The biosynthesis of shikimate metabolites
Andrew R.
Knaggs
Analytical Sciences Department, Discovery Research, GlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire, UK SG1 2NY
First published on 29th November 2002
Abstract
Covering 2000. Previous review: Nat. Prod. Rep., 2001, 18, 334
This review covers the literature published during 2000 on the biosynthesis of compounds derived wholly or partly from intermediates on the shikimate pathway. Recent developments in the enzymology and genetics of the shikimate pathway are also described. Enzymes involved in the biogenetic pathway to the aromatic amino acids are covered initially followed by sections dedicated to metabolites derived in some part from intermediates on the pathway. These include pyrrolnitrin, violacein, indole-3-acetic acid, coumarins, lignans, lignin, tannins, melanin, flavanoids, ubiquinone, TOPA quinone, PQQ, and tropanes.
 Andrew R. Knaggs | Andrew Knaggs was born in Darlington, County Durham, in 1965. He attended the University of Leeds where he received his Bachelors degree in Chemical Sciences in 1987. He remained at the University of Leeds carrying out research with Dr Richard B. Herbert into the biosynthesis of the Pseudomonas β-lactone metabolite, obafluorin. Upon completion of his PhD in 1990 he moved to the group of Professor Heinz G. Floss at the University of Washington, Seattle, where he spent 2 years investigating various aspects of the biosynthesis of the cyclic thiopeptide, nosiheptide, and the plant metabolite taxol. He returned to the UK in 1993 to take up a position in the Natural Products department at Glaxo Group Research, Greenford, where he was involved in both natural product chemistry and biotransformation projects. He is currently working in the Analytical Sciences Department at the GlaxoSmithKline Medicines Research Centre, Stevenage. |
1 Introduction
This report reviews the literature published during 2000 on the biosynthesis of compounds derived wholly or partly from intermediates on the shikimate pathway. Recent developments in the enzymology and genetics of the shikimate pathway are also described. This review will concentrate mainly on biochemical and enzymological aspects of the biosynthetic pathways to the secondary metabolites. However, in many cases descriptions of the genetic nature and organisation of the biosynthetic pathways will also be reported. Background information on many of the sections in this review can be seen in a previous review.1 I have endeavoured to be as thorough and accurate as possible in writing this review, but I realise I may have missed some relevant work. I would be very grateful for any correspondence regarding such omissions so that they may be included in next year's review.2 The shikimate pathway
The regulatory mechanisms controlling the first metabolic branch point in the shikimate pathway (Scheme 1) have been the subject of some recent investigations in Saccharomyces cerevisiae using recombinant DNA technology.2 At this metabolic node chorismate (10) can either follow a biosynthetic pathway to the aromatic amino acids L-tyrosine and L-phenylalanine through the action of chorismate mutase or follow an alternative anabolic route via the enzyme anthranilate synthase to form L-tryptophan. The activities of chorismate mutase (CM) and anthranilate synthase (AAS) are regulated by different mechanisms. The AAS complex is known to be transciptionally regulated and in addition is under allosteric control via feedback inhibition by L-tryptophan. In contrast, CM activity is not transcriptionally regulated but instead is allosterically modulated by the inhibitory effect of L-tyrosine and the activating potential of L-tryptophan. The various regulatory elements operating at the chorismate branch point have been studied in detail by means of genetic manipulation. The results of these experiments indicated that the allosteric and transcriptional control of S. cerevisiae CM and AAS are interconnected and suggest a continual coevolution of these regulatory systems might have occurred.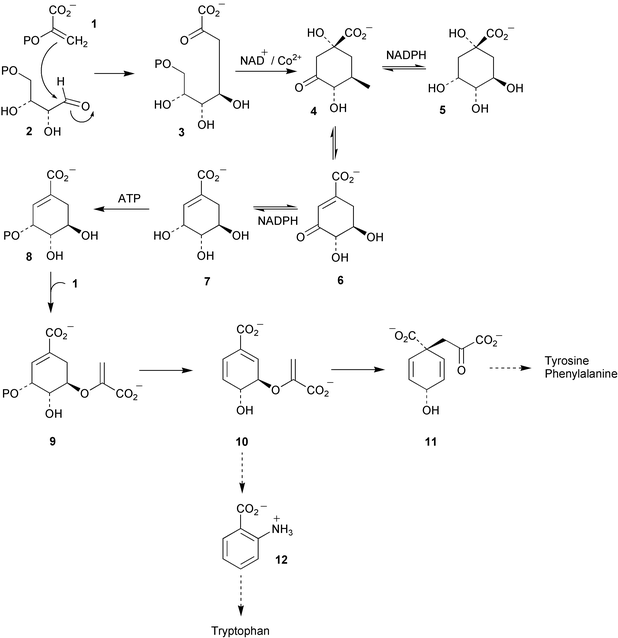 | ||
| Scheme 1 | ||
2.1 DAHP synthase
Two type II and one type I 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) synthase genes have been identified in the myxobacterium Stigmatella aurantiaca.3 Type I DAHP synthase is known to be inhibited by L-phenylalanine, L-tyrosine, and L-tryptophan whereas the corresponding type II enzyme is not inhibited by aromatic amino acids but is instead activated by L-tryptophan. Microorganisms do not usually possess both type II and type I DAHP synthases and this represents the first report of such a case. One of the type II DAHP isozymes was found to play a role in the biosynthetic pathway to the family of electron transport inhibitors called the aurachins (see aurachin A 13) known to derive from anthranilate (12) and acetate.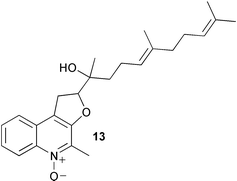
2.2 3-Dehydroquinase
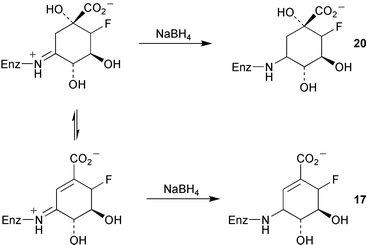
The mechanisms of type I and type II dehydroquinase have been probed in studies with the fluorinated intermediates (6R)- (14) and (6S)-6-fluoro-3-dehydroquinic acids (15, Scheme 2).4 Both were found to be substrates for the type I and II enzymes. The rate of turnover of 14 and 15 was found to be slower than that of the natural substrate (3-dehydroquinic acid, 4) and interestingly the stereochemical orientation of the fluorine was found to have a marked effect on the kinetics of the type I and II dehydroquinases. The differential effects shown in the kinetic data obtained from both a catabolic and biosynthetic type II enzyme were rationalised in terms of the stereoelectronic interaction of the fluorine atom with the developing conjugated system in the hydroxy elimination step using the currently accepted enolate mechanism (Scheme 2). The larger reduction of kcat observed for the catabolic dehydroquinase with fluorinated substrates was consistent with previous results which indicated that the hydroxy removal step is partially rate determining for this enzyme. Differential effects of 14 and 15 were also seen in sodium borohydride trapping experiments with a type I dehydroquinase. Incubation of 14 in the presence of the corresponding product 16 gave rise to trapped product imine 17 (Scheme 3). In contrast, incubations of 15 and product 18 with the type I dehydroquinase yielded a modified protein of mass consistent with the presence of reduced substrate imine 20. This represents the first indirect observation of the substrate imine in a type I dehydroquinase and has been postulated to occur due to the destabilisation of the product imine by the axial fluorine relative to the corresponding substrate imine complex.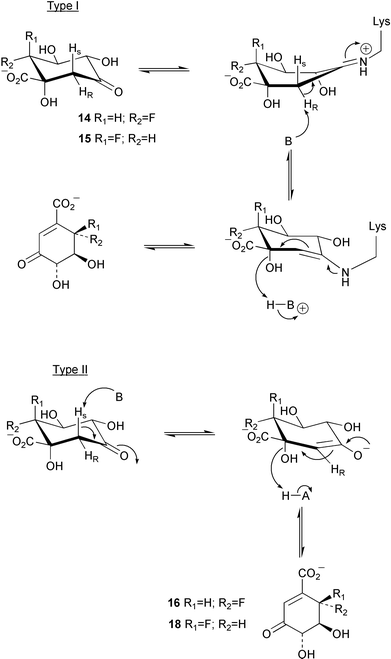 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
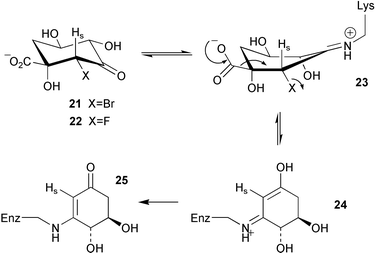
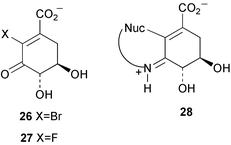
The same group have now reported the design of 3 specific irreversible inhibitors of type I dehydroquinase.5 The binding of (2R)-2-bromo-3-dehydroquinic acid (21) and (2R)-2-fluoro-3-dehydroquinic acid (22) to the enzyme via the imine 23 (Scheme 4) is thought to move the halogen atom into an anti-periplanar like orientation with respect to the carboxylate group and promotes decarbonylative elimination to produce the enol-imine intermediate 24. Subsequent tautomerisation of N then gives the vinylogous amide 25 which prevents any further catalysis by the enzyme. Both 21 and 22 are readily accepted by type II dehydroquinase and are converted smoothly, without causing inhibition, to the corresponding 2-fluoro and 2-bromo-3-dehydroshikimic acids (26 and 27 respectively). 2-Bromo-3-dehydroshikimic acid 26 was also found to cause the irreversible inhibition of type I dehydroquinase. However, in this case the mode of inactivation is thought to be via initial imine formation followed by the conjugate addition of an active site nucleophile to give the adduct 28.
 | ||
| Scheme 4 | ||
2.3 Shikimate dehydrogenase
Shikimate dehydrogenase from Escherichia coli has been overexpressed and purified.6 The enzyme was subsequently crystallised by the vapour-diffusion method with ammonium sulfate as precipitant yielding monoclinic crystals. X-Ray diffraction data were obtained using cryo-cooling as the crystals were found to be radiation sensitive and produced a data set to 2.3 Å resolution. Work is ongoing to solve the structure by multiple isomorphous replacement.2.4 Shikimate kinase
Shikimate kinase from the hyperthermophilic bacterium Aquifex aeolicus has been cloned and overproduced in E. coli.7 Uniformly 15N- and 15N/13C-labelled enzyme was produced by fermentation using 15N and 13C enriched nitrogen and carbon sources. The purified enzyme was analysed by multinuclear multidimensional NMR spectroscopy in complexation with its substrate shikimate and resulted in the almost total assignment of resonances. The assignment data have been deposited in the BioMagResBank database.2.5 5-Enolpyruvylshikimate 3-phosphate synthase
A gene encoding 5-enolpyruvylshikimate 3-phosphate (EPSP) synthase (aroA) has been cloned from Streptococcus pneumoniae and overexpressed in E. coli.8 The resulting enzyme was purified to homogeneity. The AroA protein has a mass of 45.8 kDa, contains 427 amino acid residues, and shows 53% sequence identity with B. subtilis EPSP synthase and 25% with the corresponding enzyme in E. coli. The S. pneumoniae EPSP synthase was found to be activated in the forward reaction by the presence of univalent cations, with NH4+, Rb+, and K+ being the most effective ions studied. In contrast, the sensitivity of the reverse reaction to these cations was very low. The activation observed followed saturation kinetics with the saturation concentration being at a much higher level than that of PEP (phosphoenolpyruvate, 1) and shikimate 3-phosphate (8). Glyphosate (29) was shown to be a competitive inhibitor with respect to PEP and interestingly, in the presence of NH4+ and K+, the potency of this inhibition was raised markedly. It was hypothesised that the univalent cations act by changing the conformation of EPSP synthase resulting in the modulation of substrate binding and enzyme catalysis.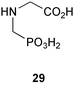
The same group have also reported the results from some detailed steady-state kinetic studies on the S. pneumoniae EPSP synthase.9 The data obtained seemed to support a random sequential mechanism for the forward reaction with respect to the binding of the substrates PEP and shikimate 3-phosphate. Interestingly, investigations into the inhibition of glyphosate on the reverse reaction indicated that a non-productive quaternary enzyme complex with EPSP (9), glyphosate, and phosphate may exist.
2.6 Chorismate synthase
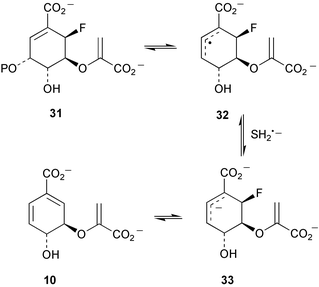
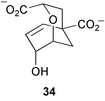
Kinetic isotope effects have been observed with incubations of [4-2H]EPSP (as 30) with chorismate synthase from Neurospora crassa and Escherichia coli.10 These have been rationalised in terms of the operation of secondary β isotope effects and indicate that cleavage of the carbon–oxygen bond at C-3 occurs prior to cleavage of the proR hydrogen at C-6 consistent with either a non-concerted E1 or radical reaction mechanism (Scheme 5). Some additional studies by the same group with E. coli chorismate synthase have now provided evidence that strongly supports the operation of a radical mechanism.11 Incubation of (6R)-6-fluoro-EPSP (31, Scheme 6) with the enzyme in the presence of the reducing agent dithionite gave rise to chorismate and was linked to the oxidation of flavin mononucleotide (FMN) to its corresponding semiquinone. It was proposed that a single electron transfer from FMN to 31 initiates the elimination of phosphate yielding the allylic radical intermediate 32. 32 is unable to transfer an electron and a proton to the oxidised flavin cofactor due to the fluorine at the 6R-position and is consequently reduced by dithionite to the allylic anion 33 which readily undergoes fluoride elimination to produce chorismate 10. The formation of flavin semiquinone was also observed during incubations with (6S)-6-fluoro-EPSP in the presence of dithionite. No enzyme activity was detected with 1,5-dihydro-5-deaza-FMN confirming the essential catalytic role of FMN in E. coli chorismate synthase and giving further support to the postulated radical mechanism. Results of thermodynamic studies on the conversion of chorismate into isochorismate catalysed by isochorismate synthase have been reported.12 At T = 311.15 K and under physiological conditions (pH = 7, pMg = 3.0, and Im = 0.25 mol kg−1) the apparent equilibrium (K′) constant was 0.83 and the standard molar transformed Gibbs energy change ΔrG′°m = 0.48 kJ mol−1.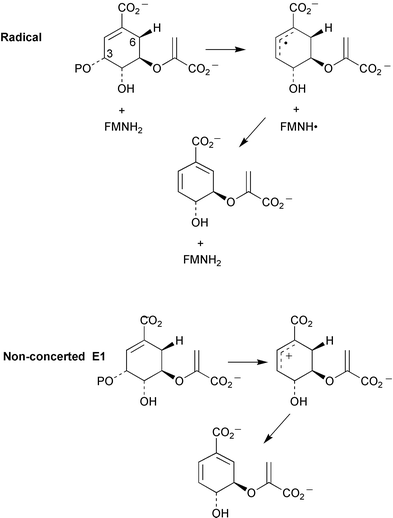 | ||
| Scheme 5 | ||
 | ||
| Scheme 6 | ||
2.7 Chorismate mutase
Hybrid quantum mechanical (QM)/molecular mechanical (MM) methods have recently been applied to the chorismate mutase reaction.13 The calculations predicted the barrier for the conversion of chorismate (10) into prephenate (11) to be 1.4 kcal mol−1 at the active site of the enzyme compared to a value of 29.1 kcal mol−1 in the gas phase. Data obtained indicate that reactant compression did not significantly contribute to catalysis, however, the interaction of substrate carboxylate groups and ether oxygen with nearby arginine residues was shown to be an important factor for catalytic activity. QM/MM studies have also been used to study conformational equilibria of chorismate in the chorismate mutase active site.14 The data obtained indicated that the major factor influencing the energetic order of the conformers studied was the deformation energy of the enzyme required to mould substrate into the active site.A gene (HAR07) encoding a chorismate mutase (CM) has been characterised and cloned from the methylotrophic yeast Hansenula polymorpha.15 The gene product is a 280 amino acid monofunctional protein showing strong similarities to the chorismate mutase of the related yeast Saccharomyces cerevisiae and thus belongs to the AroQ class of chorismate mutase. The enzyme has a higher optimum temperature for activity than S. cerevisiae CM but displays lower thermal stability. The activity of the H. polymorpha enzyme is strictly regulated by a catabolite repression system but levels of HARO7 are not effected by amino acid starvation. Tryptophan was found to be a positive effector whereas tyrosine was found to inhibit the activity of the chorismate mutase.
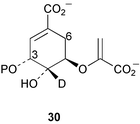
A crystal structure of the Bacillus subtilis chorismate synthase homotrimer has been determined to a resolution of 1.30 Å.16 The 3 active sites are found at the subunit interfaces with residues from 2 subunits playing active roles in each site. Under the crystallisation conditions employed one glycerol molecule and one sulfate ion were found to be present at each catalytic centre mimicking the transition-state analogue 34.
Detailed kinetic and structural studies have been carried out on functional variants of B. subtilis chorismate mutase formed through combinatorial mutagenesis.17 Removal of the conserved active site residue Arg-90 severely reduced catalytic activity. It was found that this could be recovered to a large extent by substitution of lysine at either position 90 or 88. Crystal structures of these variants showed that the conformations of these lysine residues would bring the ε-ammonium group within hydrogen-bonding distance of the substrate ether oxygen during the transition state. The results support the proposal that a strategically placed cation capable of stabilising the developing negative charge in the transition state is needed for full catalytic efficiency.
The role of the flexible C-terminal tail of B. subtilis chorismate mutase has been investigated using a novel random protein truncation strategy which essentially consisted of introducing premature termination codons into the C-terminal coding sequence.18 The resulting activities of the enzyme variants indicated that none of the last 17 residues are essential for catalysis and in fact the final 11 amino acids can be deleted without significantly altering activity. A preference however is shown for Lys-111, Ala-112, Leu-115 and Arg-116 and kinetic data suggested a potential role for the C-terminal residues to aid in the uniform binding of the substrate and transition state.
2.8 Prephenate dehydratase
A gene coding for a bifunctional chorismate mutase-prephenate dehydratase protein (CM/PDT) has been cloned and sequenced from the prokaryotic aphid endosymbiont Buchnera aphidicola.19 Detailed comparison of this gene sequence with ones derived from other organisms showed that the B. aphidicola gene lacked an attenuator region and implied the constitutive expression of the CM/PDT gene in the endosymbiont. In addition, a highly conserved PDT allosteric binding site characterised by the amino acid sequence Glu-Ser-Pro-Arg was severely altered. These findings suggested that prephenate dehydratase in B. aphidicola was not sensitive to end product inhibition by L-phenylalanine, and taken together with the apparent absence of gene regulation, indicated that the overproduction of this aromatic amino acid readily occurs in this symbiotic organism.Site-directed mutagenesis of the Escherichia coli P-protein prephenate dehydratase domain has been carried out in order to identify the key residues involved in substrate binding and catalysis.20 The data obtained confirmed the importance of Thr-278 in catalysis possibly via hydrogen bonding to prephenate or another catalytic residue. Curiously mutation of 9 conserved acidic residues, which could potentially play a role in an acid catalysed dehydration mechanism, did not cause any significant decrease in enzyme activity. It was suggested that E. coli PDT perhaps only requires a hydrogen bonding type interaction to promote the dehydration–decarboxylation of prephenate. Cys-216 and the adjacent Gln-215 were shown to have roles in binding substrate although kinetic data suggested that Gln-215 may also play a part in stabilising the transition state. Results indicated that Ser-208 and Trp-226 were also involved in prephenate binding whereas Asn-160 was likely to be a catalytic residue. Interestingly, point mutations at the acidic residues, Glu-159 and Glu-232 (mutation to alanine), gave rise to PDT with 2–3 fold greater activity and was mainly the result of improved substrate binding. It was suggested that this could arise through decreases in unfavourable ionic interactions, creation of a more accessible active site, or from conformational changes bringing about better prephenate binding.
2.9 Cyclohexadienyl dehydrogenase
A recent report details the cloning of a gene encoding a cyclohexadienyl dehydrogenase (tyrAc) from Pseudomonas stutzeri.21 The gene was subsequently sequenced, overexpressed in E. coli, and the purified enzyme fully characterised. This work represents the first detailed molecular-genetic and biochemical study of a feedback sensitive cyclohexadienyl dehydrogenase. The enzyme was shown to have much better activity with prephenate (11) than arogenate (35) and was inhibited by the products 4-hydroxyphenylpyruvate and L-tyrosine, competitively with respect to arogenate and prephenate, and non-competitively with respect to the cofactor NAD+. Several other related compounds were found to act as inhibitors and indicated that the major structural features required for efficient inhibition were an aromatic ring with a hydroxy or amino substituent. Following the comparison of amino acid sequences of related TyrA family members, including ones both sensitive and insensitive to endproduct inhibition, it was proposed that the mode of inhibition probably operates via competitive binding at the catalytic site and may not occur through interactions at a discrete allosteric domain.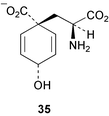
2.10 Chorismate lyase
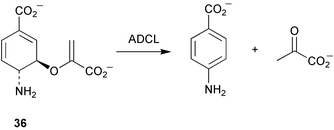
Crystallisation studies with chorismate lyase have been reported.22 Due to difficulties in crystal growth with the wild type enzyme, protein modification and mutation techniques were tested. Mutation of two cysteine residues to serine gave rise to active enzyme which crystallised in 3 different forms. One of these (triclinic) yielded diffraction data to 1.1 Å resolution. Work is now concentrating on obtaining a selenomethionyl version of chorismate lyase to enable the use of multiple wavelength anomalous dispersion (MAD) phasing for structure determination.The X-ray structure of 4-amino-4-deoxychorismate lyase (ADCL) from Escherichia coli has been solved to 2.2 Å resolution.23 ADCL is a pyridoxal phosphate dependent enzyme belonging to fold type IV (Re-face specific for proton transfer to C-4′ of cofactor) and catalyses the conversion of 4-amino-4-deoxychorismate (36) to 4-aminobenzoate and pyruvate (Scheme 7). The three-dimensional data obtained showed ADCL folded into a dimeric form with each subunit possessing 2 domains (large and small). The active site is located at a domain-subunit interface with residues from one subunit and the small domain of the other subunit participating in the catalytic cleft. The structural data allowed the confirmation of the Re-face specificity of E. coli ADCL and subsequent model studies with substrate suggested that the carboxylates of 4-amino-4-dexoychorismate form interactions with Asn-256, Arg-107, and Lys-97 and that the cyclohexadiene moiety forms Van der Waals contact with Leu-258. The Lys-159 residue forms an imine with the pyridoxal cofactor and when released on substrate binding is thought to play a role in the abstraction of the proton at C-4 of the substrate. In addition, it is proposed that Thr-38 acts to transfer a proton to the methylene carbon of the enol-pyruvyl side chain during the elimination process.
 | ||
| Scheme 7 | ||
3 Tryptophan and related compounds
3.1 Tryptophan
The results of initial biochemical analyses of a thermostable tryptophan synthase (TS) from the hyperthermophilic archaeon Thermococcus kodakaraensis have now been published.24 This represents the first report of an archaeal TS and the first biochemical analysis of a thermostable TS at high temperature. TrpA and TrpB encoding the α and β subunit of T. kodakaraenis TS were expressed independently in E. coli and the TS complex produced by heat treatment of a mixture of cell-free extracts of the corresponding subunits. The α subunit was shown to exist as a monomer, the β subunit as a dimer, and the TS complex itself as a tetramer (α2β2). Comparison of the T. kodakaraensis TS sequence with other characterised enzymes, and kinetic data obtained with the TS complex and the α (TrpA) and β (TrpB) subunits in isolation, suggested the presence of a tunnel connecting the active sites of the α and β subunits in the enzyme complex. The TS complex was found to have a higher thermal stability than the independent subunits and displayed much higher activity in the α and β reactions, implying that a cooperative folding of the α and β subunits takes place giving rise to the fully active complex. The activity of TrpB was found to markedly increase between 90 and 100 °C but at the same time a drastic decrease in thermostability was observed. It was proposed that the high temperatures induce a structural change in TrpB which results in better access to the active site for the substrate indole. In contrast, TrpA only showed low level activity at all temperatures studied. Furthermore, the observed decrease in TS activity at temperatures above 80 °C is thought to be due to denaturation of the α subunit.The trpBA gene cluster has now been isolated and sequenced from the nitrogen-fixing bacterium Azospirillum brasilense.25 The trpA start codon was found to overlap with the trpB stop codon suggesting that the translation of both genes is coupled resulting in the biosynthesis of equimolar quantities of the 2-tryptophan synthase subunits. Curiously sequences indicating the operation of attenuation or transcriptional control could not be found upstream of the trpBA cluster.
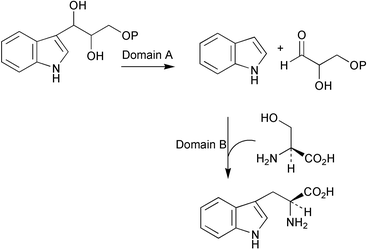
The gene encoding tryptophan synthase (trpB) in the filamentous fungus Aspergillus nidulans has been cloned and characterised.26 Interestingly, the deduced protein encoded by trpB (723 amino acids in length, calculated molecular weight 77.6 kDa) was found to possess a substantially longer connector sequence, linking domain A (catalysing the cleavage of 3-indole-D-glycerol 3-phosphate to indole and D-glyceraldehyde 3-phosphate) and domain B (catalysing the condensation of L-serine with indole) (Scheme 8), than seen in other fungal TrpB characterised to date. Regulation of trpB transcription is achieved through cross-pathway control via the binding of the regulatory protein CPCA (cross pathway control activator) to a single site. A knockout mutant for trpB showed tryptophan auxotrophy and gave rise to severe growth defects but did not affect the biosynthesis of L-tryptophan to auxin.
 | ||
| Scheme 8 | ||
The regulation of the tryptophan biosynthetic operon in Bacillus subtilis occurs at the transcriptional level via an attenuation mechanism mediated by the trp RNA-binding attenuation protein (TRAP) and with levels of tryptophan acting as a signal.27 Recent studies have now indicated that B. subtilis also possesses an operon which responds to the level of uncharged tRNATrp by the production of a transcript which limits the action of TRAP. Thus as with many other bacteria B. subtilis seems to operate control mechanisms which recognise both tryptophan levels and the availability of charged tRNATrp. The operon involved in this control mechanism contains the genes yczA and ycbK both with unknown function and at present the exact nature of their interaction with TRAP is not clear.
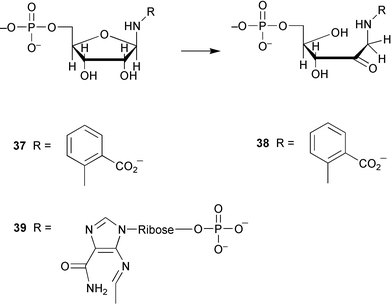
Phosphoribosylanthranilate isomerase (TrpF) catalyses the irreversible rearrangement (Amadori) of the aminoaldose 37 to the corresponding aminoketose 38 in the tryptophan biosynthetic pathway (Scheme 9). TrpF belongs to the same structural class of enzyme as HisA, N′-[(5′-phosphoribosyl)formimino]-5-aminoimidazole-4-carboxamide ribonucleotide isomerase, which catalyses an analogous rearrangement of aminoaldose 39. In some recent work an enzyme has been generated which is capable of catalysing both the HisA and TrpF reactions.28 This was achieved through the directed evolution of the HisA protein involving random mutagenesis and selection. This work suggests that HisA and TrpF may have evolved from the same ancestral enzyme possessing less substrate specificity.
 | ||
| Scheme 9 | ||
3.2 Pyrrolnitrin
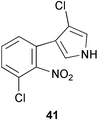
Tryptophan 7-halogenase (PrnA) which catalyses the regioselective chlorination of tryptophan to 7-chlorotryptophan (40) in the first step of the biosynthetic pathway to pyrrolnitrin (41) has now been purified to homogeneity.29 The activity of PrnA was found to be dependent on the presence of a flavin reductase and had an absolute requirement for oxygen, FAD, and NADH. A hypothetical reaction mechanism for the halogenation was proposed (Scheme 10) and involves the formation of an epoxide intermediate (42) mediated by flavin hydroperoxide species. Subsequent regioselective attack of chloride ion on 42 followed by specific elimination of water gives rise to the chlorinated product (40). The biosynthesis of pyrrolnitrin and other phenylpyrrole metabolites by bacteria has been the subject of a recent review.30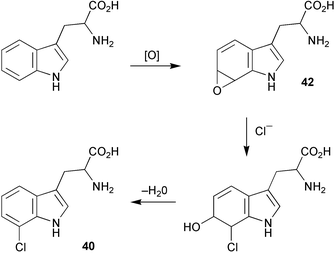 | ||
| Scheme 10 | ||
3.3 Violacein
The nature of the intramolecular rearrangement of indole and the origin of the oxindole portion of violacein (43, Scheme 11) have been probed by some elegant mixed feeding studies with singly labelled tryptophans.31 Results from feeding studies with equimolar mixtures of [2-13C]- and [indole-3-13C]tryptophan, and separately [3 13C]- and [indole-3-13C]tryptophan, provided strong evidence for the involvement of an intramolecular 1,2-shift of indole during the biosynthesis of the 5-hydroxyindole portion of violacein. In addition, incorporation studies with equimolar mixtures of [2-13C]- and [α-15N]tryptophan showed that the nitrogen in the pyrrolidone ring derives exclusively from the tryptophan molecule which forms the oxindole part of 43. This indicated that this portion of violacein originates from the intact incorporation of a tryptophan molecule (Scheme 11) with decarboxylation probably occurring at a later stage in the biosynthetic pathway (tryptamine 47 was not an efficient precursor of violacein). The exact nature of the intermediate which condenses with tryptophan to form the 5-hydroxyindole portion of violacein is not yet known although evidence obtained so far seems to exclude indolepyruvic acid.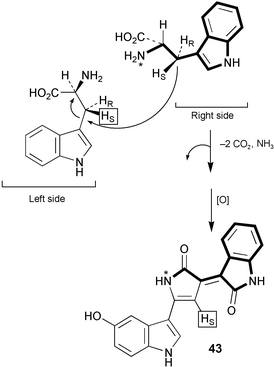 | ||
| Scheme 11 | ||
3.4 Indole-3-acetic acid
Two genes encoding cytochrome P450 enzymes have been isolated and characterised from Arabidopsis and have been shown to be capable of converting L-trytophan to indole-3-acetaldoxime (44, Scheme 12).3244 is a postulated intermediate in the tryptophan dependent indole-3-acetonitrile (46, IAN) biosynthetic pathway to indole-3-acetic acid (45, IAA) and also in indole glucosinolate biosynthesis (Scheme 12). It has been proposed that these enzymes may be responsible for the catalysis of the first step of the IAN pathway in Arabidopsis. Results from feeding experiments to shoots of Arabidopsis thaliana using [2H5]L-tryptophan have shown the efficient incorporation of label into indole-3-acetonitrile and indole-3-acetic acid and have provided additional support to the proposed intermediacy of IAN in the biosynthetic pathway from L-tryptophan to IAA.33 No evidence could be found for the presence of a tryptophan independent pathway to IAA.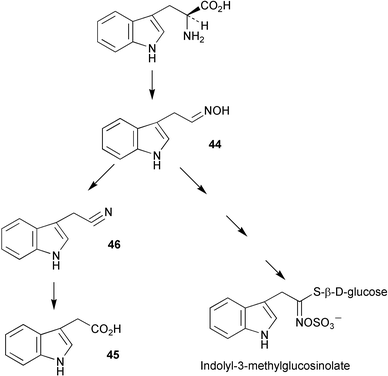 | ||
| Scheme 12 | ||
An enzyme complex capable of catalysing the synthesis of indole-3-acetic acid from L-tryptophan has been isolated from Arabidopsis thaliana.34 The complex which occurs in shoots, roots and cell cultures of A. thaliana, has a mass of 160–180 kDa, is dioxygen dependent but requires no other cofactors for activity. The oxygen atoms in the product IAA were shown to originate with water. This information together with immunological evidence obtained suggesting the presence of a nitrilase protein in the complex has led to the proposal that indole-3-acetonitrile may be an intermediate in the catalytic pathway. Work is now in progress to characterise the complex fully and gain a more detailed mechanistic picture of the enzymatic reactions in operation.
In some recent studies in Arabidopsis the levels of IAA and tryptophan (Trp) were compared between transgenic plants bearing antisense indole-3-glycerol phosphate synthase RNA and Trp biosynthetic pathway mutants.35 Levels of Trp were very much reduced in both cases compared to wild type plants. Total levels of IAA in the transgenic plants were markedly lower than in the wild type. In contrast, levels of IAA in the Trp biosynthetic mutant plants were found to be significantly higher than that found in the wild type plants. These results have led to the proposal that a tryptophan independent biosynthetic pathway to IAA exists in Arabidopsis with indole-3-glycerol phosphate (IGP) serving as the branchpoint compound. However, in light of recent findings33 regarding the degradation of indole-3-glycerol phosphate during alkaline treatment designed to liberate IAA from base-labile conjugates, it is quite conceivable that the raised levels of IAA found in the Trp biosynthetic mutant plants could be a consequence of the presence of high concentrations of IGP. A re-evaluation of experimental data is required to check for this possibility.
Evidence has now been obtained which suggests the operation of several Trp-dependent routes to IAA in the Gram-negative bacterium Azospirillum brasilense.36 Physiological data indicated the presence of biosynthetic routes to IAA via tryptamine (47) and IAN (46), as well as the main pathway involving decarboxylation of indole pyruvate. In other, more quantitative studies using the tobacco plant (Nicotiana tabacum L.) it was found that Trp-dependent IAA formation contributes less than 25% of the total biosynthesis of IAA.37 Some recent precursor feeding studies in maize kernels (Zea mays) have in contrast indicated that Trp-dependent IAA synthesis is the predominant route for auxin production in this species.38
4 Tyrosine and related compounds
4.1 Trihydroxyphenylalanine (TOPA) quinone
In a recent article several models for the biosynthesis of TOPA quinone have been discussed.39 A new mechanism for cofactor biogenesis is proposed (Scheme 13) based on the mechanism of the oxidative half reaction thought to operate in copper amine oxidases in conjunction with current knowledge of some non-heme iron containing systems. This new model predicts the oxidation state of the copper ion remains unchanged during TOPA quinone biosynthesis in line with experimental observations and is also consistent with the derivation of the C-2 oxygen from solvent and the recent finding that hydrogen peroxide is formed during the biogenetic pathway.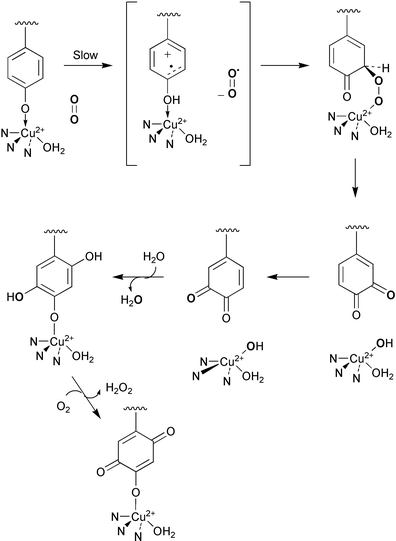 | ||
| Scheme 13 | ||
The crystal structure of a Zn2+ substituted copper amine oxidase from Hansenula polymorpha (HPAO) has been solved to 2.5 Å.40 The structural data indicated the binding of zinc to the same site as copper and importantly showed a direct ligation of the precursor tyrosine residue to zinc. This gives support to the proposed biogenetic model above whereby the binding of Tyr-405 to copper induces a radical character in the tyrosine ligand and thus decreases the kinetic barrier associated with the spin-forbidden reaction with oxygen yielding an overall increase in rate of oxygenation. In addition, the apo-Zn structure suggests that rearrangements of active site residues proposed to play a role in oxygen binding in the mature protein may explain the differences seen in the Km for dioxygen during biogenesis and catalysis. Further support for the new biogenetic model proposed comes from kinetic investigations41 and spectroscopic studies42 of intermediates formed during copper binding and TOPA quinone synthesis in wild type and active site mutants of H. polymorpha amine oxidase. The data obtained strongly suggest the formation of an activated Cu(II)-tryrosinate species during TOPA quinone biosynthesis. It is furthermore proposed that the binding of molecular oxygen at a pocket in the active site causes the displacement of the Tyr-405 ligand onto Cu(II), the subsequent reaction of oxygen with the Cu(II)-tyrosinate adduct is thought to be the rate limiting step.
In some related studies carried out by the same group on HPAO, evidence has been obtained which suggests that the catalytic role of copper during amine oxidation is merely to provide electrostatic stabilisation during the rate limiting reduction of molecular oxygen to superoxide anion and indicated the need for a discrete non-metal binding site for dioxygen.43 There was no requirement for the transfer of electrons from the TOPA quinone cofactor to copper(II) during the catalytic cycle and thus provided support to the mechanism proposed by Su and Klinman44 in which electrons are passed directly from reduced TOPA quinone to the pre-bound oxygen.
The active site quino-cofactor present in Aspergillus niger amine oxidase is thought to derive from the addition of the γ-carboxy group of a glutamic acid residue to a DOPA quinone precursor (see 48).45 This proposal has been reassessed in some recent work using model compounds.46 The spectral and redox properties of the model compound 49 and derivatives suggest that the A. niger cofactor is probably not the glutamic acid quinone adduct but most likely TOPA quinone itself. In addition, the physiochemical characteristics of 49 indicate that a TOPA quinone carboxylate ester would probably not be biologically functional.
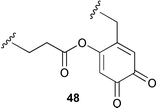
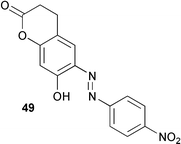
A review of novel cofactors biosynthesised via post-translational modifications of active sites has been published.47
4.2 Pyrroloquinoline quinone (PQQ)
A gene cluster encoding the pyrroloquinoline quinone (50, PQQ) biosynthetic pathway has been cloned and sequenced from the Gram-negative bacterium Gluconobacter oxydans.48 The cluster is arranged in a similar way to other species with the pqqA gene possessing its own promoter followed by an operon containing the 4 remaining genes (pqqBCDE). Spectral and kinetic studies of various forms of glucose dehydrogenase from the bacterium Acinetobacter calcoaceticus have now shown that the catalytic potential of the PQQ cofactor is directly linked to its ability to be activated with calcium ion.49 Furthermore, this result taken together with the large observed deuterium kinetic isotope effects seen with incubations of [1-2H]D-glucose, suggested a catalytic mechanism involving a Ca2+ assisted direct hydride transfer from the anomeric centre of D-glucose to C-5 of PQQ (Scheme 14). His-144 may play a role in the hydride transfer by mediating the abstraction of the proton from the anomeric hydroxy group. An X-ray structure of ethanol dehydrogenase from Psuedomonas aeruginosa has been solved to 2.6 Å by molecular replacement techniques.50 The enzyme is homodimeric and shows structural similarity to the large subunit of methanol dehydrogenase (MDH) from Methylobacterium extorquens or Methylophilus W3A however the structural data did not indicate the presence of a small subunit like that found in MDH. The PQQ cofactor was shown to be situated in the centre of a superbarrel structure coordinated to Ca2+. Interestingly, a bulky tryptophan residue found in the active site of MDH is replaced by a phenylalanine and leucine, and a leucine situated in close proximity to PQQ in MDH is substituted with a tryptophan side chain. An additional calcium ion binding site was discovered at the N-terminus of ethanol dehydrogenase which increases the stability of the enzyme. Two reviews have been published covering the physiology of quinoenzymes51 and discussing the reaction mechanism of soluble glucose dehydrogenase and methanol dehydrogenase based on structural evidence.52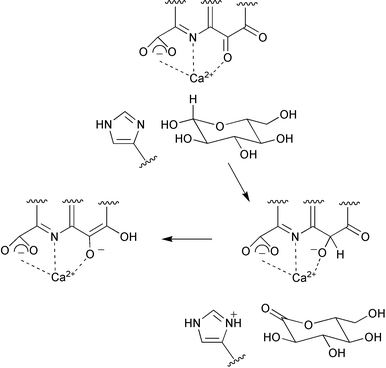 | ||
| Scheme 14 | ||
4.3 Tropanes
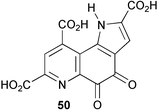
The intramolecular rearrangement of the phenyllactate appendage in littorine (51) to form hyoscyamine (52, Scheme 15) has been probed by stable isotope labelling experiments using [2′-13C, 3-2H]littorine (53), [2-13C]phenyllactate (54), and [3-2H]tropine (55).53 The levels of incorporation of label from a feeding experiment with the double labelled [2′-13C, 3-2H]littorine (53) were significantly higher than that observed following the separate administration of a mixture of singly labelled [2-13C]phenyllactate (54) and [3-2H]tropine (55). This result was consistent with the direct rearrangement of littorine to hyoscyamine and did not support an alternative pathway to 52 involving the rearrangement of phenyllactate (derived from the hydrolysis of littorine) to tropic acid followed by the subsequent condensation with tropine. Three reviews have been published on the biosynthesis, chemotaxonomy and distribution of tropane alkaloids.54–56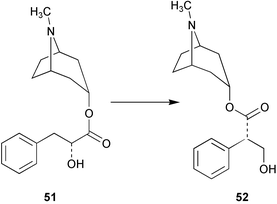 | ||
| Scheme 15 | ||
4.4 Metabolites derived from tyrosine
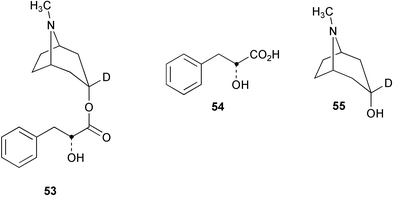
The genes encoding two cytochrome P450 enzymes involved in the biosynthetic pathway to the cyanogenic glucosides taxiphyllin (56, Scheme 16) and triglochinin (57) in Triglochin maritima have been cloned and overexpressed in Escherichia coli.57 The enzymes which belong to the CYP79 family of cytochrome P450 (designated CYP79E1 and CYP79E2) catalyse the N-hydroxylation of tyrosine to 4-hydroxyphenylacetaldoxime (58) display 94% sequence identity and share the same functionality as the CYP79A1 hydroxylase from Sorghum bicolor. The presence of these enzymes in T. maritima indicate that 58 is probably a free intermediate in the biosynthesis of 56 and 57 in addition to 4-hydroxyphenylacetonitrile (59). The enzymes were not able to metabolise L-3,4-dihydroxyphenylalanine to the corresponding aldoxime and thus did not support a route to triglochinin involving an early hydroxylation of the phenyl ring.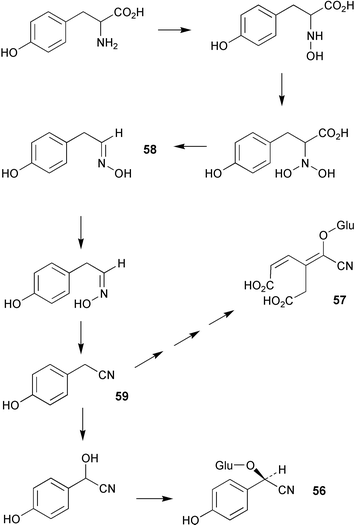 | ||
| Scheme 16 | ||
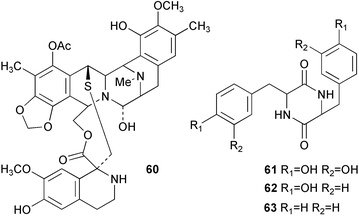
The biosynthesis of the tetrahydroquinoline alkaloid ecteinascidin 743 (60) isolated from the tunicate Ecteinascidia turbinata has been probed by feeding experiments with radiolabelled diketopiperazines 61, 62 and 63 in cell-free extracts.58 Efficient transformation of both 62 (1.4%) and 61 (11.4%) to 60 was observed which contrasted to the relatively low incorporation of L-tyrosine (0.016%). No transformation of the phenylalanine diketopiperazine 63 was detected. These results suggested a biosynthetic pathway involving the initial formation of the diketopiperazine of L-tyrosine 62 followed by oxidation of this intermediate to the corresponding DOPA derivative 61. This hypothesis was supported by additional experiments which were able to confirm the presence of hydroxylase activity in crude enzyme preparation made from E. turbinata which was capable of converting 62 into 61.
5 Phenylalanine and related compounds
5.1 Metabolites derived from phenylalanine
The nature of the biosynthetic building blocks utilised in the synthesis of the marine cyanobacterium metabolite barbamide (64) have been probed by a series of feeding experiments using stable isotope labelled compounds.59 Results suggest that the phenyl moiety derives from the intact use of L-phenylalanine which provides the phenyl ring, C-7 and C-8, C-16 of the thiazole ring, and the amide nitrogen. Indirect evidence was obtained from glycine feeding experiments that supported a proposed biosynthetic route to the thiazole ring via the combination of cysteine with the carboxylate of L-phenylalanine. Carbons 6 and 5 were shown to originate with acetate/malonate whereas C-9 and C-1 to C-4 derive from 5,5,5-trichloroleucine 65. Interestingly, results indicate that 65 is formed via the direct chlorination of the pro-R methyl group of L-leucine and occurs without any form of activation. It was proposed that a mechanism involving radical intermediates may be in operation.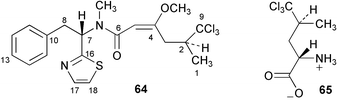
A cytochrome P450 dependent monoxygenase involved in the biosynthetic pathway to benzylglucosinolate in Arabidopsis thaliana has been cloned and functionally expressed in E. coli.60 The enzyme (CYP79A2) was shown to be an N-hydroxylase and catalyses the conversion of L-phenylalanine to phenylacetaldoxime (66). L-Tyrosine, L-tryptophan, L-methionine and DL-homophenylalanine were not turned over by the monoxygenase.
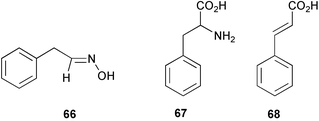
The metabolism of L-phenylalanine (67) by the fungus Bjerkandera adusta has been studied in detail by use of precursor labelling experiments.61 The results indicated that 2 major degradative pathways were in operation leading from the key intermediate trans-cinnamic acid (68) (formed from 67 by the action of phenylalanine ammonia lyase) to form both benzyl alcohol and benzaldehyde or via β-oxidation to produce benzoic acid.
6 Coumarins
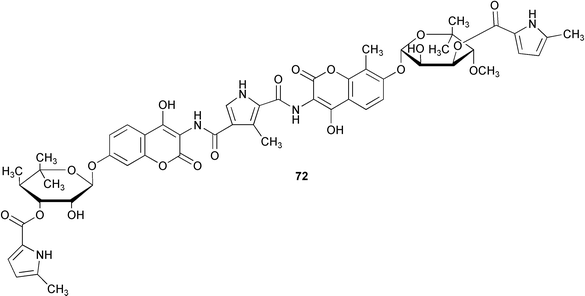
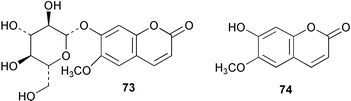
Novobiocic acid synthetase has been cloned from Streptomyces spheroides and overexpressed in E. coli. The purified enzyme was shown to catalyse the amide coupling of 3-dimethylallyl-4-hydroxybenzoic acid (69) with 3-amino-4,7-dihydroxy-8-methylcoumarin (70) to give novobiocic acid (71, Scheme 17). The enzyme is thought to activate the carboxylate of 69via an adenylate and is thus dependent on ATP, Mg2+ or Mn2+ for activity.62 The biosynthetic gene cluster of the aminocoumarin coumermycin Al (72) produced by Streptomyces rishiriensis DSM 40489 has been through use of heterologous probes from a dTDP-glucose 4,6-dehydratase gene and the aminocoumarin resistance gene.63 The cluster contains 28 complete open reading frames 17 of which have been tentatively assigned functions by comparison with sequence databases. A hydroxycoumarin 7-O-glucosyltransferase enzyme from cultures of tobacco cells has been purified to apparent homogeneity and characterised.64 The enzyme mediates in the formation of scopolin (73) from scopoletin (74) in Nicotiana tabacum although it was shown to display a broad substrate specificity accepting various hydroxycoumarins and flavonoids.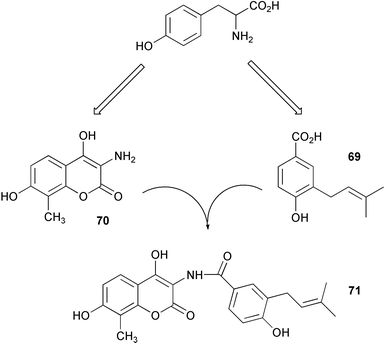 | ||
| Scheme 17 | ||
7 Lignans and lignin
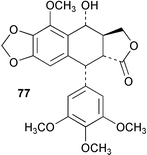
In some recent work the biosynthesis of (−)-matairesinol (75) and 5-methoxypodophyllotoxin (76) was studied in Linum flavum using feeding experiments with isotopically labelled precursors to crude and partially purified enzyme preparations.65 Results indicated that the biosynthetic pathway to 75 mirrors the route found to operate in Forsythia sp. (Scheme 18). In addition, it was found that L. flavum was able to hydroxylate 75 at the 7′-position to give 7′-hydroxymatairesinol (77) which then serves as an intermediate on the pathway to 5-methoxypodophyllotoxin (76). In some associated work, the dirigent enzyme which mediates the coupling of E-coniferyl alcohol (see 78) in Podophyllum peltatum was cloned from a rhizome cDNA library and subsequently heterologously expressed in Drosophila melanogaster cells. The expressed protein was found to cross-react with F. intermedia dirigent polyclonal antibodies.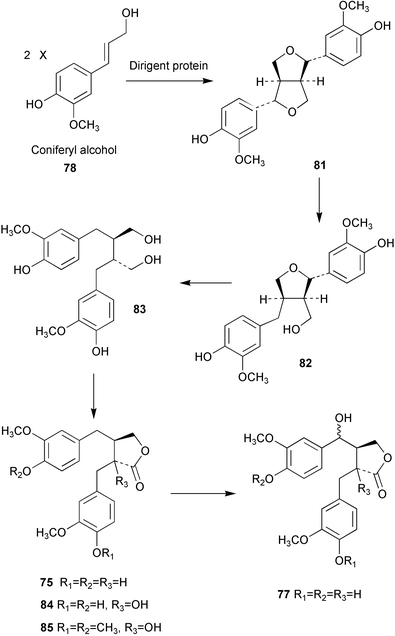 | ||
| Scheme 18 | ||
A study of the distribution and abundance of the enzyme phenylcoumaran benzylic ether reductase (PCBER) has been carried out in poplar.66 PCBER is involved in lignan biosynthesis and mediates the non-enantiospecific NADPH dependent reduction of benzylic ether moieties in 8–5′-linked lignans and thus converts dehydrodiconiferyl alcohol (79) to the ring opened alcohol 80 (Scheme 19). The results of these studies suggest a strong link between PCBER expression and distribution to cells involved in the lignin biosynthesis, consequently it has been hypothesised that this enzyme may play a role in the lignification process. Alternatively it has been proposed that the expression pattern of PCBER and the resultant production of lignans may be operating for the purpose of providing a general protection for the plant during xylogenesis.
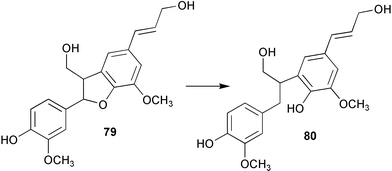 | ||
| Scheme 19 | ||
The enantiomeric composition and biosynthesis of lignans have recently been investigated in Wikstroemia sikokiana.67 Studies showed the efficient conversion of coniferyl alcohol (78) to pinoresinol (81) and the subsequent interconversion from 81 to dibenzylbutyrolactone lignans (e.g. 84 and 85) via lariciresinol (82), secoisolariciresinol (83), and matairesinol (75) (Scheme 18). The biosynthetic reaction sequence was similar to that seen in Forsythia, however, the predominant enantiomers of the Wikstroemia lignans were opposite to those isolated from Forsythia spp. apart from secoisolariciresinol (83). In addition, Forsythia spp. biosynthesise 83 in optically pure form whereas optically impure secoisolariciresinol is generated in W. sikokiana (the first optically pure lignan produced by W. sikokiana is (−)-matairesinol). These findings suggest that there are different stereochemical mechanisms in each plant and imply the operation of different enantioselective steps in the respective lignan biosynthetic pathways.
The role of caffeoyl coenzyme A O-methyltransferase (CCoAOMT) in lignin biosynthesis has been investigated in woody poplar plants.68 Repression of CCoAOMT expression via introduction of antisense DNA resulted in a marked decrease in the levels of guaiacyl and syringyl lignins and gave rise to a less condensed and cross-linked lignin in the wood. These findings were similar to those found in transgenic tobacco plants and showed the key role played by CCoAOMT in lignin biosynthesis in both herbacious and woody plant species.
Feeding studies with radiolabelled glucoside derivatives of caffeyl and 5-hydroxyconiferyl alcohols (86 and 87) has been used to investigate the pathways of lignin biosynthesis in several angiosperms.6986 was incorporated into both the syringyl (S) and guaiacyl units of lignin, however, 87 served as a precursor only to the S units. The G/S ratio calculated from the radiolabel incorporations was the same as that determined for natural abundance G and S in each tree respectively. These results indicated the operation of a cinnamyl alcohol pathway alongside the established pathways utilising cinnamic acid and cinnamoyl CoA involving analogous transformations of aromatic hydroxylation and O-methylation (see Scheme 20). However, other work with caffeate O-methyltransferase (COMT) derived from Populus termuloides has established for the first time that COMT is in fact a 5-hydroxyconiferyl aldehyde O-methyltransferase (AldOMT).70 In addition, 5-hydroxyconiferyl aldehyde (88) was found to act as an inhibitor of caffeate (89) and 5-hydroxyferulate (90) methylation. These studies together with previous findings have led to the proposal that the syringyl monolignol biosynthesis in angiosperms is directed by a coniferyl aldehyde 5-hydroxylase/5-hydroxyconiferyl aldehyde O-methyltransferase pathway rather than routes via caffeate or feruloyl CoA (91, Scheme 20).
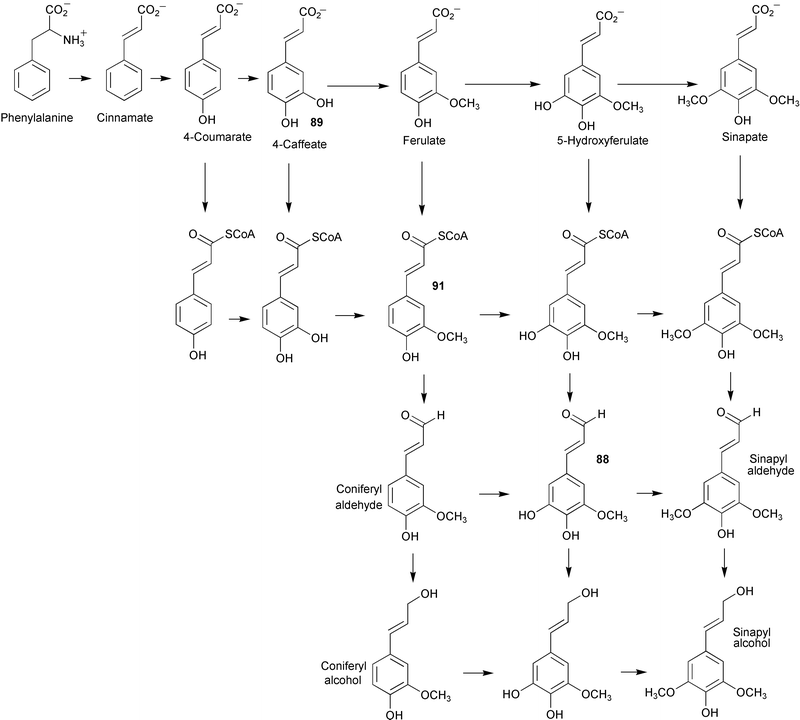 | ||
| Scheme 20 | ||
Two reviews covering the control of lignin biosynthesis71 and the discovery and nature of the dirigent proteins72 have recently been published.
8 Tannins
The key process parameters involved in the biosynthesis of tannase by Aspergillus awamori have been found to be tannic acid concentration, agitation speed and pH. These were subsequently optimised by response surface methodology.73 Tetragalloylglucose 4-O-galloyltransferase has now been purified to apparent homogeneity from leaves of pedunculate oak (Quercus robur).74 The protein was then used to produce polyclonal antibodies in rabbits. These were found to bind specifically to acyltransferase from oak and displayed no reactivity with analogous enzymes from other plant species known to biosynthesis hydrolisable tannins.9 Ubiquinone (coenzyme Q)
A gene (ubiB) required for the first hydroxylation step in coenzyme Q (92) biosynthesis (Scheme 21) has been identified in Escherichia coli.75 Disruption of ubiB gives rise to mutant strains which accumulate octaprenylphenol (93) and they are unable to synthesise 92. The fre gene previously thought to correspond to ubiB was found not to be involved in the biosynthesis of coenzyme Q. The functional role of the UbiB is unknown, however, its gene shows sequence identity with the ABCl gene in S. cerevisiae which is thought to belong to a family of protein kinases. Thus the role of UbiB may be to activate via phosphorylation biosynthetic pathway enzymes required for hydroxylation. In other work now reported76 the gene required for the conversion of 2-octaprenyl-3-methyl-6-methoxy-1,4-benzoquinol (94) to 2-octaprenyl-3-methyl-5-hydroxy-6-methoxy-1,4-benzoquinol (95) (ubiF) has been identified in Escherichia coli. The sequence of ubiF shows homology to that of ubiH (also required for a hydroxylation step in coenzyme Q biosynthesis) and codes for a 391-amino acid protein of molecular mass 42 kDa. Amino acid sequence analysis has predicted the presence of 2 strong transmembrane helices in the three-dimensional structure of the enzyme. Genetic evidence has now also been obtained in Saccharomyces cerevisiae which suggests that a mult-subunit enzyme complex is involved in coenzyme Q biosynthesis in this organism.77 Two reviews of recent advances in the biosynthesis of coenzyme Q have been published.78,79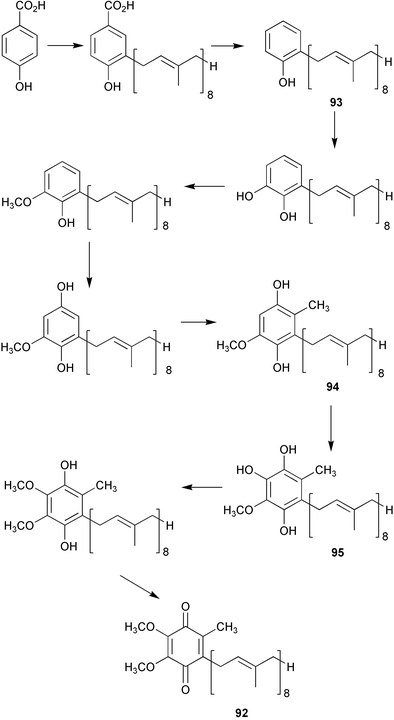 | ||
| Scheme 21 | ||
10 Melanin
Atomic force microscopy has been used to probe the structural nature of eumelanin isolated from ink sacs of cuttlefish (Sepia officinalis).80 Results indicate that stable spherical particles of eumelanin (100–200 nm in diameter) observed in previous scanning electron microscopy images are composed of yet smaller particles approximately 15–25 nm in diameter. It is proposed that the morphology of the macroscopic pigment is formed through a hierarchical self-assembly process with small units assembling into 100 nm particles which then aggregate to produce eumelanin pigment. A review of the characteristics, biosynthesis, processing and stability of several natural pigments (carotenoids, anthocyanins and betalains) has now been published.8111 Flavanoids
An inducible S-adenosyl-L-methionine naringenin 7-O-methyltransferase derived from UV-irradiated rice leaves has been purified using a combination of affinity and gel filtration chromatography.82 The mass of the enzyme was estimated to be approximately 41 kDa by SDS PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and was shown to be inhibited by a range of divalent metal ions as well as ethylenediamine tetraacetic acid (EDTA). A cDNA clone from Arabidopsis thaliana previously assigned83 on the basis of deduced amino acid sequence as encoding a caffeic acid/5-hydroxyferulic acid O-methyltransferase has now been functionally expressed in Escherichia coli. The enzyme was found to use the flavonol quercetin (96, Scheme 22) as its preferred substrate whilst showing very little activity toward caffeic or 5-hydroxyferulic acids. On the basis of these new findings the enzyme has been reclassified as being a flavonol 3′-O-methyltransferase.84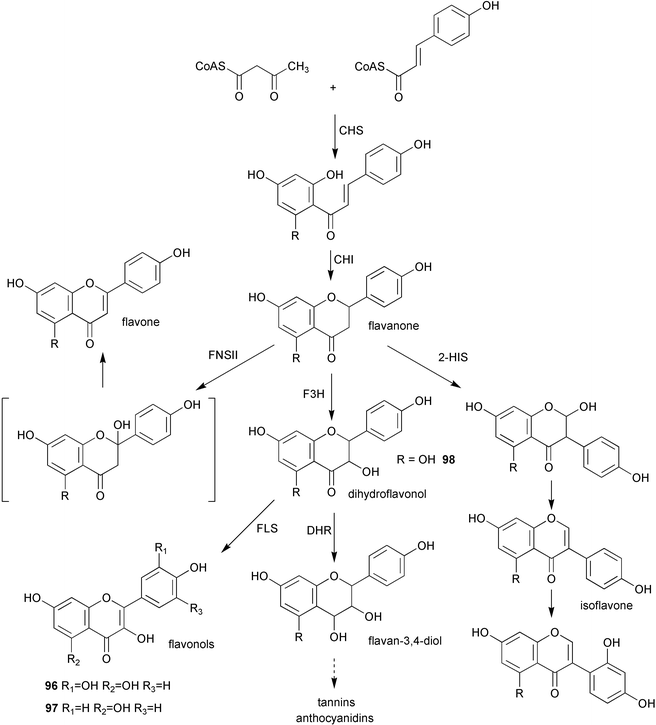 | ||
| Scheme 22 | ||
The gene encoding flavonoid 3′-hydroxylase (F3′H) has now been identified in Arabidopsis thaliana.85 The enzyme was expressed in yeast and shown to catalyse the hydroxylation of both naringenin (97) and dihydrokaempferol (98) to the corresponding 3′,4′-hydroxylated derivatives. This activity was dependent on the addition of NADPH and thus supported the proposed nature of F3′H as a P450-dependent monooxygenase. A 2-oxoglutarate dependent dioxygenase which catalyses the 6-hydroxylation of partially O-methylated flavonols has been purified to near homogeneity from the semi-aquatic weed Chrysosplenium americanum.86 Data indicated that the enzyme had a molecular mass of approximately 42 kDa, exists in a monomeric form, and displays cofactor dependence of a typical plant dioxygenase. Kinetic studies found that the enzyme utilises an ordered, sequential reaction mechanism with 2-oxoglutarate binding first followed by oxygen and the flavonol substrate. The product is then released followed by CO2 and succinate.
Recombinant flavanone 3β-hydroxylase (FHT) from Petunia hybrida has now been purified using a rapid two-step method involving a combination of size exclusion and anion-exchange chromatography giving protein of high specific activity and in approximately 5% yield.87 Previous attempts to purify this enzyme have been hampered by its severe proteolytic instability. Other investigations have shown a tendency of the enzyme to oligomerise, however, results suggest that the monomeric polypeptide is the catalytically active FHT in P. hybrida.88 In subsequent work the active site serine residue (Ser-290) was subject to site-directed mutagenesis. The catalytic activity of the Thr-290 and Ala-290 mutants was 20 and 8% respectively whereas the Val-290 mutant showed negligible turnover (1%).89 In addition, kinetic analysis of these mutants compared with wild type enzyme indicated a possible functional role for Ser-290 in the binding of 2-oxoglutarate. The three-dimensional structure of FHT was also compared to isopenicillin N synthase by circular dichroism (CD) spectroscopy and revealed a close structural relationship. No significant changes were seen in the CD spectra on addition of ferrous ion, 2-oxoglutarate either individually or in combination.
In some recent work with chalcone synthase (CHS) derived from Pueraria lobata utilising both chemical modification and site-directed mutagenesis evidence has been obtained which indicates that an imidazolium-thiolate ion pair (His-303/Cys164) is present at the active site of CHS.90 Similar studies carried out on a stilbene synthase from Arachis hypogaea have shown that only Cys-164 participates in the STS reaction contrasting to earlier work which implied roles for both Cys-60 and Cys-164 in CoA transfer reactions. Three chalcone synthase cDNAs (CHS1, 2 and 3) have been cloned from immature Ruta graveolens flowers.91 CHS1 was subsequently functionally expressed in Escherichia coli and substrate specificity compared to two recombinant acridone synthase (ACS1 and 2) cloned from Ruta cell cultures. 4-Coumaroyl-CoA was the preferred substrate for CHS1 but N-methylanthraniloyl-CoA (the preferred substrate of ACS) was not accepted. In contrast, ACS1 and 2 showed activity with both these compounds giving further support to the proposed evolutionary origin of ACS from CHS.
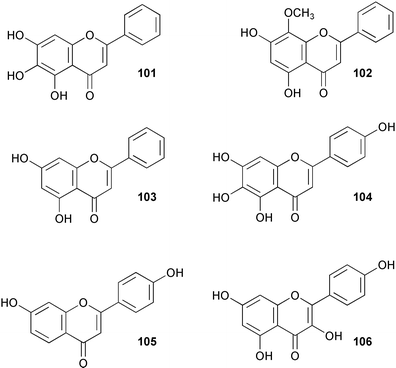
A cDNA encoding hydroxycinnamoyl-CoA: anthocyanin 3-O-glucoside-6″-O-acyltransferase has been isolated from Perilla frutescens and functionally expressed in E. coli.92 The recombinant enzyme showed activity with cyanidin 3-glucoside (99) and cyanidin 3,5-diglucoside (100, Scheme 23). In addition, results from chemical modification studies indicated the importance of histidine and cysteine residues for catalytic function. The characteristics of the enzyme parallel those found for the corresponding anthocyanin 5-O-glucoside-6″-O-acyltransferase. A cDNA encoding UDP-glucose:baicalein 7-O-glucosyltransferase (see baicalein, 101) was isolated from hairy root cultures of Scutellaria baicalensis and functionally expressed in E. coli.93 The resultant protein showed activity towards a wide range of flavonoids including wogonin (102), apigenin (103), scutellarein (104), 7,4′-dihydroxyflavone (105) and kaempferol (106). Investigations into the nature of dimethylallyl diphosphate: naringenin 8-dimethylallyltransferase (see naringenin, 97) present in cell-free extracts of Sophora flavescens suspension cultures have shown that the enzyme is enantiospecific for (−)-(2S)-naringenin and uses 3,3-dimethylallyl diphosphate (DMAP) as sole prenyl donor.94 There is a requirement of Mg2+ for activity which has a pH optimum of 9–10. Apparent Km's were determined as 120 and 36 µM for DMAP and naringenin respectively. Other results indicate that the sequence and regiospecificity of prenyl transfer is modified upon 2′-hydroxylation of naringenin.
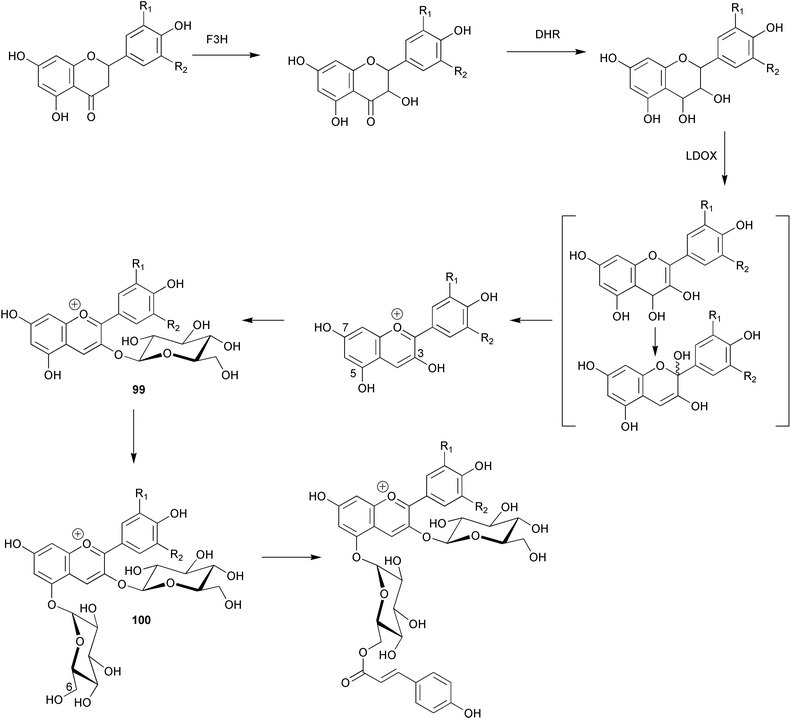 | ||
| Scheme 23 | ||
12 Isoflavanoids
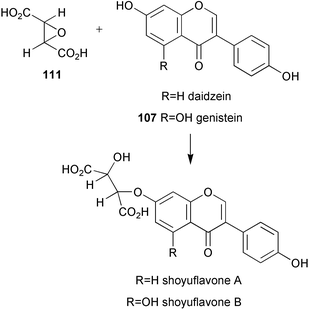
Two soybean genes encoding isoflavone synthase (IFS1 and 2) have now been identified and IFS1 functionally expressed in transgenic Arabidopsis thaliana resulting in the production of genistein (107).95 These genes were subsequently used to isolate homologous genes from red clover, white clover, hairy vetch, mung bean, alfalfa, lentil, snow pea, lupine, and sugarbeet. These genes were found to be both structurally and functionally similar to IFSl. Formononetin biosynthesis has been studied in a combined system of heterologously expressed isoflavone synthase and Glycyrrhiza echinata cell-free extract.96 Results obtained have led to a new proposed pathway in which 2,7,4′-trihydroxyisoflavanone (108, Scheme 24) rather than daidzein (109) acts as the methyl acceptor from S-adenosylmethionine, and implies the existence of a dehydratase enzyme which is specific for 2,7-dihydroxy-4′-methoxyisoflavonone (110), but which does not use 2,7,4′-trihydroxyisoflavonone (108) as substrate. The biosynthesis of shoyuflavones (Scheme 25) has been probed in culture extracts of Aspergillus oryzae.97 Preliminary findings have suggested that the ether linkage is formed via a novel enzyme catalysed process from (±)-trans-epoxysuccinic acid (111) and isoflavones. The exact nature of this conjugating enzyme is not known.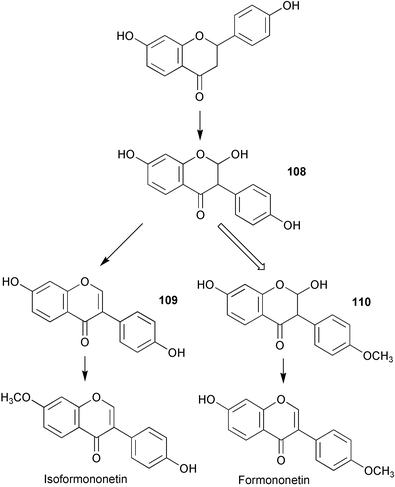 | ||
| Scheme 24 | ||
 | ||
| Scheme 25 | ||
References
- A. R. Knaggs, Nat. Prod. Rep., 1999, 16, 525 RSC.
- S. Krappmann, W. N. Lipscomb and G. H. Braus, Proc. Natl. Acad. Sci. USA, 2000, 97, 13585 CrossRef CAS.
- B. Silakowski, B. Kunze and R. Muller, Arch. Microbiol., 2000, 173, 403 CrossRef CAS.
- E. J. Parker, C. G. Bello, J. R. Coggins, A. R. Hawkins and C. Abell, Bioorg. Med. Chem. Lett., 2000, 10, 231 CrossRef CAS.
- C. G. Bello, J. M. Harris, M. K. Manthey, J. R. Coggins and C. Abell, Bioorg. Med. Chem. Lett., 2000, 10, 407 CrossRef CAS.
- J. Maclean, S. A. Campbell, K. Pollock, S. Chackrewarthy, J. R. Coggins and A. J. Lapthorn, Acta Crystallogr Sect. D: Biol. Crystallogr., 2000, 56, 512 CrossRef.
- Q. Liu, Y. Li, Y. Wu and H. Yan, J. Biomol. NMR, 2000, 17, 277 CrossRef CAS.
- W. Du, N. G. Wallis, M. J. Mazzulla, A. F. Chalker, L. Zhang, W.-S. Liu, H. Kallender and D. J. Payne, Eur. J. Biochem., 2000, 267, 222 CrossRef CAS.
- W. Du, N. G. Wallis and D. J. Payne, J. Enzyme Inhib., 2000, 15, 571 Search PubMed.
- S. Bornemann, M.-E. Theoclitou, M. Brune, M. R. Webb, R. N. F. Thorneley and C. Abell, Bioorg. Chem., 2000, 28, 191 CrossRef CAS.
- A. Osborne, R. N. F. Thorneley, C. Abell and S. Bornemann, J. Biol. Chem., 2000, 275, 35825 CrossRef CAS.
- Y. B. Tewari, A. M. Davis, P. Reddy and R. N. Goldberg, J. Chem. Thermodyn., 2000, 32, 1057 CrossRef CAS.
- R. J. Hall, S. A. Hindle, N. A. Burton and I. A. Hillier, J. Comput. Chem., 2000, 21, 1433 CrossRef CAS.
- S. Marti, J. Andres, V. Moliner, E. Silla, I. Tunon and J. Bertran, J. Phys. Chem. B, 2000, 104, 11308 CrossRef CAS.
- S. Krappmann, R. Pries, G. Gellissen, M. Hiller and G. H. Braus, J. Bacteriol., 2000, 182, 4188 CrossRef CAS.
- J. E. Ladner, P. Reddy, A. Davis, M. Tordova, A. J. Howard and G. L. Gilliland, Acta Crystallogr. Sect. D: Biol. Crystallogr., 2000, 56, 673 CrossRef.
- P. Kast, C. Grisostom, I. A. Chen, S. Li, U. Krengel, Y. Xue and D. Hilvert, J. Biol. Chem., 2000, 275, 36832 CrossRef CAS.
- M. Gamper, D. Hilvert and P. Kast, Biochemistry, 2000, 39, 14087 CrossRef CAS.
- N. Jimenez, F. Gonzalez-Candelas and F. J. Silva, J. Bacteriol., 2000, 182, 2967 CrossRef CAS.
- S. Zhang, D. B. Wilson and B. Ganem, Biochemistry, 2000, 39, 4722 CrossRef CAS.
- G. Xie, C. A. Bonner and R. A. Jensen, Comp. Biochem. and Physiol. C: Pharmacol. Toxicol. Endocrinol., 2000, 125, 65 Search PubMed.
- C. Stover, M. P. Mayhew, M. J. Holden, A. Howard and D. T. Gallagher, J. Struct. Biol., 2000, 129, 96 CrossRef CAS.
- T. Nakai, H. Mizutani, I. Miyahara, K. Hirotsu, S. Takeda, K.-H. Jhee, T. Yoshimura and N. Esaki, J. Biochem., 2000, 128, 29 Search PubMed.
- X.-F. Tang, S. Ezaki, H. Atomi and T. Imanaka, Eur. J. Biochem., 2000, 267, 6369 CrossRef CAS.
- F. Dosselaere, M. Lambrecht and J. Vanderleyden, DNA Sequence, 2000, 11, 287 Search PubMed.
- S. E. Eckert, E. Kubler, B. Hoffmann and G. H. Braus, Mol. Gen. Genet., 2000, 263, 867 Search PubMed.
- J. P. Sarsero, E. Merino and C. Yanofsky, Proc. Natl. Acad. Sci. USA, 2000, 97, 2656 CrossRef CAS.
- C. Jurgens, A. Strom, D. Wegener, S. Hettwer, M. Wilmanns and R. Sterner, Proc. Natl. Acad. Sci. USA, 2000, 97, 9925 CrossRef CAS.
- S. Keller, T. Wage, K. Hohaus, M. Holzer, E. Eichhorn and K.-H. van Pee, Angew. Chem., Int. Ed., 2000, 39, 2300 CrossRef CAS.
- K.-H. Van Pee and J. M. Ligon, Nat. Prod. Rep., 2000, 17, 157 RSC.
- A. Z. M. R. Momen and T. Hoshino, Biosci. Biotechnol. Biochem., 2000, 64, 539 Search PubMed.
- A. K. Hull, R. Vij and J. L. Celenza, Proc. Natl. Acad. Sci. USA, 2000, 97, 2379 CrossRef CAS.
- A. Muller and E. W. Weiler, Planta, 2000, 211, 855 CrossRef CAS.
- A. Muller and E. W. Weiler, Biol. Chem., 2000, 381, 679 Search PubMed.
- J. Ouyang, X. Shao and J. Li, Plant J., 2000, 24, 327 CrossRef CAS.
- R. Carreno-Lopez, N. Campos-Reales, C. Elmerich and B. E. Baca, Mol. Gen. Genet., 2000, 264, 521 Search PubMed.
- F. Sitbon, C. Astot, A. Edlund, A. Crozier and G. Sandberg, Planta, 2000, 211, 715 CrossRef CAS.
- E. Glawischnig, A. Tomas, W. Eisenreich, P. Spiteller, A. Bacher and A. Gierl, Plant Physiol., 2000, 123, 1109 CrossRef CAS.
- N. K. Williams and J. P. Klinman, J. Mol. Catal. B Enzym., 2000, 8, 95 CrossRef CAS.
- Z.-W. Chen, B. Schwartz, N. K. Williams, R. Li, J. P. Klinman and F. S. Mathews, Biochemistry, 2000, 39, 9709 CrossRef CAS.
- B. Schwartz, J. E. Dove and J. P. Klinman, Biochemistry, 2000, 39, 3699 CrossRef CAS.
- J. E. Dove, B. Schwartz, N. K. Williams and J. P. Klinman, Biochemistry, 2000, 39, 3690 CrossRef CAS.
- S. A. Mills and J. P. Klinman, J. Am. Chem. Soc., 2000, 122, 9897 CrossRef CAS.
- Q. Su and J. P. Klinman, Biochemistry, 1998, 37, 12513 CrossRef CAS.
- I. Frebort, Biochim. Biophys. Acta, 1996, 1295, 59 CAS.
- C. R. Melville, E. L. Green, J. Sanders-Loehr and J. P. Klinman, Biochemistry, 2000, 39, 7589 CrossRef CAS.
- N. M. Okeley and W. A. van der Donk, Chem. Biol., 2000, 7, 8159 CrossRef CAS.
- M. Felder, A. Gupta, V. Verma, A. Kumar, G. N. Qazi and J. Cullum, FEMS Mirobiol. Lett., 2000, 193, 231 Search PubMed.
- A. R. Dewanti and J. A. Duine, Biochemistry, 2000, 39, 9384 CrossRef CAS.
- T. Keitel, A. Diehl, T. Knaute, J. J. Stezowski, W. Hohne and H. Gorisch, J. Mol. Biol., 2000, 297, 961 CrossRef CAS.
- T. E. Stites, A. E. Mitchell and R. B. Rucker, J. Nutr., 2000, 130, 719 Search PubMed.
- A. Oubrie and B. W. Dijkstra, Protein Sci., 2000, 9, 1265 CAS.
- R. Duran-Patron, D. O'Hagan, J. T. G. Hamilton and C. W. Wong, Phytochemistry, 2000, 53, 777 CrossRef CAS.
- D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC.
- T. Hemscheidt, Biosynthesis: Aromatic polyketides, isoprenoids, alkaloids, 2000, 209, 175 Search PubMed.
- W. J. Griffin and G. D. Lin, Phytochemistry, 2000, 53, 623 CrossRef CAS.
- J. S. Nielsen and B. L. Moller, Plant Physiol., 2000, 122, 1311 CrossRef CAS.
- S. Jeedigunta, J. M. Krenisky and R. G. Kerr, Tetrahedron, 2000, 56, 3303 CrossRef CAS.
- N. Sitachitta, B. L. Marquez, R. T. Williamson, J. Rossi, M. A. Roberts, W. H. Gerwick, V.-A. Nguyen and C. L. Willis, Tetrahedron, 2000, 56, 9103 CrossRef CAS.
- U. Wittstock and B. A. Halkier, J. Biol. Chem., 2000, 275, 14659 CrossRef CAS.
- C. Lapadatescu, C. Ginies, J.-L. Le Quere and P. Bonnarme, Appl. Environ. Microbiol., 2000, 66, 1517 CrossRef CAS.
- M. Steffensky, S.-M. Li and L. Heide, J. Biol. Chem., 2000, 275, 21754 CrossRef CAS.
- Z-X. Wang, S.-M. Li and L. Heide, Antimicrob. Agents Chemother., 2000, 44, 3040 CrossRef CAS.
- G. Taguchi, H. Imura, Y. Maeda, R. Kodaira, N. Hayashida, M. Shimosaka and M. Okazaki, Plant Sci., 2000, 157, 105 CrossRef CAS.
- Z.-Q. Xia, M. A. Costa, J. Proctor, L. B. Davin and N. G. Lewis, Phytochemistry, 2000, 55, 537 CrossRef CAS.
- K. V. Mijnsbrugge, H. Beeckman, R. De Rycke, M. Van Montagu, G. Engler and W. Boerjan, Planta, 2000, 211, 502 CrossRef.
- T. Okunishi, T. Umezawa and M. Shimada, J. Wood Sci., 2000, 46, 234 Search PubMed.
- R. Zhong, W. H. Morrison III, D. S. Himmelsbach, F. L. Poole II and Z.-H. Ye, Plant Physiol., 2000, 124, 563 CrossRef CAS.
- N. Matsui, F. Chen, S. Yasuda and K. Fukushima, Planta, 2000, 210, 831 CrossRef CAS.
- L. Li, J. L. Popko, T. Umezawa and V. L. Chiang, J. Biol. Chem., 2000, 275, 6537 CrossRef CAS.
- J. H. Christensen, M. Baucher, A. O'Connell, M. Van Montagu, and W. Boerjan in Molecular Biology of Woody Plants, eds. S. M. Jain and S. C. Minocha, Vol. 1, 2000, 227 Search PubMed.
- L. B. Davin and N. G. Lewis, Plant Physiol., 2000, 123, 453 CrossRef CAS.
- M. Seth and S. Chand, Process Biochem., 2000, 36, 39 CrossRef CAS.
- P. Grundhofer and G. G. Gross, Z. Naturforsch. C: Biosci., 2000, 55, 582 Search PubMed.
- W. W. Poon, D. E. Davis, H. T. Ha, T. Jonassen, P. N. Rather and C. F. Clarke, J. Bacteriol., 2000, 182, 5139 CrossRef CAS.
- O. Kwon, A. Kotsakis and R. Meganathan, FEMS Microbiol. Lett., 2000, 186, 157 CrossRef CAS.
- A. Y. Hsu, T. Q. Do, P. T. Lee and C. F. Clarke, Biochim. Biophys. Acta, 2000, 1484, 287 CrossRef CAS.
- A. Szkopinska, Acta Biochim. Pol., 2000, 47, 469 Search PubMed.
- C. F. Clarke, Protoplasma, 2000, 213, 134 Search PubMed.
- C. M. R. Clancy, J. B. Nofsinger, R. K. Hanks and J. D. Simon, J. Phys. Chem. B, 2000, 104, 7871 CrossRef CAS.
- F. Delgado-Vargas, A. R. Jimenez and O. Paredes-Lopez, Crit. Rev. Food Sci. Nutr., 2000, 40, 173 Search PubMed.
- R. Rakwal, G. K. Agrawal, M. Yonekura and O. Kodama, Plant Sci., 2000, 155, 213 CrossRef CAS.
- H. Zhang, J. Wang and H. M. Goodman, Biochem. Biophys. Acta, 1997, 1353, 199 Search PubMed.
- I. Muzac, J. Wang, D. Anzellotti, H. Zhang and R. K. Ibrahim, Arch. Biochem. Biophys., 2000, 375, 385 CrossRef CAS.
- C. Schoenbohm, S. Martens, C. Eder, G. Forkmann and B. Weisshaar, Biol. Chem., 2000, 381, 749 Search PubMed.
- D. Anzellotti and R. K. Ibrahim, Arch. Biochem. Biophys., 2000, 382, 161 CrossRef CAS.
- R. Lukacin, I. Groning, E. Schiltz, L. Britsch and U. Matern, Arch. Biochem. Biophys., 2000, 375, 364 CrossRef CAS.
- R. Lukacin, C. Urbanke, I. Groning and U. Matern, FEBS Lett., 2000, 467, 353 CrossRef CAS.
- R. Lukacin, I. Groning, U. Pieper and U. Matern, Eur. J. Biochem., 2000, 267, 853 CrossRef CAS.
- D.-Y. Suh, J. Kagami, K. Fukuma and U. Sankawa, Biochem. Biophys. Res. Commun., 2000, 275, 725 CrossRef CAS.
- K. Springob, R. Lukacin, C. Ernwein, I. Groning and U. Matern, Eur. J. Biochem., 2000, 267, 6552 CrossRef CAS.
- K. Yonekura-Sakakibara, Y. Tanaka, M. Fukuchi-Mizutani, H. Fujiwara, Y. Fukui, T. Ashikari, Y. Murakami, M. Yamaguchi and T. Kusumi, Plant Cell Physiol., 2000, 41, 495 Search PubMed.
- M. Hirotani, R. Kuroda, H. Suzuki and T. Yoshikawa, Planta, 2000, 210, 1006 CrossRef CAS.
- H. Yamamoto, M. Senda and K. Inoue, Phytochem., 2000, 54, 649 Search PubMed.
- W. Jung, O. Yu, S.-M. C. Lau, D. P. O'Keefe, J. Odell, G. Fader and B. McGonigle, Nat. Biotech., 2000, 18, 208 Search PubMed.
- T. Akashi, Y. Sawada, T. Aoki and S.-I. Ayabe, Biosci. Biotechnol. Biochem., 2000, 64, 2276 Search PubMed.
- E. Kinoshita, S. Murakami and T. Aishima, J. Agric. Food Chem., 2000, 48, 2149 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |