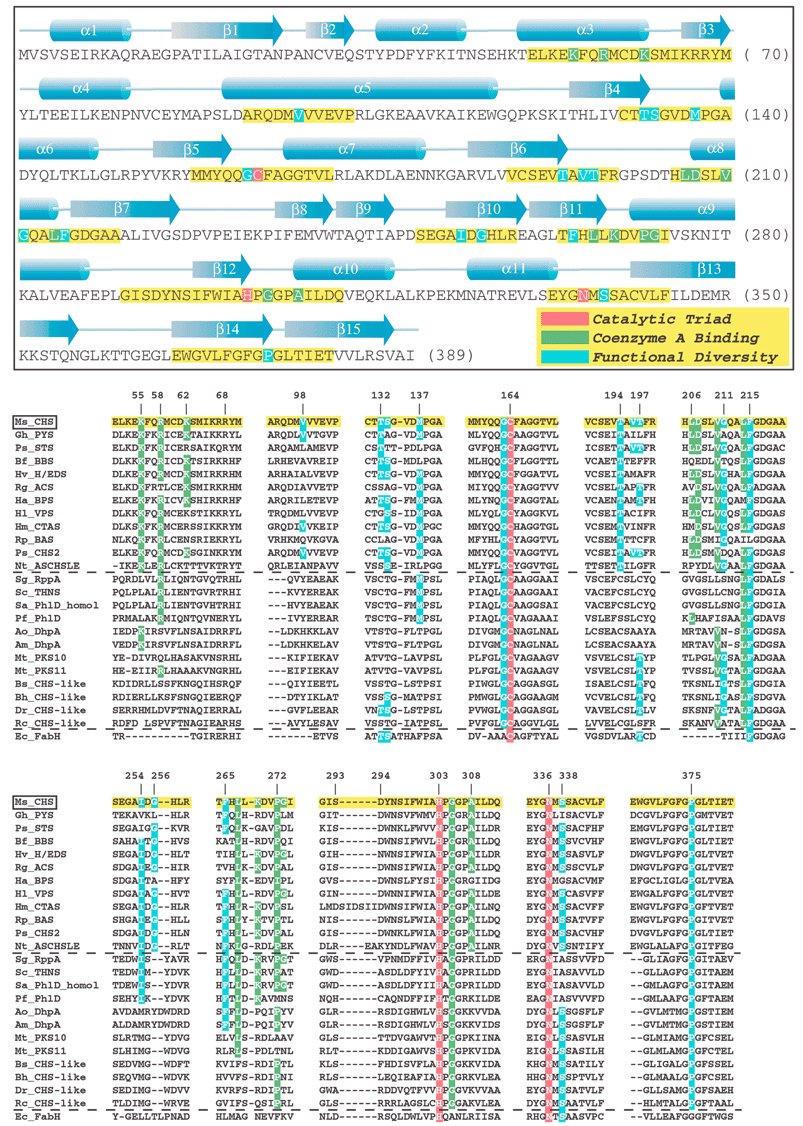 | ||
| Fig. 11 (see previous page) Annotated sequence and secondary structure of alfalfa CHS. Key sections, highlighted in yellow, are aligned against the functionally divergent plant and bacterial type III PKSs, bacterial type III PKSs of unknown function (see text), as well as the evolutionarily related KAS III enzyme (FabH) from E. coli. CHS's catalytic triad, residues that contact bound CoA, and other residues important for functional diversity are highlighted in red, green, and blue, respectively. For clarity, only identical residues in the equivalent positions of the aligned sequences are highlighted, even though in some cases conservative substitutions may play an equivalent mechanistic role. Plant sequences:Ms_CHS = Medicago sativa (alfalfa) chalcone synthase, accession P30074; Gh_PYS (a.k.a. 2-PS) = Gerbera hybrida (daisy) methylpyrone synthase, accession CAA86219; Ps_STS = Pinus sylvestris (scots pine) pinosylvin-forming stilbene synthase, accession CAA43165; Bf_BBS = Bromheadia finlaysoniana (orchid) bibenzyl synthase, accession CAA10514; Hv_H/EDS = Hordeum vulgare subsp. vulgare (barley) homoeriodictyol/eriodictyol chalcone synthase, accession CAA70435; Rg_ACS = Ruta graveolens (common rue) acridone synthase, accession S60241; Ha_BPS = Hypericum androsaemum (tutsan) benzophenone synthase, accession AAL79808; Hl_VPS = Humulus lupulus (hop) valerophenone synthase (a.k.a. phloroisovalerophenone synthase), accession BAB12102; Hm_CTAS = Hydrangea macrophylla var. thunbergii (hydrangea) coumaroyl triacetic acid synthase, accession BAA32733; Rp_BAS = Rheum palmatum (rhubarb) benzalacetone synthase (a.k.a. p-hydroxyphenylbutenone synthase), accession AAK82824; Ps_CHS2 = Pinus strobus (white pine) CHS-like protein, accession CAA05214); Nt_ASCHSLE = Nicotiana sylvestris anther-specific CHS-like enzyme, accession CAA74847. Bacterial sequences: Sg_RppA = Streptomyces griseus 1,3,6,8-tetrahydroxynaphthalene synthase, accession BAA33495; Sc_THNS = Streptomyces coelicolor 1,3,6,8-tetrahydroxynaphthalene synthase, accession CAC01488; Sa_PhlD_homol = Streptomyces avermitilis putative PhlD homologue, accession BAB69299; Pf_PhlD = Pseudomonas fluorescens PhlD (see text), accession AAB48106, Ao_DhpA =Amycolatopsis orientalis dihydroxyphenylacetate synthase, accession T17474; Am_DhpA =Amycolatopsis mediterranei dihydroxyphenylacetate synthase, accession CAC48378; Mt_PKS10 = Mycobacterium tuberculosis CHS-like protein, accession CAB06631; Mt_PKS11 = Mycobacterium tuberculosis CHS-like protein, accession CAB09101; Bs_CHS-like = Bacillus subtilis CHS-like protein, accession NP_390087; Bh_CHS-like = Bacillus halodurans CHS-like protein, accession NP_241483; Dr_CHS-like = Deinococcus radiodurans CHS-like protein, accession AAF11641; Rc_CHS-like = Rhodospirillum centenum “CHSA” CHS-like protein, accession AAD43969. KAS III sequence: Ec_FabH = Escherichia coli KAS III (see text), accession NP_415609. | ||
This steric modulation model was supported by the crystallographic elucidation of the structure of 2-pyrone synthase (2-PS; Section 3.2.1), a type III PKS that differs from CHS in starter usage, elongation steps, and cyclization chemistry. Just as a CHS-based homology model predicted,44 the only significant structural difference between CHS and 2-PS, which share 74% sequence identity, is the reduction of active site volume in 2-PS by the introduction of bulkier side-chains at three key positions.46 Notably, this reduction in volume is achieved without rearrangement of the protein main-chain. As further proof, introduction of these three mutations into alfalfa CHS fully converted that enzyme into a functional 2-PS.46 To further test the effects of active site volume on specificity, the steric bulk of a single CHS side-chain (position 256) was modulated by site-directed mutagenesis, and the mutants assayed with various starters. As expected, the observed trends in starter usage and number of extensions correlated with active site volume.109
While this work validates the simple steric modulation model for type III PKS functional elaboration, the repeated emergence of STS (Section 3.2.2) activity from CHS99 does not fit this general paradigm, suggesting additional routes for the evolution of type III PKS mechanistic diversity. STS and CHS generate the same tetraketide intermediate, but differ in their cyclization mechanism and specificity. Alignments and CHS-derived homology models of STS failed to identify an STS consensus sequence or any structural basis for this divergent activity. Crystal structures of two STSs accompanied by mutagenic modulation of CHS cyclization specificity by the authors (manuscripts in preparation) confirm that alterations of the backbone position not predicted by homology modeling are the causative agent for cyclization changes. Brought about by amino acid substitutions remote from the active site, this backbone rearrangement subtly alters the position and hydrogen-bonding pattern of Thr132 and Glu192, two active-site residues conserved in both enzymes. Rather than using steric changes to achieve a different productive folding of the intermediate, as was previously assumed, these changes instead appear to drive the aldol-based cyclization mechanism of STS by facilitating a thioesterase-like hydrolysis activity.
Although 2-PS and STS are the only divergent type III PKS enzymes that have been structurally characterized to date, their examples illustrate both the value and pitfalls of using homology models to attempt to explain the structural basis for type III PKS functional diversity. However, the failure of STS homology models to predict important differences appears to be the exception rather than the rule. For each of the other divergent enzymes discussed below, sequence alignments and homology models highlight active site differences that are likely to affect specificity (Fig. 11). While these substitutions may not alone be sufficient to interconvert the various type III activities, they represent obvious starting points for mutagenic experimentation aimed at exploring the underlying evolutionary and mechanistic principles governing type III PKS functional divergence.
3.2 Plant type III PKS activities associated with divergent products
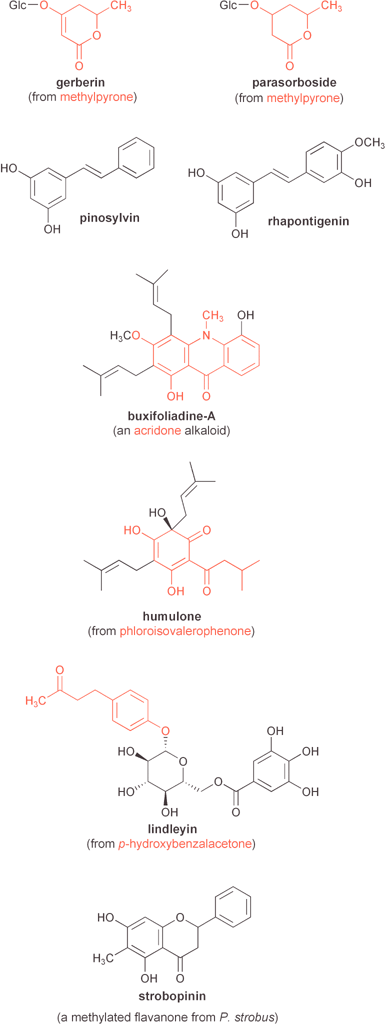 | ||
| Fig. 12 Examples of several natural products mentioned in the text that are derived from plant type III polyketide synthases. PKS product-derived portions are highlighted in red. | ||
Using gchs2 as a probe, a number of similar enzymes were cloned from G. hybrida and four other species. Comparison of these sequences to CHS indicates that this 2-pyrone synthase (2-PS) subfamily of type III PKS enzymes probably arose from a single chs gene duplication event, prior to the evolutionary diversification of the large Asteraceae family of angiosperms.101
The 2-PS enzyme encoded by the gchs2 gene, renamed g2ps1 to reflect its true biochemical function, has been extensively characterized, yielding new information about the mechanism of functional divergence in type III PKS enzymes. A number of small hydrophobic CoA-thioester starters, including propionyl-CoA, butyryl-CoA, and isovaleryl-CoA, are accepted in vitro by 2-PS. Although smaller than the rejected phenylpropanoid starter by only two carbons, benzoyl-CoA is also utilized by 2-PS in vitro, undergoing two acetyl additions followed by lactonization to produce 6-phenyl-4-hydroxy-2-pyrone. This latter compound is of interest as a scaffold for a family of HIV-1 protease inhibitors.111
As discussed in Section 3.1.5, a 2-PS homology model based on the alfalfa CHS structure predicted a smaller active site cavity due to the increased steric bulk of three active site residues, with no other significant changes in structure, despite the hundred or so amino acid differences between CHS and 2-PS.44 The atomic resolution crystal structure of the G. hybrida 2-PS was determined in our laboratory, alone and with acetoacetyl-CoA bound.46 The structure confirmed the validity of the homology model's active site predictions by revealing the active site cavity of 2-PS to possess only a third of the volume observed in CHS. Otherwise, the 2-PS and CHS structures are remarkably similar, except for the minor rearrangement of a three-residue solvent-exposed loop at the mouth of the active site, which the 2-PS homology model failed to predict. The resulting interaction of this loop with the adenine ring and phosphates of CoA, as well as other interactions with CoA not observed in CHS, make CoA binding more extensive in 2-PS.46
However, the mutation of three CHS active site cavity residues to their 2-PS counterparts is sufficient to make alfalfa CHS functionally identical, both in terms of specificity and kinetics, to 2-PS in in vitro assays.46 This CHS triple mutant consists of the following changes at positions that form a triangle in the CHS active site cavity: T197L, G256L, and S338I. An increase in steric bulk at positions 197 and 338 effectively closes off the coumaroyl-binding pocket, excluding large substrates such as phenylpropanoid thioesters, while the increased bulk at the 256 position decreases the size of the putative cyclization lobe of the active site cavity.46 The remarkable functional conversion of CHS into 2-PS by changing less then 1% of their differing residues supports an intuitively simple model of the steric modulation hypothesis, while discounting the relevance of the observed CoA-binding differences to 2-PS's functional divergence from CHS.
While the many differences between the 2-PS and CHS reactions complicate the analysis of this mutagenic conversion, single point mutations of alfalfa CHS at these three positions proved useful in the dissection of their relative contributions to the observed functional differences.46 Not unexpectedly, each of the mutations comprising the triple mutant, when isolated, abrogate CHS's ability to make chalcone. More surprising is the ability of each of the T197L and S338I point mutants, but not the G256L mutant, to catalyze three acetyl extensions of p-coumaroyl-CoA. Paradoxically, these changes nearer the putative starter-binding pocket influence the terminating cyclization to a much greater extent than starter molecule specificity. Similarly, all three-point mutants exhibit increased utilization of acetyl-CoA as a starter, relative to wild-type CHS.46 These results serve as an important reminder of the interconnectedness of specificity for starter usage, chain extension, and cyclization chemistry.
There are a few other important lessons to be learned from these experimental results. First, as suggested by bioinformatic analysis of the natural diversity within the CHS superfamily, the type III PKS condensing machinery is remarkably unaffected by modifications to surrounding active site residues. Moreover, the number of extensions catalyzed is more dependent upon the volume of the active site cavity than on its specific shape. This point is reinforced by the observation that assorted CHS point mutants retain the ability to form the appropriate tetraketide intermediate, but are unable to carry out the terminal intramolecular Claisen cyclization step. In keeping with the observed range of CHS products synthesized in vitro from a diverse collection of starters,103–106 the correct choice of starter molecule seems to be crucial for achieving the conformation of the polyketide intermediate leading to the intramolecular Claisen condensation, but much less important for polyketide chain extension. And finally, the substrate preferences of 2-PS, CHS, and the intermediate CHS point mutants46 demonstrate that type III PKS starter molecule specificity has both upper and lower size limitations. Exclusion of bulky phenylpropanoids from the small 2-PS active site is less surprising than the low affinity of CHS for small starters like acetyl-CoA. The ineffective transfer of small starters to CHS's catalytic cysteine might be, as previously suggested,46 due to their increased conformational flexibility within the large p-coumaroyl-binding pocket. Alternatively, the inability of smaller substrates to exclude nucleophilic water molecules from the active site might be a source of the problem. For whatever reason, the various starter-binding pockets of type III PKS enzymes, like Goldilocks, seem to prefer substrates that are neither too large nor too small.
The first purification of STS from induced peanut cell cultures in 1984 confirmed that the STS and CHS reactions, although similar, are catalyzed by different enzymes.115 Cloning and further analysis revealed significant sequence homology between CHS and peanut STS.116 Apart from the divergent cyclization specificity, no significant mechanistic differences between CHS and STS were detected.91,92 The subsequent cloning of STSs from pine117 (a gymnosperm) and grapevine118 (an angiosperm) facilitated a phylogenetic comparison of STS sequences with each other and with various CHSs, all of which are 60–90% identical at the amino acid level. This study indicated that STS activity has evolved from CHS on more than one occasion, but failed to identify the STS consensus sequence that could be linked functionally with the mechanistic basis for such divergent cyclization activity.99 In contrast to the case of 2-PS, STS homology models based on the CHS structure reveal no significant differences. Although no evidence for a “steric modulation” explanation for the emergence of aldol cyclization specificity in STS enzymes exists, it had been widely assumed that divergence was achieved through folding of the tetraketide intermediate into an alternative conformation conducive to aldol condensation. Recently, using scaled-up in vitro assays and careful analysis of the resultant products, Sankawa and co-workers found that both STS and CHS produce small amounts (1–5% of major product yields) of each other's cyclization product. Additionally, this study demonstrated that STS, like CHS, also generates styrylpyrone side products.119
STSs and their products have recently attracted much attention, because of the agricultural and medicinal properties of their small molecule products. STS enzymes occur naturally in a subset of plants, but synthesize the antifungal phytoalexins resveratrol and pinosylvin using substrates common to all higher plants. Given this, STS encoding genes are an obvious target for transgenic crop enhancement. Resveratrol synthases from peanut and grapevine have been expressed in tobacco and alfalfa, respectively. In both cases, considerable resistance to pathogenic fungi was conferred upon the heterologous host plants.120,121 In terms of disease prevention in humans, resveratrol has been shown to possess a number of beneficial medicinal activities, including copper chelation, anti-oxidant scavenging of free radicals, inhibition of both platelet aggregation and lipid peroxidation, anti-inflammatory activity, vasodilation, and anti-cancer properties.122,123 Positive health effects of resveratrol are often attributed to the moderate consumption of red wine, the so-called “French paradox”.124 Red wines generally contain higher amounts of resveratrol due to inclusion of the phytoalexin-rich grape skins and stems in the fermentation process for longer periods of time.
Very recently, these authors determined the crystal structures of both the pinosylvin-forming STS from P. sylvestris (pine) and the resveratrol-forming STS from A. hypogaea (peanut) (manuscripts in preparation). Both enzymes, although independently evolved from CHS through different sets of amino acid changes, exhibit similar main-chain conformational changes with respect to CHS. Structure-guided mutagenic conversion of CHS into STS confirmed that STS's alternative cyclization specificity is achieved by a conformational difference in the main chain of a short, buried loop that spans the dimer interface between the two active sites (residues 132–137). This movement is caused by various amino acid substitutions at buried positions adjacent to this loop, most notably the replacement of Val98 with a bulkier side-chain, and results in an altered active site hydrogen-bonding network around the slightly repositioned Thr132. This active site residue (conserved in both CHS and STS) changes its bonding interaction with Glu192 (conserved in all type III PKSs), and forms a hydrogen bond with a well-ordered water molecule adjacent to the catalytic cysteine. A three dimensional comparison of the CHS and STS active sites casts doubt upon models that attribute these enzymes' cyclization differences to alternative productive conformations of their shared linear tetraketide intermediate, achieved by steric differences between the CHS and STS active site cavities. Our preliminary results suggest instead that subtler active site changes, apparently of an electronic rather than steric nature, favor an alternative cyclization mechanism within the context of a quite similarly folded tetraketide intermediate. This latter model is also consistent with the cross-reactivity of CHS and STS previously observed by Ebizuka's group.119
Directed changes in three areas of primary sequence were required to achieve near-wild-type levels of STS activity in the converted CHS mutant. Considering that the separately evolved STS subfamilies vary considerably in each of these three areas of primary sequence, it is not surprising that previous (non-crystallographic) efforts in our laboratory and others have failed to identify all of these crucial changes, and have in some cases mistakenly identified spurious residues as mechanistically important. These examples contain valuable lessons for the interpretation of structure/function relationships. First, conclusions based on mutations of an STS/CHS chimera were reported in the same paper where the repeated evolution of STS was elucidated.99 In this study, which predates any type III PKS crystal structure, a chimera consisting of the first 107 residues of S. alba CHS and 287 C-terminal residues of peanut STS was shown to lack activity. Mutating three residues in the N-terminal CHS-derived portion of this chimera to resemble STS (Q100E, V103M and V105R) resulted in very low STS-like activity, and the further mutation of G23T increased this activity to 25% that of wild-type STS. Position 103 corresponds to alfalfa CHS Val98, confirmed by our later structural work to be a crucial area of specificity determination for the conversion of CHS into STS. However, in retrospect the G23T mutation appears unrelated to any of the important architectural differences between STS and CHS, and is probably important only in the context of the folded and stabilized conformation of this artificial chimera. This illustrates an important point: within a conserved fold, the distribution and importance of some residues that confer rigidity or flexibility drifts in a compensatory fashion, as do various other compensatory changes in regions of primary sequence that associate in the folded protein. Such differences between divergent enzymes often reflect alternative ways to stabilize the same tertiary fold, and often have little to do with enzymatic differences. However, taken out of the context of the protein in which such changes evolved, these sequence differences can have seemingly important mechanistic effects, as the chimera's G23T change reflects.99
In another example, Pro375, conserved in all type III PKSs, was mutated to glycine in both CHS and STS, resulting in impairment of both solubility and activity in both enzymes.125 Mechanistic conclusions were argued for, citing the differential effects of this mutation on CHS and STS product distributions. Drawing conclusions about functional divergence from mutation of a conserved residue is risky, especially when overall activity or protein stability is greatly decreased by that mutation. The apparently greater effect of the P375G mutation on STS solubility as well as activity125 is a warning that the extent to which this proline stabilizes the type III PKS fold has also diverged in these two enzymes.
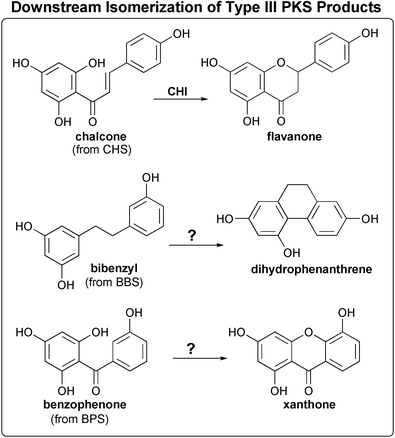 | ||
| Fig. 13 The similar (post-PKS) cyclization reactions of three plant type III PKS natural products (see text for details). | ||
Stressed or wounded orchid tissues, especially those infected by endomycorrhizal fungi, accumulate bibenzyl stilbenes and their tricyclic derivatives, 9,10-dihydrophenanthrenes (Fig. 13), presumably as antifungal agents.126 An enzyme producing the bibenzyl product was purified, characterized, and subsequently cloned from a Phalaenopsis orchid, along with an O-methyltransferase enzyme found to be essential for the further conversion of bibenzyl into 9,10-dihydrophenanthrene.127,128 This BBS clearly belonged to the CHS/STS superfamily, but preferred dihydro- m-coumaroyl-CoA as a starter over p-coumaroyl-CoA (6 vs. 0.2 pkat mg−1 respectively, compared to 3 vs. 58 pkat mg−1 for peanut STS), in in vitro assays.128 Although the steady-state kinetics associated with type III PKS enzymes for their preferred substrates are usually quite similar, BBS's activity towards dihydro- m-coumaroyl-CoA is five-fold less than that of STS towards its preferred starter, p-coumaroyl-CoA.129 These starters differ in both the position of ring hydroxylation and in the saturation of their propanoid moiety. Assays using substrate analogues demonstrated that BBS selects for each of these modifications over the more commonly used p-coumaroyl starter.127
A study utilizing both mutant and BBS/STS chimeric enzymes attempted to elucidate the structural basis for this unusual substrate preference.129 Chimeras were made by joining the C-terminal portion of grape STS to the N-terminal portion of BBS and vice versa, with residue 222 serving as the junction point. Although both chimeras exhibited 50-fold reductions in steady-state activity, likely due to mismatched pairing of drifting compensatory mutations as discussed in Section 3.2.2, the N-terminal segment appeared to dictate starter selection. These authors (in the absence of a three-dimensional structure) also made an STS mutant by replacing STS residues 230–233 with their BBS counterparts. In the context of a 25% loss of resveratrol synthase activity, this mutant exhibited a disproportionate increase in selectivity for the reduced starter. The mutant's rate of conversion of m-hydroxyphenylpropionyl-CoA into the bibenzyl product was 11% of its rate of conversion of p-coumaroyl-CoA into resveratrol (compared to only 0.35% in wild-type STS).129 Notably, these surface-exposed loop residues are in fact located on the opposite side of the core αβαβα domain from the active site, and would seem unlikely to be important for determining specificity. The 25% loss of wild-type activity caused by these changes, which are remote from the active site, suggests that in the context of the STS protein they may be destabilizing to the proper, catalytically active fold.
However, a structure-based examination of the BBS sequence reveals a few residues that might directly influence starter molecule preference. CHS Val98 is replaced in BBS by an alanine, a substitution also found in other divergent type III PKS enzymes known to utilize starters possessing m-substituted phenyl rings (discussed in Sections 3.2.4 and 3.2.5). The substitution of CHS Met137 by leucine, also seen in pine STS, may work synergistically with Ala98. A third notable active site feature of BBS is the use of threonine in place of CHS's Gly211. This residue lines the front of the active site cavity, just underneath the gatekeeper phenylalanines located at the end of the CoA-binding tunnel. While this bulky substitution is unlikely to translate into increased specificity for m-substituted phenylpropanoid starters, it presumably changes the shape of BBS's starter-binding pocket. Substrates with reduced propanoid moieties possess increased conformational flexibility, which should give them an advantage over more rigid substrates in binding to a distorted starter-binding pocket. This advantage might be the basis of BBS's selectivity for reduced phenylpropanoid substrates.
The presence of dihydropinosylvins in pines is more difficult to explain at the current time, as a distinct dihydropinosylvin synthase (DPS) has not been conclusively identified. Dihydropinosylvins share the saturation of the bibenzyl linker with BBS products, but are derived from an unsubstituted dihydro-cinnamoyl starter, rather than an m-substituted starter. A putative DPS cloned from P. sylvestris and expressed in E. coli appeared to prefer dihydro-substrates,117 but better expression and purification revealed the enzyme to be a pinosylvin synthase (a pinosylvin-forming STS, Section 3.2.2).114 Moreover, the addition of P. sylvestris extract to the in vitro assay selectively inhibited the DPS activity of this enzyme even further.114 However, no further characterization of the reaction-modifying element contained in the pine extract accompanied these experiments, and the biosynthesis of dihydropinosylvins in pines remains a mystery.
Two more clues to the puzzle come from experiments designed to elucidate the cause of the stilbene cyclization specificity. First, the STS/CHS chimera with four additional mutations that exhibited 25% of the native STS enzyme's cyclization specificity, discussed in Section 3.2.2, preferred dihydro- p-cinnamoyl-CoA over p-coumaroyl-CoA, and was thus technically a dihydropinosylvin synthase rather than a resveratrol or pinosylvin synthase.99 Since the STS template was from peanut, which makes only resveratrol stilbenes, this cannot be considered a physiologically relevant characteristic of the chimeric enzyme, but it may still offer insight into the basis of bibenzyl specificity. The second clue comes from another set of STS-probing experiments, where the −2 and −3 residues (relative to the catalytic Cys 164) were mutated to test their effect on cyclization specificity.130 This 161–2 sequence is Gln-Gln in CHS (and in many plant type III PKSs), but Gln-His in pine STS and His-Gln in peanut STS. While mutagenic exchanges of these residues did not alter cyclization specificity as hoped, the peanut STS Gln-Gln mutant showed selectively reduced activity towards p-coumaroyl-CoA, while retaining dihydro-cinnamoyl-CoA activity. Thus, the peanut STS was effectively transformed into a dihydropinosylvin synthase by the H161Q mutation.130 Interestingly, this residue is sandwiched at the dimer interface between the dimer's active site-spanning loops (residues 132–137) whose conformation differs in CHS and STS.
Taken together, these similar results with two different mutants of peanut STS implicate slight changes near the active site cavity that selectively reduce wild-type activity, with no harm to the dihydropinosylvin synthase activity. These results are consistent with the model proposed for BBS, where a flexible substrate compensates for a perturbed active site cavity. Aside from the issue of starter molecule binding, the additional flexibility of the tetraketide intermediate due to saturation of the starter molecule's propanoid moiety may help it achieve a folded conformation suitable for cyclization within a slightly contorted active site cavity.
 | ||
| Fig. 14 Phenylpropanoid CoA thioesters commonly found in plants (discussed in Sections 3.1.4 and 3.2.4). | ||
Danish researchers have cloned an interesting chs-like gene (78% amino acid identity with the constitutively expressed barley CHS1) from a library prepared from Hordeum vulgare (barley) leaves infected with the fungus Blumeria graminis f.sp. hordei (bgh).132 Expression of the resulting HvCHS2 enzyme occurs 24–36 h after bgh inoculation, corresponding with a 500% increase in the eriodictyol-derived phytoalexin lutonarin. In vitro, HvCHS2 was shown to prefer the di-substituted caffeoyl- and feruloyl-CoA starters at physiological pH, with only minimal activity towards p-coumaroyl-CoA or cinnamoyl-CoA (Fig. 14).132 This unusual substrate specificity is the inverse of that shown by typical CHS enzymes. Activity profiles with all four of these CoA thioesters show that both enzymes exhibit decreasing activity towards substrates that are increasingly different in size from their preferred substrate (i.e. too large in CHS1 or too small in HvCHS2).132 Although HvCHS2 synthesizes pentahydroxychalcone from caffeoyl- and feruloyl-CoA (O-methylated at position 3 when feruloyl-CoA is the starter), the enzyme was labeled a homoeriodictyol/eriodictyol synthase (H/EDS) after the flavanone derivatives of these pentahydroxychalcone natural products.
These same authors, working before any type III PKS crystal structure was available, cited 27 positions where HvCHS2 differed from the CHS consensus sequence, and pointed out three changes that they found particularly notable: Q95H, A166G and G168A (alfalfa CHS numbering).132 Comparison with the CHS crystal structure, however, reveals two additional and perhaps more functionally important differences. First, substitution in the active site of an alanine for CHS's Thr197 very likely results in the larger starter-binding pocket needed to achieve HvCHS2's unusual specificity for bi-substituted phenylpropanoid-CoA starter molecules. This contrasts with 2-PS's use of a bulkier residue at this same position to decrease the 2-PS active site volume (Section 3.21).46 While the T197A mutation is probably the most relevant change in HvCHS2, other mutations may reinforce the enzyme's preference for larger starters. One likely candidate is a V98A substitution, also observed in other plant type III PKSs that prefer meta- and ortho-substituted aromatic starter molecules (see Sections 3.2.3 and 3.2.5).
The three-ring acridone skeleton is made by acridone synthase (ACS), a CHS-like enzyme that catalyzes three condensations of malonyl-CoA to an N-methylanthraniloyl-CoA starter, followed by a CHS-like intramolecular Claisen cyclization of the tetraketide intermediate (Fig. 10).133 Unlike the CHS reaction, formation of the heterocyclic middle ring is likely to occur prior to aromatization of the new phenolic ring possibly involving formation of a Schiff base, which is accompanied by elimination of a water molecule to produce the 1,3-dihydroxy-N-methylacridone product.136 Initially purified from cell suspension cultures of Ruta graveolens (common rue),133 several ACS isozymes have since been cloned from elicited or irradiated cell cultures and from immature flowers.135–137 Although ACS has the unusual characteristic of eluting from gel filtration columns with an apparent molecular weight equal to between one and two monomers, sedimentation equilibrium experiments clearly show the active form to be a homodimer.137
Interestingly, while neither Ruta CHS nor any other known CHS or STS can utilize N-methylanthraniloyl-CoA, wild-type ACS retains some CHS-like activity in vitro, producing naringenin chalcone from p-coumaroyl-CoA (with about 15% of the activity seen with N-methylanthraniloyl-CoA).136 Like most divergent plant type III PKSs, the ACS protein sequence differs from CHS in about 100 places. Three of these changes (T132S, S133A, and F265V) are in the active site (Figs. 9 and 11), and were the subject of some recent and enlightening mutagenesis experiments. Mutation of these three amino acids in the ACS active site to their corresponding CHS residues seriously impaired the mutant ACS's ability to utilize its normal N-methylanthraniloyl-CoA starter, while greatly increasing CHS-like activity towards p-coumaroyl-CoA.138 The corresponding reverse CHS triple mutant has not yet been reported, but the F265V mutation alone does not confer any ACS-like activity on alfalfa CHS.107 When the single mutation of V265F was made in ACS, thus restoring the second CHS gatekeeper phenylalanine, ACS-like activity was impaired, but CHS-like activity did not improve.138 Clearly, all three changes were important for the evolution of ACS from CHS, but since this ACS triple mutant still possesses marginal ACS-like activity, whereas wild-type CHS does not, additional differences must also be important for allowing the unusually wide N-methylanthraniloyl-CoA starter into the active site. Other ACS mutations made by these authors (“MS1” = R146K, M147L, N151R, M157F and I159M; and “MS2” = P203D, D204T, and A205H) did not significantly alter ACS specificity.138
Some other second-tier residues not noted in this study may nonetheless be important for mechanistic differences between CHS and ACS. Interestingly, mutation of Phe215 (the other “gatekeeper” phenylalanine, absolutely conserved in both FAS KAS III and known type III PKS enzymes other than benzalacetone synthase, Section 3.2.9) to serine in alfalfa CHS unexpectedly results in the only known CHS mutant that can turn-over N-methylanthraniloyl-CoA (yielding a lactone product).107 The ability of this non-physiological mutant to accept the bulkier N-methylanthraniloyl-CoA substrate is most likely due to the enlargement of the active site entrance (analogous to the F265V change in ACS). However, ACS does have an unusual substitution in the residue adjacent to position 215. Gly216 (Fig. 9), conserved in almost all other plant type III PKSs, is an alanine in ACS (Fig. 11). While the α-carbon of position 216 is buried and faces away from the active site cavity, insertion of a methyl group into this closely packed environment may subtly reposition the main-chain lining the active site surface. However, as mentioned in Sections 3.2.3 and 3.2.4, the as-yet untested second-tier difference most likely to affect ACS substrate specificity is the substitution of CHS Val98 for alanine, also observed in the other two type III PKS enzymes known to prefer meta- or ortho-substituted phenyl substrates.
Examination of the only BPS sequence deposited in the database (from H.androsaemum) reveals a few unusual, yet subtle changes in the active site. Ser338, conserved in most plant type III PKS enzymes that catalyze Claisen or aldol cyclization reactions, is replaced in BPS with a glycine. CHS's second gatekeeper phenylalanine, Phe265, is conservatively mutated in BPS to a tyrosine. CHS's Gly256, known to reduce active site volume when mutated to a bulkier residue,46,109 is a slightly larger alanine in BPS, and finally, position 216, adjacent to the other gatekeeper, Phe215, is occupied by a serine in BPS (Figs. 9 and 11). As discussed in Section 3.2.5 in the context of the ACS active site, introduction of a bulky side-chain on the buried 216 α-carbon may translate into a slight decrease in volume of the starter-binding pocket.
These minor active site changes alone seem unlikely to fully explain BPS's reported substrate specificity. Presumably these changes are complemented by additional mutations further from the active site cavity, yet sequence analysis does not suggest which changes might be functionally relevant. Unfortunately, while relative activities of BPS against various substituted and unsubstituted benzoyl-CoAs are published, the limited BPS literature fails to report a single assay using CHS's preferred phenylpropanoid-CoA starters. Conversely, at least one CHS enzyme has been shown in vitro to synthesize benzophenone when provided benzoyl-CoA as a starter.105 These considerations raise the possibility that this BPS enzyme may in fact be a dual-purpose CHS, possessing relaxed starter specificity in order to accommodate two distinct physiological starter molecules, rather than being a fully committed BPS. While this scenario does not match the standard model for the functional divergence of type III PKS enzymes, where gene duplication provides redundancy prior to functional divergence (discussed in Section 3.1.1), the existence of separate cinnamoyl-, p-coumaroyl-, and benzoyl-CoA ligase enzymes in xanthone-producing plants139 may provide the upstream regulation necessary to make a dual-purpose CHS enzyme physiologically feasible.
Comparison of the cloned hop VPS amino acid sequence with the CHS structure reveals a number of interesting differences near the starter-binding pocket. Two active site threonines at positions 132 and 197, conserved in CHS, are replaced with glycine and isoleucine in hops VPS. However, a second VPS has just been discovered in the primitive vascular plant Psilotum nudum (See Section 3.1.1). This enzyme uses serines at positions 132 and 197, and a valine at position 338 (Figs. 9 and 11). As discussed in the previous section, the residue at position 338 is a serine in CHSs, hops VPS, and all other plant type III enzymes known to catalyze the CHS-like intramolecular Claisen cyclization (other than BPS). As for the STS reaction (Section 3.2.2), there seems to be multiple active site configurations leading to the VPS reaction. Interestingly, the second-tier V98L mutation that our recent work has implicated in the conversion of CHS into STS is also seen in both VPS enzymes.
 | ||
| Fig. 15 Polyketide natural products found in Hydrangea (see Section 3.2.8). | ||
Sequence analysis of CTAS reveals a substitution in the active site cavity that explains its unusual activity. An asparagine residue replaces the CHS-conserved Thr197 in CTAS (Figs. 9 and 11). We saw in Section 3.2.1 how an alfalfa CHS T197L point mutant displayed exactly the same mechanistic phenotype, producing CTAL as a major product when the reaction mixture was acidified.46 As discussed in Section 2.2.2, Thr197 appears to be essential for achievement of the intramolecular Claisen cyclization in CHS. Another interesting feature of CTAS is a six-residue insertion (after CHS residue 290) relative to most plant type III PKS enzymes (Fig. 11). VPS and anther-specific CHS-like enzymes (Sections 3.2.7 and 3.2.11) have four- and two-residue insertions, respectively, in this solvent-exposed loop on the top of the monomer, remote from the active site. While unlikely to directly affect the mechanistic fate of enzyme-bound intermediates, this insertion is positioned to alter interactions between monomers through proximity to the adjacent and entwined N-terminal helices. In turn, such effects may influence dimerization and/or the formation of (putative) multi-enzyme complexes.
The first purification of BAS from raspberries was complicated by extensive contamination by CHS, but enhanced activity in the presence of either β-mercaptoethanol or ethylene glycol (both inhibit CHS) and a higher affinity for the bulkier feruloyl-CoA than for p-coumaroyl-CoA (Fig. 14), convinced researchers that the BAS reaction was not merely a derailment of the normal CHS pathway.146 The more recent cloning of BAS from rhubarb ( Rheum palmatum) revealed its amino acid sequence, and allowed better biochemical characterization after purification from CHS-free E. coli expression cultures.147 Interestingly, the double bond that is eliminated by the downstream reductase is absent in the hydroxyphenylpropionyl-CoA starters used by the STS-like bibenzyl synthase (BBS) enzymes (see Section 3.2.3), but in vitro, BAS shows no activity with these reduced starters nor various aliphatic starters accepted in vitro by CHS.147
The cloning of BAS followed the alfalfa CHS crystal structure, so Abe and coworkers were able to identify a curious substitution in a BAS active site residue. Phe215 (alfalfa CHS numbering), absolutely conserved in all other known type III PKS enzymes, is a leucine in BAS (Figs. 9 and 11). While normally this mutation would be considered conservative, this hydrophobic phenylalanine is proposed to assist in the malonyl decarboxylation and extension reactions, based upon its position in the active site, and conservation and mutational studies.44,45 However, the presence of a second unorthodox BAS active site mutation not remarked upon in the literature makes the exact role of the F215L mutation unclear. CHS's Thr197, sterically altered in a number of divergent type III PKS enzymes, is exchanged for a reactive cysteine in BAS. While no other plant type III PKS uses a second active site cysteine, biosynthetic thiolases use such a cysteine to activate acetyl-CoA by the abstraction of a proton (Sections 1.3.2 and 1.5). Perhaps BAS utilizes its second active site cysteine to carry out its unusual decarboxylation of the diketide intermediate? This could involve cleavage of the diketide's thioester bond to CoA (likely to precede the decarboxylation reaction), or donation of a proton to the diketide to facilitate diketide decarboxylation. The nearby Leu215 residue may actively assist in this novel activity, but, conversely, the loss of the conserved gatekeeper phenylalanine might instead only minimize iterative CHS-like elongation of the diketide intermediate, by decreasing the enzyme's more typical decarboxylation activity toward malonyl-CoA, as observed in mutants of CHS Phe215.45 Regardless of which mechanistic strategy BBS utilizes to execute its unusual reaction, it seems clear that chemical modifications to the type III PKS iterative machinery are involved, in contrast to the simple steric constrictions of the active site seen in both 2-PS (Section 3.2.1)46 and CHS model studies.109 Further study of this divergent mechanism is sure to produce valuable insight into the active site machinery of all type III PKS enzymes.
Specificity for both a diketide starter and a single turnover catalytic event, rather than use of methylmalonyl-CoA, are the in vitro characteristics that distinguish PStrCHS2 from typical type III PKS enzymes. These factors are independent of any hypothetical in vivo complex or channeling, and so must arise from some of the approximately 50 amino acid differences between PStrCHS2 and PStrCHS1, a closely related typical CHS cloned by the same authors.149 One obvious active site difference in PstrCHS2 is the substitution of CHS Gly211 for a reactive Asp residue (Figs. 9 and 11). The iterative nature of BBS, the only other plant PKS enzymes with a residue of any significant size or polarity at this position (threonine, see Section 3.2.3), suggests that this change in PStrCHS2 is more likely to influence substrate selection than modularity. On the other hand, residue 211 is spatially positioned near the conserved Phe215, whose mutation to leucine in BAS is likely to prevent iterative activity by that enzyme (Section 3.2.9), so it is possible that this single change might be responsible for both of PstrCHS2's unusual in vitro characteristics. On the other hand, this mutation might be accompanied by reorientation of the side-chain at position 211 away from the active site cavity, in which case homology modeling of PstrCHS2 would be misleading.
Comparison of the ASCL consensus sequence150 to CHS reveals conservation of the catalytic triad and most of the other residues lining the active site, although the low overall sequence identity in surrounding areas increases the likelihood that second-tier interactions may subtly reshape the active site cavity (Fig. 11). However, CHS's conserved Thr197 is replaced by a glycine, a drastic difference similar to that observed in HEDS (see Section 3.2.4), where it is used for selection of larger (increasingly hydroxylated) phenylpropanoid starters. Overall, the ASCL active site thus seems most similar to HEDS, but a further substitution of Thr132 for a serine, as well as other unusual substitutions in the CHS/STS aldol cyclization specificity-determining area (see Section 3.2.2), preclude any exact homology-based prediction of ASCHSLE substrate or product specificity. Interestingly, the major component of exine is sporopollenin, an intractable polymer made up of both aliphatic and aromatic constituents, and exine is known to contain both p-coumaric acid and ferulic acid moieties.153 The modeled ASCL active site appears capable of loading and elongating either starter. Thus it seems plausible, as Atanassov and co-workers suggest, that this CHS-like enzyme may synthesize a monomer for exine biosynthesis.150 However, active site similarity to HEDS suggests that the substrates and intermediates (if not the product) of the anther-specific CHS-like enzymes may not be as novel as previously anticipated.
3.3 Plant natural product pathways likely to utilize unknown type III PKSs
Several other plant natural products are very likely to be synthesized by a CHS-like enzyme, but their in vitro biosynthesis has yet to be demonstrated. There are two possible reasons for this. The simplest explanation is that the responsible enzyme has yet to be isolated and characterized. Alternatively, the enzyme may be known, but might behave differently in vitro, thus obscuring its true biological function. In a few natural product cases, the latter option seems more likely, which leads us to consider the potential causes of such a discrepancy. The in vitro absence of some essential in vivo protein partner that modifies either the enzyme or a reaction intermediate seems the most plausible explanation. Thus far, the reduction of CHS intermediate products by chalcone reductase (CHR) is the only known example of another protein modulating a CHS-like reaction (aside from variability in the availability of starters). Although a long CoA-binding tunnel limits access to the buried type III PKS active site, the cavity's cyclization pocket is separated from the outside solution (orthogonal to the dimer interface) by a single, looped β-strand finger (Fig. 1). It is easy to imagine how formation of some multi-enzyme complex could cause a conformational change along this thin wall, thus reshaping the active site cavity. Interestingly, most of the following natural products appear to utilize a ketoreduction step during their biosynthesis.In hydrangea, the stilbenecarboxylic product hydrangic acid (Fig. 15) appears to be derived from an STS-like cyclization of a reduced (at the C5 carbonyl group) tetraketide intermediate, but with retention (unlike STS) of the terminal C1 carboxyl moiety as a substituent of the new ring.96 CTAS might catalyze the formation of hydrangic acid in the presence of the correct PKR to suitably modify the substrate, but sequence analysis of this gene viewed in light of our recent unpublished studies of STSs suggest that CTAS is more CHS-like than STS-like.
Besides dihydrostilbene natural products (bibenzyls, see Section 3.2.3) leading to three-ringed phenanthrenes (Fig. 13) and dimerized bis-bibenzyls, liverworts also make stilbenecarboxylic acids such as lunularic acid (Fig. 16).154 Like hydrangic acid, lunularic acid appears to be synthesized from a C5 reduced tetraketide intermediate, but it also appears to derive from a reduced dihydro- p-coumaroyl starter, like the dihydrostilbenes. Prelunularic acid (Fig. 16), a cyclized precursor retaining the reduced C5 hydroxyl on its as yet non-aromatized new ring, has been isolated from these primitive plants,154 but no liverwort type III PKS enzymes or putative ORFs have yet been reported in the sequence database or the literature. Thus, it is difficult to speculate on the mechanistic details of liverwort polyketide biosynthesis.
 | ||
| Fig. 16 Additional known stilbenecarboxylic acids discussed in Section 3.3.1 (see also Fig. 15) from: (a) liverworts, (b) marijuana, and (c) poison ivy and related species. | ||
The psychotropic tetrahydrocannabinol (THC) natural products in Cannabis sativa (Fig. 16) are synthesized via the condensation of a stilbenecarboxylic acid (olivetolic acid) to an isoprenoid (geranyl pyrophosphate), followed by rearrangement and cyclization of the isoprenoid-derived portion (reviewed in Ref. 155). Feeding experiments in Cannabis with C13-labeled glucose implicate a polyketide synthase in the biosynthesis of the olivetolic acid precursor,156 although the participation of a CHS-like enzyme has not yet been demonstrated. Nonetheless, a type III PKS mechanism involving three condensations of malonyl-CoA to a hexanoyl-CoA starter, followed by an STS-like cyclization (C2→C7) and aromatization, but with retention of the C1 carboxyl group as a ring substituent, would produce olivetolic acid (Fig. 16). Unlike the other stilbenecarboxylic acids discussed, olivetolic acid appears to be synthesized via an unreduced tetraketide intermediate, and also appears to aromatize spontaneously upon cyclization (no non-aromatic cyclized intermediates are known). Some THC-containing natural products lack the carboxylic acid derived from olivetolic acid, but in these cases decarboxylation seems to occur after olivetolic acid is joined to the isoprenoid moiety, as opposed to an STS-like loss of CO2 concurrent with cyclization.155
Various plants, predominantly from the Anicardiaceae family, synthesize natural products that cause contact dermatitis in pests (including humans).157 Anacardic acids are stilbenecarboxylic acids resulting from cyclization and aromatization of three acetate additions (with one ketoreduction step) to a monounsaturated 16- or 18-carbon acyl-CoA starter (Fig. 16).158 The sap of lacquer trees,159 as well as the shells of cashew nuts,160 contain these allergenic compounds. Urushiols, responsible for the allergenic effects of poison ivy, poison oak, and related Toxicodendron species, apparently result from the decarboxylation and hydroxylation of anacardic acids.158 However, none of the responsible biosynthetic enzymes have been identified.
Anacardic acids are also found outside of the Anicardiaceae family, in geraniums. Geranium resistance to spider mites and aphids results from production of monounsaturated anacardic acids.161 Δ9-desaturation of a saturated 14-carbon fatty acid precursor leads to the unusual 16:1Δ11 and 18:1Δ13 fatty acids that pest-resistant geraniums incorporate into anacardic acids.162
 | ||
| Fig. 17 An allelopathic phytoalexin from Sorghum bicolor (see Section 3.3.2). | ||
4 Bacterial type III PKS enzymes
4.1 Introduction to bacterial type III PKSs
Unlike plant type III PKS enzymes, which usually share 60–95% amino acid identity, bacterial CHS-like sequences typically share only 25–50% identity with each other and with plant type III PKS enzymes. Functional characterization of these extremely divergent bacterial enzymes has begun only recently, but some interesting and complicated reactions have already been discovered, and are the subject of a recent review.166 While no bacterial type III PKS crystal structures are yet available, homology modeling predicts much more active site diversity than that seen in plant CHS-like enzymes, suggesting we still have much to learn about type III polyketide synthases.4.2 Bacterial type III enzymes of known function
RppA, a type III PKS cloned from Streptomyces griseus by Horinouchi's group in 1995,171 and the first bacterial CHS-like protein to be functionally characterized, is also a THN synthase. This ∼85 kD homodimer was found to catalyze the formation of THN from five molecules of malonyl-CoA (Fig. 18).172S. griseus knockouts of rppa are albino, and overexpression of RppA in E. coli confers pigment production ( via auto-oxidation of THN to form flaviolin) to this normally colorless heterologous host.172 THN is also thought to be incorporated into a family of prenylated naphthoquinone natural products found in various Streptomyces species.172 Consistent with this hypothesis, one of three chs-like sequences in S. coelicolor is highly homologous to S. griseus rppa, and also encodes a THN synthase.173
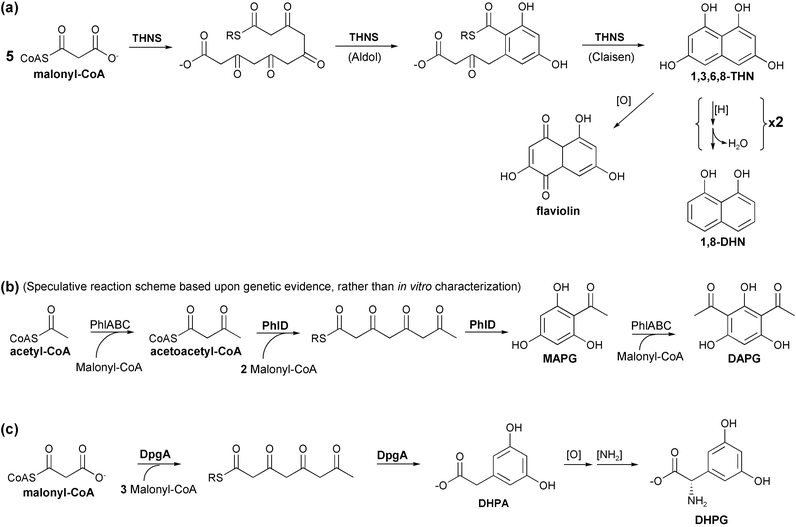 | ||
| Fig. 18 Known bacterial type III PKS enzyme reactions (see Sections 4.2.1 to 4.2.3). Type III PKS enzymes (THNS, PhlD, and DpgA) are shown in boldface. (a) Reaction of THNS leading to 1,3,6,8-tetrahydroxynaphthalene. Derivation of both flaviolin and 1,8-dihydroxynaphthalene natural products from 1,3,6,8-THN are also shown (see text). (b) PhlD's apparent role in the biosynthesis of monoacetylphloroglucinol (MAPG) and diacetylphloroglucinol (DAPG). (c) Reaction of DpgA leading to dihydroxyphenylacetate (DHPA). Additional enzymes further modify this product to form the unusual amino acid dihydroxyphenylglycine (DHPG), which is then incorporated into vancomycin-related antibiotics (see Fig. 19). | ||
Two unusual features of the THNS reaction stand out. Unlike other CHS-like enzymes, THNS forms two new six-carbon rings, and does so using different intramolecular cyclization mechanisms. Although the intermediates of cyclization are unknown, comparison of the symmetric THN product to a linear polyketide implicates an STS-like aldol condensation followed by a CHS-like Claisen condensation. The second unusual aspect of the THNS reaction is its condensation of five acetate units. No other known type III PKS enzymes catalyzes more than three extension steps, presumably due to active site volume or the reactive nature of the resulting tetraketide intermediate. THNS may form the first of two rings prior to making a pentaketide, thus eliminating the potential for a number of unproductive side reactions. However, when provided with the non-physiological octanoyl-CoA starter, the THNS from S. griseus (RppA) makes a hexaketide, with lactone formation due to cyclization of only the final three acetate units.174
A homology model of S. coelicolor THNS based on the alfalfa CHS crystal structure reveals a number of unusual changes in the active site. Conservation of the catalytic triad, both gatekeeper phenylalanines, and the absolutely conserved Pro375 speak to the reliability of the structural alignment, despite the low sequence similarity between the two enzymes (Fig. 11). In contrast to plant PKSs, which have a single active site cysteine (or two in the case of BAS, see Section 3.2.9), this model reveals four additional cysteines in the THNS active site. One of these, at CHS position 211, is a serine in the S. griseus THNS (RppA). The other three cysteines, at CHS positions 132, 194, and 197, are conserved among Streptomyces THNS enzymes, and occur at positions known to be important for modulating both substrate specificity and proper cyclization of polyketide intermediates (see Section 2.2.2) in plant type III PKS enzymes. It is a matter of speculation whether the potential reactivity of the cysteine rich active site in THNS plays a role in either promoting the proper folding of intermediate polyketides or in preventing formation of improper polyketide conformations that would otherwise lead to truncation products. A number of additional non-conservative substitutions, both in and out of the active site, further complicate our understanding of the THNS reaction.
Sequence gazing yields another interesting observation, concerning an arginine residue (at CHS position 308) on the surface of both known Streptomyces THNS sequences. The equivalent residue in the ancestral KAS III enzymes is also an absolutely conserved arginine, whose side-chain has been shown to facilitate KAS III ACP binding.175 Likewise, the ACP-utilizing KS domains of both type I DEBS and type II actinorhodin PKS enzymes also possess this arginine residue. Conversely, CHS and all plant CHS-like enzymes possess an absolutely conserved alanine at this position, while all other known bacterial CHS-like sequences have a lysine here. It is tempting to speculate that some bacterial type III PKS enzymes, especially THNS, may have an as yet undiscovered specificity for ACP-thioester substrates. To the best of our knowledge, this possibility has not previously been considered for any CHS-like bacterial enzymes, since CoA specificity is a defining feature of the well-characterized plant enzymes. Two features of ACP-utilizing condensing enzymes that might obscure an unsuspected ACP specificity include the tendency of condensing enzymes to discriminate between various ACP isozymes within a species (see Section 1.5), as well as in vitro utilization of non-physiological CoA substrates.
Bangera and Thomashow isolated and characterized four clustered genes ( phlACBD) in Pseudomonas fluorescens responsible for the biosynthesis of DAPG (Fig. 18).180 Trios of predicted gene products from Pyrococcus furiosus and Methanococcus jannaschii show homology to the predicted gene products of PhlA, PhlC, and PhlB, but the CHS-like nature of the PhlD enzyme was unexpected. PhlA is homologous to E. coli FabH (KAS III), but like the KS-β domains of type II PKSs (Section 1.4) it lacks the active site cysteine. PhlC somewhat resembles thiolase, and PhlB shows no significant homology with any known protein. 2,4-Diacetylphloroglucinol is produced by transfer of an acetyl group to a monoacetylphloroglucinol (MAPG) intermediate, which could result from a CHS-like C6-to-C1 intramolecular cyclization of a tetraketide intermediate. Phenotypes of selective in vivo knockouts suggest that PhlA, PhlB, and PhlC function collectively in the acetylation of MAPG to form DAPG, and also demonstrate that all four gene products are necessary for the production of the MAPG intermediate (Fig. 18).180 This latter result is surprising, as the synthesis and cyclization of a tetraketide from four malonyl-CoA molecules seems within the scope of an independent type III PKS homodimer. Bangera and Thomashow speculate that PhlA–C provide PhlD with an activated starter such as acetoacetyl-CoA. Another possibility is that PhlD does not function as an independent homodimer, but rather in some complex with PhlA–C. The in vitro characterization of these enzymes will undoubtedly answer these questions and provide further insights.
Like the Streptomyces THN synthases, sequence analysis of P. fluorescens PhlD suggests it has the same overall fold as the plant type III PKS enzymes, as well as conservation of the Cys-His-Asn catalytic trio and both gatekeeper phenylalanines in an otherwise quite divergent active site. Also like THNS, CHS's Ser338 is replaced by alanine, and additional cysteines are found in the active site, aligning with CHS threonines 132 and 197 (Fig. 11). Other putative PhlD active site residues differ from both THNS and CHS, presumably providing a unique steric shape to the PhlD active site cavity. On the basis of these differences, a Streptomyces avermitilis sequence in the database (accession BAB69299.1) that is annotated as a PhlD homologue appears instead to be another THNS.
 | ||
| Fig. 19 Structure of vancomycin. Red denotes the location, in antibiotics of the vancomycin class, of atoms derived from the type III PKS natural product DHPA (see also Fig. 18). | ||
Subsequently, an analogous type III PKS from Amycolatopsis mediterranei, which produces the similar antibiotic balhimycin, was shown to be one of five proteins (DpgA-D and Pgat) involved in the biosynthesis of intermediates leading to DHPG.186 A knockout of the CHS-like dpgA abrogated balhimycin production in this organism, but it could be rescued by supplementation with dihydroxyphenylacetate (DHPA), a presumed intermediate in DHPG biosynthesis. Further, heterologous expression of DpgA in Streptomyces lividans resulted in the production of DHPA, whereas expression of DpgA–D in this host enhanced DHPA synthesis and further resulted in the production of another DHPG intermediate, 3,5-hydroxyphenylglyoxylate. Following heterologous expression in Streptomyces lividans, assays of extracts using either radiolabeled acetyl- or malonyl-CoA implicated malonyl-CoA as the only efficiently incorporated starter molecule for this reaction. Transamination of this latter intermediate by phenylglycine aminotransferase (Pgat), which is also involved in the biosynthesis of the other unusual amino acid incorporated into these antibiotics, completes the synthesis of DHPG.186,187
Another group was able to purify soluble protein from the expression in E. coli of the analogous DpgA-D enzymes from A. orientalis, although the multimeric state of each purified enzyme and the tendency of mixtures to form complexes were unfortunately omitted from their report.183 This study confirmed the ability of DpgA to synthesize DHPA from four molecules of malonyl-CoA, but found the reaction rate to be very slow with DpgA alone. Although neither DpgB nor DpgD show any activity with malonyl-CoA by themselves, DpgA's rate of DHPA production is increased 17-fold by the inclusion of DpgB in reaction mixtures, and the addition of DpgD to this mixture results in another two-fold increase in rate.183 The exact role of DpgB and DpgD is not clear, but these crotonase superfamily enzymes appear to utilize their hydratase/dehydratase activities to facilitate the aromatization of the cyclized DHPA skeleton. However, this study firmly establishes DpgC's role in DHPG biosynthesis. DpgC oxidizes the methylene α-carbon of DHPA-CoA to a carbonyl, and also acts as a thioesterase, releasing the 3,5-dihydrophenylglyoxylate product as a free acid.183
Uncertainty regarding the mechanistic details of the PKS's cyclization of the polyketide intermediate complicates analysis of the exact role of DpgB and DpgD. A C8-to-C3 intramolecular aldol cyclization of the linear intermediate has been presumed, as it produces the proper skeleton while maintaining the thioester at C1,186 and use of CoA thioester substrate analogues by all three downstream Dpg enzymes supports this interpretation.183 C8 of the proposed tetraketide derives from the first malonyl-CoA to bind the enzyme. If DpgA decarboxylates this starter before loading it, the resulting tetraketide's C8 is a methyl group, whose suitability as a nucleophile would be increased by a thiolase-like abstraction of a proton. As in other bacterial type III PKS enzymes, sequence alignments predict the presence of an additional cysteine in the DpgA active site (this time only at CHS position 194), making a thiolase-like methyl activation a distinct possibility. On the other hand, if the malonyl starter is loaded intact, the resulting linear polyketide possesses a free carboxylate group that can activate C8 as a nucleophile through a coupled decarboxylation mechanism. Precedence for this interpretation comes from mechanistic studies of plant type III PKS enzymes that show impaired decarboxylation of malonyl-CoA when no starter is bound to Cys 164.94 Presumably, either of these mechanisms could outcompete the typical CHS-like C6-to-C1 Claisen cyclization, favored by the labile thioester bond at C1.
A final consideration is DpgA's apparent difficulty in aromatizing its product's new ring.183 Interestingly, the linear portion of both the proposed cyclic intermediate and aromatized DHPA product lacks the π-bond conjugation with the new ring seen in most type III PKS products. Although sequence alignments reveal DpgA to have other unusual active site features that might explain its lack of aromatase activity, including an alanine at CHS positions 197 (Fig. 11), it is possible that this impaired aromatization instead reflects the unique nature of the C8-to-C3 cyclization product.
5 Conclusions and future directions
Although comparison of CHS-like enzymes to their KAS III ancestors is informative, the details of type III PKS emergence remain obscure. And while much is known about CHS, some mechanistic questions remain unanswered (see Section 2.2.3). The last few years have revealed a number of new type III PKS superfamily members, and future efforts will undoubtedly discover more. Homology models suggest that much of the functional diversity of plant CHS-like enzymes has evolved by conservation of the decarboxylation and condensing machinery accompanied by simple steric modulation of other active site residues, as observed for 2-PS (Section 3.2.1). Bacterial CHS-like enzymes are much more divergent than their plant counterparts, however, and, like STS (Section 3.2.2), are likely to have accumulated some main-chain movements relative to CHS, thus complicating homology-based analyses of these latter gene products. The comparative analysis of type III PKS sequences given here, with the differences most likely to serve important mechanistic roles highlighted, should assist researchers to better annotate newly discovered CHS-like sequences. The mutagenic interconversion and in vitro characterization of type III PKS enzymes will continue to be a valuable tool for confirming functionally relevant differences within this superfamily, with crystallography providing answers where homology modeling fails to predict the structural and mechanistic consequences of crucial amino acid substitutions.As our understanding of the biosynthesis of biologically and medicinally important natural products grows, the benefits of harnessing this awesome machinery become apparent. In Section 3.2.2, we saw how antifungal resveratrol biosynthesis was introduced into plants lacking this useful pathway. Altered flower color, nitrogen fixation, increased digestability of forage crops, and increased production of health-promoting natural products in food crops188 are other goals of past and present flavonoid pathway engineering efforts (reviewed in Refs. 57,189). The combination of flavonoid pathway enzymes from different plants,190 as well as the combinatorial application of enzymes for hydroxylation, methylation, prenylation, glycosylation and acylation,188 promises to yield even greater chemical diversity. While these experiments, as well as extensive domain-swapping in various type I and II PKSs,19 demonstrate the utility of mixing and matching naturally evolved enzymes to achieve new pathways, our increasing understanding of individual enzymes will allow a more sophisticated approach. The ability to engineer substrate and product specificities (natural or unnatural) into native enzymes or domains will increasingly remove our dependence upon nature to provide desirable activities.
CHS-like enzymes are beginning to be appreciated for their natural functional diversity and architectural simplicity: two qualities that facilitate the elucidation of the determinants of that diversity, as examined in this review. The increasing ability to modulate specificity in type III PKS enzymes46,107,109,138 complements parallel efforts to exploit other aspects of flavonoid biosynthesis. Beyond controlling substrate and product specificity, a number of other type III PKS engineering projects seem worthwhile. Besides the use of substituted malonyl moieties to diversify acetate units, Schröder's characterization of the divergent methylmalonyl-CoA utilizing enzyme in Section 3.2.10 suggests the possibility of engineering modularity into these normally iterative enzymes. Using separate active sites for individual steps should allow increased diversity and optimization of each catalytic step. CHS's recruitment of the CHR ketoreductase implies that other enzymes (hydratases for example) could be selectively employed to further modify diffusable intermediates. Synthetic NAC thioesters (introduced in Section 3.2.10) have proven useful for introducing unnatural starters and intermediate analogs into various PKS systems.149 CHS-like enzymes might be redesigned to optimize activities with these synthetic thioesters. Another approach is to re-engineer the upstream CoA ligase (Fig. 7) to catalyze the activation of unnatural starters. Alternatively, an ACP-utilizing type III PKS might allow the coupling of type III activities to the reactions of their larger and more-complicated cousins (PKS types I and II). Aside from the potential utility of these hypothetical divergent activities, experimentation toward these engineering objectives will undoubtedly shed even more light on type III PKS enzymes and mechanisms. However, these engineering flights of fancy should not distract us from appreciating the rich and beautiful tapestry of evolutionary and ecological interactions illuminated by our examination of the natural chemical diversity generated by the plant and bacterial type III PKS superfamily.
References
- B. J. Rawlings, Nat. Prod. Rep., 1998, 15, 275 RSC.
- C. C. DiRusso, P. N. Black and J. D. Weimar, Prog. Lipid Res., 1999, 38, 129 CrossRef CAS.
- C. O. Rock and J. E. Cronan, Biochim. Biophys. Acta, 1996, 1302, 1 CrossRef CAS.
- R. J. Heath and C. O. Rock, J. Biol. Chem., 1995, 270, 15531 CrossRef CAS.
- V. S. Rangan, A. K. Joshi and S. Smith, Biochemistry, 2001, 40, 10792 CrossRef CAS.
- P. G. Roughan and J. B. Ohlrogge, Plant Physiol., 1996, 110, 1239 CAS.
- K. H. Choi, L. Kremer, G. S. Besra and C. O. Rock, J. Biol. Chem., 2000, 275, 28201 CAS.
- M. Mathieu, Y. Modis, J. P. Zeelen, C. K. Engel, R. A. Abagyan, A. Ahlberg, B. Rasmussen, V. S. Lamzin, W. H. Kunau and R. K. Wierenga, J. Mol. Biol., 1997, 273, 714 CrossRef CAS.
- M. Mathieu, J. P. Zeelen, R. A. Pauptit, R. Erdmann, W. H. Kunau and R. K. Wierenga, Structure, 1994, 2, 797 CrossRef CAS.
- C. T. Walsh, Enzymatic Reaction Mechanisms, ed. A. C. Bartlett and L. W. McCombs, W. H. Freeman and Company, San Francisco, 1979 Search PubMed.
- Y. Modis and R. K. Wierenga, J. Mol. Biol., 2000, 297, 1171 CrossRef CAS.
- S. Masamune, C. T. Walsh, A. J. Sinskey and O. P. Peoples, Pure Appl. Chem., 1989, 61, 303 CAS.
- Y. Modis and R. K. Wierenga, Struct. Fold Des., 1999, 7, 1279 Search PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380 RSC.
- B. J. Rawlings, Nat. Prod. Rep., 1999, 16, 425 RSC.
- B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 231 RSC.
- B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 190 RSC.
- B. Shen, in Biosynthesis of Aromatic Polyketides, ed. F. J. Leeper and J. C. Vederas, Springer-Verlag, Berlin, New York, 2000 (Top. Curr. Chem., 209, 1–51) Search PubMed.
- C. Khosla, R. S. Gokhale, J. R. Jacobsen and D. E. Cane, Annu. Rev. Biochem., 1999, 68, 219 CrossRef CAS.
- R. Bentley and J. W. Bennett, Annu. Rev. Microbiol., 1999, 53, 411 CrossRef CAS.
- D. A. Hopwood and D. H. Sherman, Annu. Rev. Genet., 1990, 24, 37 CrossRef CAS.
- T. A. Holak, M. Nilges, J. H. Prestegard, A. M. Gronenborn and G. M. Clore, Eur. J. Biochem., 1988, 175, 9 CAS.
- T. A. Holak, M. Nilges and H. Oschkinat, FEBS Lett., 1989, 242, 218 CrossRef CAS.
- G. Y. Xu, A. Tam, L. Lin, J. Hixon, C. C. Fritz and R. Powers, Structure (Camb), 2001, 9, 277 Search PubMed.
- M. P. Crump, J. Crosby, C. E. Dempsey, J. A. Parkinson, M. Murray, D. A. Hopwood and T. J. Simpson, Biochemistry, 1997, 36, 6000 CrossRef CAS.
- S. C. Tsai, L. J. Miercke, J. Krucinski, R. Gokhale, J. C. Chen, P. G. Foster, D. E. Cane, C. Khosla and R. M. Stroud, Proc. Natl. Acad. Sci. USA, 2001, 98, 14808 CrossRef CAS.
- X. Qiu, C. A. Janson, W. W. Smith, M. Head, J. Lonsdale and A. K. Konstantinidis, J. Mol. Biol., 2001, 307, 341 CrossRef CAS.
- R. Welle and H. Grisebach, FEBS Lett., 1988, 236, 221 CrossRef CAS.
- G. Forkmann, Genetics of Flavonoids, in The Flavonoids, ed. J. B. Harborne, Chapman & Hall, London, 1994, pp. 537–564 Search PubMed.
- R. Welle, G. Schröder, E. Schiltz, H. Grisebach and J. Schröder, Eur. J. Biochem., 1991, 196, 423 CAS.
- M. Siggaard-Andersen, Prot. Seq. Data Anal., 1993, 5, 325 Search PubMed.
- W. Huang, J. Jia, P. Edwards, K. Dehesh, G. Schneider and Y. Lindqvist, Embo J., 1998, 17, 1183 CrossRef CAS.
- J. G. Olsen, A. Kadziola, P. von Wettstein-Knowles, M. Siggaard-Andersen, Y. Lindquist and S. Larsen, FEBS Lett., 1999, 460, 46 CrossRef CAS.
- J. G. Olsen, A. Kadziola, P. von Wettstein-Knowles, M. Siggaard-Andersen and S. Larsen, Structure (Camb), 2001, 9, 233 Search PubMed.
- A. C. Price, K. H. Choi, R. J. Heath, Z. Li, S. W. White and C. O. Rock, J. Biol. Chem., 2001, 276, 6551 CrossRef CAS.
- M. Moche, G. Schneider, P. Edwards, K. Dehesh and Y. Lindqvist, J. Biol. Chem., 1999, 274, 6031 CrossRef CAS.
- X. Qiu, C. A. Janson, A. K. Konstantinidis, S. Nwagwu, C. Silverman, W. W. Smith, S. Khandekar, J. Lonsdale and S. S. Abdel-Meguid, J. Biol. Chem., 1999, 274, 36465 CrossRef CAS.
- C. Davies, R. J. Heath, S. W. White and C. O. Rock, Struct. Fold Des., 2000, 8, 185 Search PubMed.
- M. Moche, K. Dehesh, P. Edwards and Y. Lindqvist, J. Mol. Biol., 2001, 305, 491 CrossRef CAS.
- J. N. Scarsdale, G. Kazanina, X. He, K. A. Reynolds and H. T. Wright, J. Biol. Chem., 2001, 276, 20516 CrossRef CAS.
- K. A. McGuire, M. Siggaard-Andersen, M. G. Bangera, J. G. Olsen and P. von Wettstein-Knowles, Biochemistry, 2001, 40, 9836 CrossRef CAS.
- D. Val, G. Banu, K. Seshadri, Y. Lindqvist and K. Dehesh, Struct. Fold Des., 2000, 8, 565 Search PubMed.
- A. Abbadi, M. Brummel, B. S. Schutt, M. B. Slabaugh, R. Schuch and F. Spener, Biochem. J., 2000, 345(Pt 1), 153 CrossRef CAS.
- J. L. Ferrer, J. M. Jez, M. E. Bowman, R. A. Dixon and J. P. Noel, Nat. Struct. Biol., 1999, 6, 775 CrossRef CAS.
- J. M. Jez, J. L. Ferrer, M. E. Bowman, R. A. Dixon and J. P. Noel, Biochemistry, 2000, 39, 890 CrossRef CAS.
- J. M. Jez, M. B. Austin, J. Ferrer, M. E. Bowman, J. Schröder and J. P. Noel, Chem. Biol., 2000, 7, 919 CrossRef CAS.
- H. Grisebach, in Proceedings of the IVth International Congress of Biochemistry, Wien 1958, Vol. II, pp. 56–70, Pergamon Press, London, 1959 Search PubMed.
- F. Kreuzaler and K. Hahlbrock, FEBS Lett., 1972, 28, 69 CrossRef CAS.
- F. Kreuzaler and K. Hahlbrock, Arch. Biochem. Biophys., 1975, 169, 84 CAS.
- W. Heller and K. Hahlbrock, Arch. Biochem. Biophys., 1980, 200, 617 CAS.
- U. Reimold, M. Kroeger, F. Dreuzaler and K. Hahlbrock, Embo J., 1983, 2, 1801 CAS.
- R. E. Koes, C. E. Spelt, P. J. van den Elzen and J. N. Mol, Gene, 1989, 81, 245 CrossRef CAS.
- R. Wingender, H. Rohrig, C. Horicke, D. Wing and J. Schell, Mol. Gen. Genet., 1989, 218, 315 Search PubMed.
- C. J. Lamb, M. A. Lawton, M. Dron and R. A. Dixon, Cell, 1989, 56, 215 CrossRef CAS.
- B. Weisshaar and G. I. Jenkins, Curr. Opin. Plant Biol., 1998, 1, 251 CrossRef CAS.
- J. B. Harborne and C. A. Williams, Phytochemistry, 2000, 55, 481 CrossRef CAS.
- B. Winkel-Shirley, Plant Physiol., 2001, 126, 485 CrossRef CAS.
- R. A. Dixon and N. L. Paiva, Plant Cell, 1995, 7, 1085 CrossRef CAS.
- T. A. Holton and E. C. Cornish, Plant Cell, 1995, 7, 1071 CrossRef CAS.
- J. B. Harborne and C. A. Williams, Nat. Prod. Rep., 2001, 18, 310 RSC.
- S. R. Long, Cell, 1989, 56, 203 CrossRef CAS.
- X. Perret, C. Staehelin and W. J. Broughton, Microbiol. Mol. Biol. Rev., 2000, 64, 180 Search PubMed.
- H. K. Stafford, Plant Physiol., 1991, 96, 680 CAS.
- J. B. Harborne and H. Baxter, Handbook of Natural Flavonoids, Wiley-VCH, Weinheim, 1999 Search PubMed.
- J. M. Jez, M. E. Bowman, R. A. Dixon and J. P. Noel, Nat. Struct. Biol., 2000, 7, 786 CrossRef CAS.
- H. K. Dooner, T. P. Robbins and R. A. Jorgensen, Annu. Rev. Genet., 1991, 25, 173 CrossRef CAS.
- D. P. Ormrod, L. G. Landry and P. L. Conklin, Physiologia Plantarum, 1995, 93, 602 Search PubMed.
- N. R. Guz, F. R. Stermitz, J. B. Johnson, T. D. Beeson, S. Willen, J. Hsiang and K. Lewis, J. Med. Chem., 2001, 44, 261 CrossRef CAS.
- F. R. Stermitz, P. Lorenz, J. N. Tawara, L. A. Zenewicz and K. Lewis, Proc. Natl. Acad. Sci. USA, 2000, 97, 1433 CrossRef CAS.
- V. Reynolds, A. J. Plumptre, J. Greenham and J. B. Harborne, Oecologia, 1998, 115, 331 CrossRef.
- M. Haribal and J. A. Renwick, Phytochemistry, 1996, 41, 139 CrossRef CAS.
- C. Ramirez-Tortosa, O. M. Andersen, P. T. Gardner, P. C. Morrice, S. G. Wood, S. J. Duthie, A. R. Collins and G. G. Duthie, Free Radical Biol. Med., 2001, 31, 1033 CrossRef CAS.
- R. Li, G. L. Kenyon, F. E. Cohen, X. Chen, B. Gong, J. N. Dominguez, E. Davidson, G. Kurzban, R. E. Miller, E. O. Nuzum, P. J. Rosenthal and J. H. McKerrow, J. Med. Chem., 1995, 38, 5031 CAS.
- K. Yamaguchi, H. Honda, C. Wakisaka, A. Tohei and H. Kogo, Jpn. J. Pharmacol., 2001, 87, 67 Search PubMed.
- Y. Iwase, Y. Takemura, M. Ju-ichi, T. Mukainaka, E. Ichiishi, C. Ito, H. Furukawa, M. Yano, H. Tokuda and H. Nishino, Cancer Lett., 2001, 173, 105 CrossRef CAS.
- M. L. Edwards, D. M. Stemerick and P. S. Sunkara, J. Med. Chem., 1990, 33, 1948 CrossRef CAS.
- F. Herencia, M. L. Ferrandiz, A. Ubeda, I. Guillen, J. N. Dominguez, J. E. Charris, G. M. Lobo and M. J. Alcaraz, FEBS Lett., 1999, 453, 129 CrossRef CAS.
- M. E. Zwaagstra, H. Timmerman, A. C. van de Stolpe, F. J. de Kanter, M. Tamura, Y. Wada and M. Q. Zhang, J. Med. Chem., 1998, 41, 1428 CrossRef CAS.
- M. E. Zwaagstra, H. Timmerman, M. Tamura, T. Tohma, Y. Wada, K. Onogi and M. Q. Zhang, J. Med. Chem., 1997, 40, 1075 CrossRef CAS.
- M. E. Burow, S. M. Boue, B. M. Collins-Burow, L. I. Melnik, B. N. Duong, C. H. Carter-Wientjes, S. Li, T. E. Wiese, T. E. Cleveland and J. A. McLachlan, J. Clin. Endocrinol. Metab., 2001, 86, 1750 Search PubMed.
- Z. Gyorgypal and A. Kondorosi, Mol. Gen. Genet., 1991, 226, 337 Search PubMed.
- P. W. Groundwater, K. R. Solomons, J. A. Drewe and M. A. Munawar, Prog. Med. Chem., 1996, 33, 233 Search PubMed.
- G. W. Peet and J. Li, J. Biol. Chem., 1999, 274, 32655 CrossRef CAS.
- S. Ghosh, M. J. May and E. B. Kopp, Annu. Rev. Immunol., 1998, 16, 225 CrossRef CAS.
- R. Stoll, C. Renner, S. Hansen, S. Palme, C. Klein, A. Belling, W. Zeslawski, M. Kamionka, T. Rehm, P. Muhlhahn, R. Schumacher, F. Hesse, B. Kaluza, W. Voelter, R. A. Engh and T. A. Holak, Biochemistry, 2001, 40, 336 CrossRef CAS.
- M. Dai, Y. Feng and P. J. Tonge, J. Am. Chem. Soc., 2001, 123, 506 CrossRef CAS.
- R. Erdmann and W. H. Kunau, Yeast, 1994, 10, 1173 CAS.
- F. Kreuzaler and K. Hahlbrock, Eur. J. Biochem., 1975, 56, 205 CAS.
- F. Kreuzaler, R. J. Light and K. Hahlbrock, FEBS Lett., 1978, 94, 175 CrossRef CAS.
- F. Kreuzaler, H. Ragg, W. Heller, R. Tesch, I. Witt, D. Hammer and K. Hahlbrock, Eur. J. Biochem., 1979, 99, 89 CAS.
- T. Lanz, S. Tropf, F. J. Marner, J. Schröder and G. Schröder, J. Biol. Chem., 1991, 266, 9971 CAS.
- S. Tropf, B. Kärcher, G. Schröder and J. Schröder, J. Biol. Chem., 1995, 270, 7922 CrossRef CAS.
- J. M. Jez and J. P. Noel, J. Biol. Chem., 2000, 275, 39640 CrossRef CAS.
- D. Y. Suh, J. Kagami, K. Fukuma and U. Sankawa, Biochem. Biophys. Res. Commun., 2000, 275, 725 CrossRef CAS.
- T. Kortemme and T. E. Creighton, J. Mol. Biol., 1995, 253, 799 CrossRef CAS.
- T. Akiyama, M. Shibuya, H. M. Liu and Y. Ebizuka, Eur. J. Biochem., 1999, 263, 834 CrossRef CAS.
- C. Beckert, C. Horn, J.-G. Schnitzler, A. Lehning, W. Heller and M. Veit, Phytochemistry, 1997, 44, 275 CrossRef CAS.
- Y. Yamazaki, D. Y. Suh, W. Sitthithaworn, K. Ishiguro, Y. Kobayashi, M. Shibuya, Y. Ebizuka and U. Sankawa, Planta, 2001, 214, 75 CAS.
- S. Tropf, T. Lanz, S. A. Rensing, J. Schröder and G. Schröder, J. Mol. Evol., 1994, 38, 610 CrossRef CAS.
- M. L. Durbin, G. H. Learn, Jr., G. A. Huttley and M. T. Clegg, Proc. Natl. Acad. Sci. USA, 1995, 92, 3338 CAS.
- Y. Helariutta, M. Kotilainen, P. Elomaa, N. Kalkkinen, K. Bremer, T. H. Teeri and V. A. Albert, Proc. Natl. Acad. Sci. USA, 1996, 93, 9033 CrossRef CAS.
- V. Oberholzer, M. L. Durbin and M. T. Clegg, Gen. Genet. Syst., 2000, 75, 1 Search PubMed.
- G. Hrazdina, F. Kreuzaler, K. Hahlbrock and H. Grisebach, Arch. Biochem. Biophys., 1976, 175, 392 CAS.
- K. W. Zuurbier, J. Leser, T. Berger, A. J. Hofte, G. Schröder, R. Verpoorte and J. Schröder, Phytochemistry, 1998, 49, 1945 CrossRef CAS.
- H. Morita, Y. Takahashi, H. Noguchi and I. Abe, Biochem. Biophys. Res. Commun., 2000, 279, 190 CrossRef CAS.
- I. Abe, H. Morita, A. Nomura and H. Noguchi, J. Am. Chem. Soc., 2000, 122, 11242 CrossRef CAS.
- J. M. Jez, M. E. Bowman and J. P. Noel, Proc. Natl. Acad. Sci. USA, 2002, 99, 5319 CrossRef CAS.
- K. H. Choi, R. J. Heath and C. O. Rock, J. Bacteriol., 2000, 182, 365 CrossRef CAS.
- J. M. Jez, M. E. Bowman and J. P. Noel, Biochemistry, 2001, 40, 14829 CrossRef CAS.
- Y. Helariutta, P. Elomaa, M. Kotilainen, R. J. Griesbach, J. Schröder and T. H. Teeri, Plant Mol. Biol., 1995, 28, 47 CrossRef CAS.
- S. Eckermann, G. Schröder, J. Schmidt, D. Strack, R. Edrada, Y. Helariutta, P. Elomaa, M. Kotilainen, I. Kilpelainen, P. Proksch, T. H. Teeri and J. G. Schröder, Nature, 1998, 396, 387 CrossRef CAS.
- J. Schröder and G. Schröder, Z. Naturforsch. C, 1990, 45, 1 Search PubMed.
- N. Rupprich, H. Hildebrand and H. Kindl, Arch. Biochem. Biophys., 1980, 200, 72 CAS.
- S. Schanz, G. Schröder and J. Schröder, FEBS Lett., 1992, 313, 71 CrossRef CAS.
- A. Schoppner and H. Kindl, J. Biol. Chem., 1984, 259, 6806 CAS.
- G. Schröder, J. W. Brown and J. Schröder, Eur. J. Biochem., 1988, 172, 161 CAS.
- J. Fliegmann, G. Schröder, S. Schanz, L. Britsch and J. Schröder, Plant Mol. Biol., 1992, 18, 489 CrossRef CAS.
- F. Melchior and H. Kindl, FEBS Lett., 1990, 268, 17 CrossRef CAS.
- T. Yamaguchi, F. Kurosaki, D. Y. Suh, U. Sankawa, M. Nishioka, T. Akiyama, M. Shibuya and Y. Ebizuka, FEBS Lett., 1999, 460, 457 CrossRef CAS.
- R. Hain, H. J. Reif, E. Krause, R. Langebartels, H. Kindl, B. Vornam, W. Wiese, E. Schmelzer, P. H. Schreier, R. H. Stocker and K. Stenzel, Nature, 1993, 361, 153 CrossRef CAS.
- J. D. Hipskind and N. L. Paiva, Mol. Plant Microbe Interact., 2000, 13, 551 Search PubMed.
- L. Fremont, Life Sci., 2000, 66, 663 CrossRef CAS.
- G. J. Soleas, E. P. Diamandis and D. M. Goldberg, Clin. Biochem., 1997, 30, 91 CrossRef CAS.
- E. H. Siemann and L. L. Creasy, Am. J. Enol. Vitic., 1992, 43, 49 Search PubMed.
- D. Y. Suh, K. Fukuma, J. Kagami, Y. Yamazaki, M. Shibuya, Y. Ebizuka and U. Sankawa, Biochem. J., 2000, 350((Pt 1)), 229 CrossRef CAS.
- R. Gehlert and H. Kindl, Phytochemistry, 1991, 30, 457 CrossRef CAS.
- T. Reinecke and H. Kindl, Phytochemistry, 1994, 35, 63 CrossRef CAS.
- R. Preisig-Muller, P. Gnau and H. Kindl, Arch. Biochem. Biophys., 1995, 317, 201 CrossRef CAS.
- R. Preisig-Muller, R. Gehlert, F. Melchior, U. Stietz and H. Kindl, Biochemistry, 1997, 36, 8349 CrossRef CAS.
- G. Schröder and J. Schröder, J. Biol. Chem., 1992, 267, 20558 CAS.
- N. A. M. Saleh, H. Fritsch, F. Kreuzaler and H. Grisebach, Phytochemistry, 1978, 17, 183 CrossRef CAS.
- A. B. Christensen, P. L. Gregersen, J. Schröder and D. B. Collinge, Plant Mol. Biol., 1998, 37, 849 CrossRef CAS.
- A. Baumert, W. Maier, D. Gröger and R. Deutzmann, Z. Naturforsch., C, 1994, 49, 26 Search PubMed.
- V. S. Parmar, S. C. Jain, S. Gupta, S. Talwar, V. K. Rajwanshi, R. Kumar, A. Azim, S. Malhotra, N. Kumar, R. Jain, N. K. Sharma, O. D. Tyagi, S. J. Lawrie, W. Errington, O. W. Howarth, C. E. Olsen, S. K. Singh and J. Wengel, Phytochemistry, 1998, 49, 1069 CrossRef CAS.
- K. T. Junghanns, R. E. Kneusel, A. Baumert, W. Maier, D. Gröger and U. Matern, Plant Mol. Biol., 1995, 27, 681 CrossRef CAS.
- K. Springob, R. Lukacin, C. Ernwein, I. Groning and U. Matern, Eur. J. Biochem., 2000, 267, 6552 CrossRef CAS.
- R. Lukacin, K. Springob, C. Urbanke, C. Ernwein, G. Schröder, J. Schröder and U. Matern, FEBS Lett., 1999, 448, 135 CrossRef CAS.
- R. Lukacin, S. Schreiner and U. Matern, FEBS Lett., 2001, 508, 413 CrossRef CAS.
- A. M. A. Abd El-Mawla and L. Beerhues, Planta, 2002, 214, 727 CrossRef CAS.
- L. Rocha, A. Marston, M. A. Kaplan, H. Stoeckli-Evans, U. Thull, B. Testa and K. Hostettmann, Phytochemistry, 1994, 36, 1381 CrossRef CAS.
- (a) L. Beerhues, FEBS Lett., 1996, 383, 264 CrossRef CAS; (b) S. Peters, W. Schmidt and L. Beerhues, Planta, 1998, 204, 64 CrossRef CAS.
- W. Schmidt and L. Beerhues, FEBS Lett., 1997, 420, 143 CrossRef CAS.
- M. C. Wildermuth, J. Dewdney, G. Wu and F. M. Ausubel, Nature, 2001, 414, 562 CrossRef CAS.
- N. B. Paniego, K. W. Zuurbier, S. Y. Fung, R. van der Heijden, J. J. Scheffer and R. Verpoorte, Eur. J. Biochem., 1999, 262, 612 CrossRef CAS.
- Y. Okada and K. Ito, Biosci. Biotechnol. Biochem., 2001, 65, 150 Search PubMed.
- W. Borejsza-Wysocki and G. Hrazdina, Plant Physiol., 1996, 110, 791 CAS.
- I. Abe, Y. Takahashi, H. Morita and H. Noguchi, Eur. J. Biochem., 2001, 268, 3354 CrossRef CAS.
- J. Schröder, Trends Plant Sci., 1997, 2, 373 CrossRef.
- J. Schröder, S. Raiber, T. Berger, A. Schmidt, J. Schmidt, A. M. Soares-Sello, E. Bardshiri, D. Strack, T. J. Simpson, M. Veit and G. Schröder, Biochemistry, 1998, 37, 8417 CrossRef CAS.
- I. Atanassov, E. Russinova, L. Antonov and A. Atanassov, Plant Mol. Biol., 1998, 38, 1169 CrossRef CAS.
- I. E. Burbulis, M. Iacobucci and B. W. Shirley, Plant Cell, 1996, 8, 1013 CrossRef CAS.
- B. Ylstra, M. Muskens and A. J. Van Tunen, Plant Mol. Biol., 1996, 32, 1155 CrossRef CAS.
- J. Rozema, R. A. Broekman, P. Blokker, B. B. Meijkamp, N. de Bakker, J. van de Staaij, A. van Beem, F. Ariese and S. M. Kars, J. Photochem. Photobiol. B, 2001, 62, 108 Search PubMed.
- J. Gorham, in ‘Bryophytes: Their Chemistry and Chemical Taxonomy’, Proceedings of The Phytochemical Society of Europe, Vol. 29, pp. 171–200, Oxford University Press, 1990.
- R. Mechoulam and S. Ben-Shabat, Nat. Prod. Rep., 1999, 16, 131 RSC.
- M. Fellermeier, W. Eisenreich, A. Bacher and M. H. Zenk, Eur. J. Biochem., 2001, 268, 1596 CrossRef CAS.
- H. Baer, Clin. Dermatol., 1986, 4, 152 CrossRef CAS.
- P. M. Dewick, Medicinal Natural Products: a Biosynthetic Approach, Wiley, Chichester, 1997 Search PubMed.
- D. H. Hong, S. B. Han, C. W. Lee, S. H. Park, Y. J. Jeon, M. J. Kim, S. S. Kwak and H. M. Kim, Arch. Pharm. Res., 1999, 22, 638 Search PubMed.
- T. Rosen and D. B. Fordice, South Med. J., 1994, 87, 543 Search PubMed.
- R. Grazzini, D. Hesk, E. Yerger, D. Cox-Foster, J. Medford, R. Craig and R. O. Mumma, J. Am. Soc. Hortic Sci., 1995, 120, 343 Search PubMed.
- D. J. Schultz, E. B. Cahoon, J. Shanklin, R. Craig, D. L. Cox-Foster, R. O. Mumma and J. I. Medford, Proc. Natl. Acad. Sci. USA, 1996, 93, 8771 CrossRef CAS.
- B. E. Scheffler, S. O. Duke, F. E. Dayan and E. Ota, in Regulation of Phytochemicals by Molecular Techniques, Chapter 12, p. 257, Pergamon Press, Amsterdam, 2001 Search PubMed.
- C. I. Nimbal, J. F. Pederson, C. N. Yerkes, L. A. Weston and S. C. Weller, J. Agric. Food Chem., 1996, 44, 1343 CrossRef CAS.
- G. Fate, M. Chang and D. G. Lynn, Plant Physiol., 1990, 93, 201 CAS.
- B. S. Moore and J. N. Hopke, ChemBioChem, 2001, 2, 35 CrossRef CAS.
- J. M. Henson, M. J. Butler and A. W. Day, Annu. Rev. Phytopathol., 1999, 37, 447 Search PubMed.
- M. J. Butler and A. J. Day, Can. J. Microbiol., 1998, 44, 1115 CrossRef CAS.
- I. Fujii, Y. Mori, A. Watanabe, Y. Kubo, G. Tsuji and Y. Ebizuka, Biochemistry, 2000, 39, 8853 CrossRef CAS.
- H.-F. Tsai, Y. C. Chang, R. G. Washburn, M. H. Wheeler and K. J. Kwon-Chung, J. Bacteriol., 1998, 180, 3031 CAS.
- K. Ueda, K. M. Kim, T. Beppu and S. Horinouchi, J. Antibiot. (Tokyo), 1995, 48, 638 Search PubMed.
- N. Funa, Y. Ohnishi, I. Fujii, M. Shibuya, Y. Ebizuka and S. Horinouchi, Nature, 1999, 400, 897 CrossRef CAS.
- B. S. Moore, personal communication, 2001.
- N. Funa, Y. Ohnishi, Y. Ebizuka and S. Horinouchi, J. Biol. Chem., 2002, 277, 4628 CrossRef CAS.
- Y. M. Zhang, M. S. Rao, R. J. Heath, A. C. Price, A. J. Olson, C. O. Rock and S. W. White, J. Biol. Chem., 2001, 276, 8231 CrossRef CAS.
- C. Keel, U. Schnider, M. Maurhofer, C. Voisard, J. Laville, U. Burger, P. Wirthner, D. Haas and G. Defago, Mol. Plant Microbe Interact., 1992, 5, 4 Search PubMed.
- J. M. Raaijmakers, D. M. Weller and L. S. Thomashow, Appl. Environ. Microbiol., 1997, 63, 881 CAS.
- C. Picard, F. Di Cello, M. Ventura, R. Fani and A. Guckert, Appl. Environ. Microbiol., 2000, 66, 948 CrossRef CAS.
- U. F. Walsh, J. P. Morrissey and F. O'Gara, Curr. Opin. Biotechnol., 2001, 12, 289 CrossRef CAS.
- M. G. Bangera and L. S. Thomashow, J. Bacteriol., 1999, 181, 3155 CAS.
- S. J. Hammond, M. P. Williamson, D. H. Williams, L. D. Boeck and G. G. Marconi, J. Chem. Soc., Chem. Commun., 1982, 344 RSC.
- O. Choroba, D. Williams and J. Spencer, J. Am. Chem. Soc., 2000, 122, 5389 CrossRef CAS.
- H. Chen, C. C. Tseng, B. K. Hubbard and C. T. Walsh, Proc. Natl. Acad. Sci. USA, 2001, 98, 14901 CrossRef CAS.
- A. M. van Wageningen, P. Kirkpatrick, D. Williams, B. Harris, J. Kershaw, N. Lennard, M. Jones, S. Jones and P. Solenberg, Chem. Biol., 1998, 5, 155 CrossRef.
- T.-L. Li, O. Choroba, H. Hong, D. Williams and J. Spencer, Chem. Commun., 2001, 20, 2156 RSC.
- V. Pfeifer, G. J. Nicholson, J. Ries, J. Recktenwald, A. B. Schefer, R. M. Shawky, J. G. Schröder, W. Wohlleben and S. Pelzer, J. Biol. Chem., 2001, 276, 38370 CrossRef CAS.
- B. K. Hubbard, M. G. Thomas and C. T. Walsh, Chem. Biol., 2000, 7, 931 CrossRef CAS.
- S. R. Muir, G. J. Collins, S. Robinson, S. Hughes, A. Bovy, C. H. Ric De Vos, A. J. van Tunen and M. E. Verhoeyen, Nat. Biotechnol., 2001, 19, 470 CrossRef CAS.
- R. A. Dixon, C. J. Lamb, S. Masoud, V. J. Sewalt and N. L. Paiva, Gene, 1996, 179, 61 CrossRef CAS.
- X. Dong, E. L. Braun and E. Grotewold, Plant Physiol., 2001, 127, 46 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |