The C7N aminocyclitol family of natural products
Taifo
Mahmud
Department of Chemistry, University of Washington, Seattle, WA 98195-1700, USA
First published on 6th December 2002
Abstract
Covering: 1970–July 2002
This review covers microbial secondary metabolites classified in the family of C7N aminocyclitols, a relatively new class of natural products that is increasingly gaining recognition due to their significant biomedical and agricultural uses. Their discovery and structure determinations, their biosynthetic origin, biological properties, chemical synthesis, as well as their further development for pharmaceutical uses are described. The literature from 1970 to July 2002 is reviewed, with 269 references cited.
 Taifo Mahmud | Dr Mahmud received his undergraduate and professional education in pharmacy from the University of North Sumatra. He then joined Osaka University in Japan where he received his PhD in 1997, working with Professors Isao Kitagawa and Motomasa Kobayashi on bioactive natural products from plants and marine organisms. After 3 years of postdoctoral work on natural products biosynthesis, aiming to understand the art of chemical synthesis in nature, with Professor Heinz G. Floss, he joined the faculty of the University of Washington in the Fall of 2000 as a Research Assistant Professor of Chemistry. His research interests are broadly in bioorganic and natural products chemistry, biosynthesis of bioactive secondary metabolites of terrestrial and marine microorganisms, and the interface of combinatorial biosynthesis and chemistry to generate new pharmacologically active leads. |
1 Introduction
The C7N aminocyclitol family is a relatively new class of natural products that is increasingly gaining recognition due to their significant biomedical and agricultural uses. These secondary metabolites are exclusively produced by microorganisms, particularly soil bacteria of the order of Actinomycetes. Their chemical structures most commonly contain an unsaturated aminocyclitol moiety, valienamine (1), which is known to be responsible for their biological activities, although in several compounds the epoxy, hydroxy or dihydro forms of valienamine are present instead. Structurally similar groups of natural products, mycosporines and mycosporine-like amino acids, e.g., mycosporine-Gly (2), have been isolated from marine organisms, terrestrial cyanobacteria and fungi. However, these compounds are classified as an independent family of natural products due mainly to their divergent biosynthetic origin, as well as their distribution and functions in nature. An extensive review on mycosporine-like amino acids and related gadusols has been published recently.1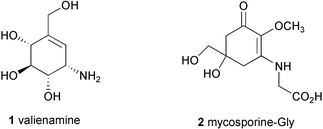
The anti-fungal antibiotic validamycin A (3), the first C7N aminocyclitol-containing compound reported in the early 1970s,2 and the α-glucosidase inhibitor acarbose (26), which was discovered several years later, have become prominent members of this family of natural products. Their chemical and biological properties have been studied in considerable detail. A review of the chemistry and biochemistry of several microbial α-glucosidase inhibitors, particularly those of the acarbose series, was published by Truscheit et al. in 1981;3 however, new discoveries and continued research on this class of natural products have been reported since then. Although aminocyclitols are often associated with pseudooligosaccharide-type bacterial secondary metabolites that possess sugar hydrolysis inhibitory activity, more examples of their diversity in structures and biological activities have been recorded.
During the past two decades, dozens of synthetic methods leading to total and semi-syntheses of aminocyclitols have been developed. In parallel, hundreds of analogs have been synthesized and their biological activities have been evaluated, resulting in the discovery of a number of pharmaceutical leads including a current clinically used potent anti-diabetic drug, voglibose (AO-128).
While studies on the biosynthetic origin of the C7N aminocyclitols in some members of this family were first carried out in the mid 1980s, it was not until recently that more thorough biosynthetic studies on these compounds have been reported.
The present review describes the discovery and characterization of compounds classified in the C7N aminocyclitol family as well as their biosynthetic origin, their biochemistry and biological properties and their further development for pharmaceutical uses.
2 C7N aminocyclitol-containing compounds
2.1 Validamycins
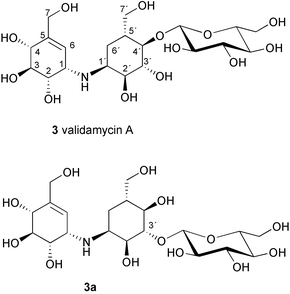
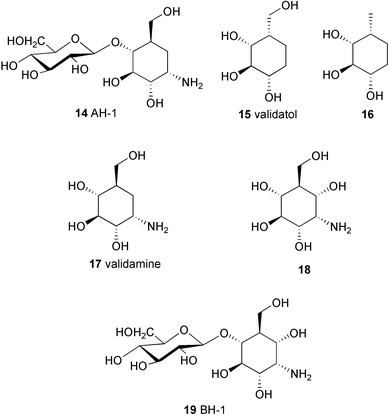
During the course of screening for new antibiotics, researchers at Takeda Chemical Company discovered from the fermentation culture of Streptomyces hygroscopicus subsp limoneus a complex of weakly basic, water-soluble antibiotics that exhibited remarkable therapeutic effects against plant diseases.2,4 These compounds were found to be effective for the control of sheath blight of rice plants caused by the fungus Pellicularia sasakii, and also to prevent dumping-off of cucumber seedlings caused by Rhizoctonia solani KUHN4,5 (in the later literature6,7P. sasakii has been asserted to be a synonym of R. solani). Further investigations on this complex mixture led to the isolation of two related compounds, validamycins A (3) and B (4).8 Validamycin A (3), the main component of the validamycin complex, is a pseudotrisaccharide consisting of a core moiety, validoxylamine A (11), and glucose. The core is assembled from two C7N aminocyclitols, valienamine (1) and validamine (17), which are connected through a single nitrogen bridge shared by both units. Validamycin B (4) has almost the same structure as validamycin A (3), except for the second aminocyclitol unit, which is a hydroxyvalidamine (18) instead of validamine. Hydrogenolysis of validamycin A over platinum oxide yielded a mixture of several degradation products,9 compound AH-1 (14) (about 55%), and two neutral cyclitols, designated as validatol (15) (about 40%) and deoxyvalidatol (16) (about 15%). AH-1 (14) gave D-glucose and validamine (17)10 upon acid hydrolysis, while hydrolysis of validamycin A in 0.5M sulfuric acid heated on a steam bath gave rise to two products, validoxylamine A (11) and D-glucose. Hydrogenolysis of validoxylamine A (11) yielded validamine (17), validatol (15) and deoxyvalidatol (16). Similarly, validamycin B (4) gave compound BH-1 (19), validatol (15), and deoxyvalidatol (16) by hydrogenolysis.9 BH-1 (19) then gave D-glucose and hydroxyvalidamine (18) on acid hydrolysis, while validamycin B (4) was hydrolyzed to validoxylamine B (12) and D-glucose. Interestingly, none of the above chemical degradations gave valienamine (1) as the product. It was later found that cleavage of the C–N bond of validoxylamine A (11) with N-bromosuccinimide (NBS) in aqueous N,N-dimethylformamide gives rise to valienamine (1) and validamine (17), as well as some keto-derivatives.11,12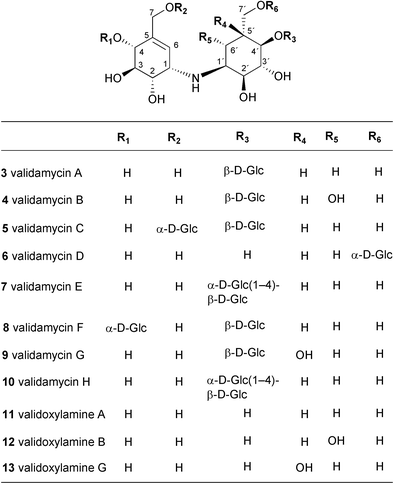
The complete chemical structure of validamycin A was first proposed by Horii and Kameda13 to be N-[(1S)-(1,4,6/5)-3-hydroxymethyl-4,5,6-trihydroxycyclohex-2-enyl][O-β-D-glucopyranosyl-(1→3)-(1S)-(1,2,4/3,5)-2,3,4-trihydroxy-5-hydroxymethylcyclohexyl]amine (3a), based on the NMR, MS, chemical degradation, and X-ray crystallography of the degradation product, validamine, as its monohydrobromide.14 This assignment, however, was later revised by Suami and co-workers15 after their synthetic product aiming for the two right-hand side residues of the proposed structure, 3-O-β-D-glucopyranosylvalidamine, was different from an authentic sample derived by hydrogenolysis of validamycin A.16,17 Further synthetic results suggested that the glucopyranosyloxy group is attached to C-4 of the validamine moiety, instead of C-3 as previously proposed.18,19 This result was in good agreement with the complete assignments of the 1H and 13C NMR spectra of 3 carried out by Rinehart and co-workers, by use of 2D NMR (COSY) and single frequency off-resonance decoupling (SFORD) experiments, as well as comparison studies with model compounds.20
A number of minor components of the validamycins complex, namely validamycins C (5), D (6), E (7), F (8), G (9) and H (10), have also been isolated from the culture broth of the same producing organism.21–24 Validamycins C–F (5–8) and H (10) contain validoxylamine A as the common core unit in their molecules, but differ from one another in at least one of the following features; the position of a glucosidic linkage, the number of D-glucose molecules and the configuration of the anomeric center of the glucose. Similar to the case of 3, upon acid hydrolysis 5, 6, 7, 8 and 10 gave validoxylamine A (11) and D-glucose. Under milder acid conditions, validamycins C (5), E (7), and F (8) partially hydrolyzed to validamycin A (3).
In contrast to other validamycins, validamycin G (9) contains validoxylamine G (13) as its core structure, which is assembled from valienamine (1) and valiolamine (20).23 Both validamycin G and validoxylamine G are less active against R. solani by the dendroid-test method;5 however, unlike other validamycins and validoxylamines, validamycin G (9) and validoxylamine G (13) were somewhat active against porcine intestinal α-glucosidases.
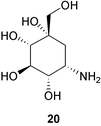
Together with the validamycins and the validoxylamines, simple aminocyclitol molecules, such as valienamine (1), validamine (17), hydroxyvalidamine (18), and valiolamine (20), have also been detected in the fermentation broth of S. hygroscopicus subsp. limoneus IFO12703.25 In contrast to the validamycins, which have no α-glucosidase inhibitory activity, the simple aminocyclitols appear to be active against a number of sugar hydrolases. Valiolamine (20) was found to have a more potent α-glucosidase inhibitory effect against porcine intestinal sucrase, maltase, and isomaltase than was exhibited by the other aminocyclitols.25
Validamycins have been shown to be susceptible to microbial degradation.26–30 A large number of microorganisms were found capable of selectively hydrolyzing the α- or β-glucosidic linkages of validamycins A and D. Validamycins A and E were transformed into validoxylamine A by the selective microbial hydrolysis of their β-glucosidic linkage, whereas validamycins C, E and F were transformed into validamycin A by the selective hydrolysis of their α-glucosidic linkage. Pseudomonas denitrificans and Flavobacterium saccharophilum are two microorganisms found capable of cleaving validamycins completely to aminocyclitols.26,28 The microbial degradation of validamycins by P. denitrificans is non-specific for α- and β-glucosidic linkages. Therefore, validamycins A, C, D, E, F, and H all are hydrolyzed to D-glucose and validoxylamine A in the first step, and the validoxylamine A is degraded further to validamine and valienamine. This microbial degradation process has been used as the method of choice for the preparation of valienamine and validamine.
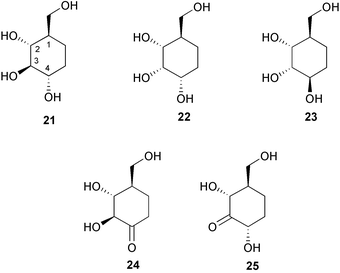
Degradation of validamycin A by Flavobacterium saccharophilum, on the other hand, yields not only validamine and valienamine, but remarkably also 1-epi-validatol (21), 1,3-epi-validatol (22), 1,3,4-epi-validatol (23), 4-keto-1-epi-validatol (24), and 3-keto-1-epi-validatol (25).28,29 Intensive studies on the degradation pathways of validamycin A on the enzymatic cleavage of its C–N linkage by F. saccharophilum have led to the conclusion that two enzymes, a dehydrogenase and a C–N lyase, participate in the cleavage process of the C–N linkage of validoxylamine A.30
2.2 Acarbose and amylostatins
Among members of the C7N aminocyclitol family of natural products, the α-glucosidase inhibitor acarbose (26)31 is recognized as one of the most clinically important compounds. Acarbose is a useful drug for the treatment of type II insulin-independent diabetes32 and has been used to treat this ailment in European countries for many years. It is, however, relatively new and is the first α-glucosidase inhibitor available in the US (known as Precose, Bayer Corporation). It was approved by the Food and Drug Administration (FDA) in September 1995 for use as monotherapy in patients with non-insulin-dependent diabetes mellitus (NIDDM).33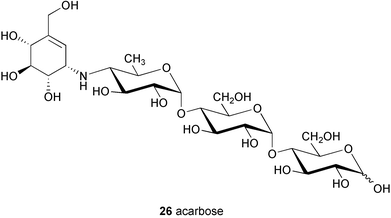
The chemistry and biochemistry of acarbose and its analogs were reviewed by Truscheit et al. in 1981.3 Found as one of the components in a complex mixture of pseudooligosaccharides isolated from culture broths of the Actinoplanes strain SE 50/110, acarbose (26) (BAY g 5421) shows potent inhibitory activity against sucrase, maltase, dextrinase, and glucoamylase.3 It also shows inhibitory activity, although to a relatively lower extent, against α-amylase and other amylose-degrading enzymes.
Structurally, acarbose represents a pseudotetrasaccharide, consisting of an unsaturated aminocyclitol, valienamine, a deoxyhexose and maltose. The pseudodisaccharide core structure, known as acarviosine, which is composed of valienamine (1) and the deoxyhexose, is postulated to be essential for its biological activity. Acarviosine itself has been isolated only in the form of the methyl glycoside, since free acarviosine readily undergoes a rearrangement to a tricyclic pyrrolo[2,1-b]benzoxazole derivative (27) under the conditions for hydrolysis of the glycoside linkage. The core unit acarviosine is also linked to a variable number of glucose residues resulting in several other components in the complex mixture of acarbose.3 The formation of these components is highly dependent on the composition of the carbon source available in the culture medium.31 Media containing glucose and maltose will result in a specifically high yield of acarbose and the lower components, while media with high concentration of starch will yield longer pseudooligosaccharide species.3 The transglycosylation involved in this process was proposed to be catalyzed by an extracellular enzyme activity, namely acarviosyl transferase (ATase), found in the culture supernatant of the acarbose producer.34 ATase, encoded by a gene, acbD, catalyzes the transfer of the acarviosyl moiety of acarbose to malto-oligosaccharides. It has been demonstrated that ATase recognizes a wide range of substrates in vitro, which suggests its central role for the formation of the observed diversity of acarbose-related compounds by Actinoplanes sp.34
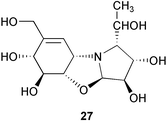
Chemical structures of 26 and its derivatives were determined by degradation reactions, derivatization and spectroscopic analysis.3,35 Complete hydrolysis of 26 gave D-glucose and a tricyclic pyrrolo[2,1-b]benzoxazole derivative (27) that was further degraded into validatol (15) and (2R,3S,4S)-2-[(1R)-1-hydroxyethyl]-pyrrolidine-3,4-diol (28) by sodium borohydride reduction and subsequent catalytic hydrogenation (Pt/H2). Methanolysis of acarbose yielded α and β glucosides 29 (methyl acarviosine) and 30, respectively, which were cleaved by hydrogenation to give 15 and methylviosamine (31). When acarbose (26) was directly hydrogenated using Pd/C or Pt as catalysts, a complex mixture including validatol (15), a basic trisaccharide 32 and traces of dihydroacarbose (33 and 34) were obtained. Trisacccharide 32 was found to have no inhibitory effects against α-amylases or sucrases, suggesting that the amino sugar alone is not critical for the inhibitory activity. In a controlled reduction of acarbose with Raney nickel as catalyst it was possible to saturate only the double bond in acarbose to produce mainly the two dihydro analogs, 33 (gluco isomer) and 34 (ido isomer).36 The cyclitol moiety in 33 (gluco isomer) adopts a 4C1 conformation; however, in the ido-isomer (34), the 1C4 conformation predominates with the hydroxy groups axial and the CH2OH and N-R substituents equatorial.
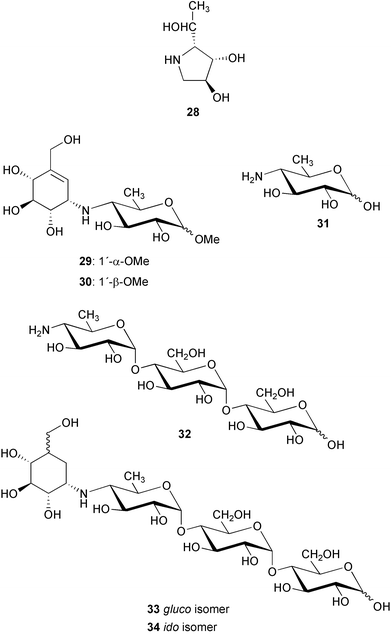
The solution conformations of acarbose and its dihydro derivatives were found to be strongly dependent on pH.37 Upon protonation, changes in the amino linkage conformation at the pseudoglycosidic bond between the C7N unit and the 4-amino-4,6-dideoxy-D-glucopyranose unit in acarbose and the dihydro isomers have been observed by NMR studies (mainly 1H NMR and proton NOE) in combination with empirical minimum energy calculations and modeling with the Discover force field.36 For acarbose and the gluco isomer, two major minimal energy conformers exist in equilibrium in solution with one [ϕH (H-1, C-1, N, C-4′), ψH (C-1, N, C-4′ , H-4′) −51, −10 for acarbose (26) and −44, −27 for the gluco isomer (33)] preponderating at basic pH, while another minimum (ϕH, ψH −26, 176 for 26 and −22, 179 for 33) appears to be populated to a significant degree at acidic pH.36 The same observation was also reported for methyl α-acarviosine (29).38,39
At nearly the same time that acarbose was discovered, Murao and co-workers independently reported the isolation of an amylase inhibitor, referred to as S-AI, from Streptomyces diastaticus subsp. amylostaticus No. 2476.40 The structure of S-AI has been considered to be G-G-G-G-G-(S-AI-X)-G, where G is glucose and S-AI-X is unknown. Later on, a bioassay-guided separation following glucoamylase inhibitory activity led to the isolation of a complex of new amylase inhibitors, the amylostatins,41 from the culture of the same bacteria. The amylostatins are also reportedly present in Streptomyces diastaticus subsp. amylostaticus No. 9410,42 a close relative of strain No. 2476. Although it was estimated that more than twenty amylostatin compounds were present in the complex, after a treatment with β-amylase the hydrolysate was found to have only six compounds. Following a successful separation by preparative HPLC, their chemical structures have been determined, based mainly on chemical degradation, NMR and mass spectral analysis, to be XG (35), XGG (36), XGGG (37), GXG (38), GXGG (39) and GXGGG (40).42
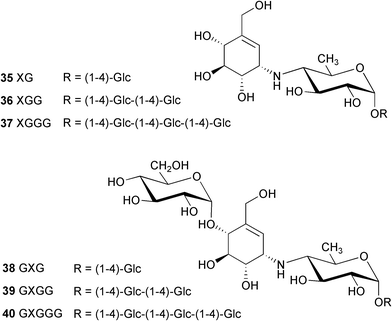
A complete acid hydrolysis (with 2M hydrochloric acid at 160 °C for 2 h) of all isolated fractions containing basic metabolites provided a common product, compound X′ (C13H21NO7, MW. 303),41 which is identical with the tricyclic pyrrolo[2,1-b]benzoxazole derivative (27).3 Reduction of X′ with sodium borohydride gave X′H (41, C13H23NO7, MW 305). A milder acid hydrolysis (with 2M trifluoroacetic acid at 100 °C for 2 h) of the same fractions yielded XG (35) as a major product, together with a small amount of compound X′. Reduction of XG with sodium borohydride gave compound XGH (43), which on hydrolysis with 2M hydrochloric acid gave compound X′ and sorbitol.41
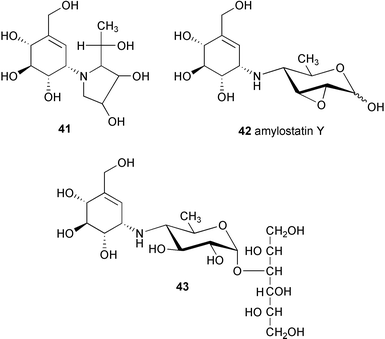
The isolation of amylostatin Y (42) and other amylostatins from Streptomyces diastaticus subsp. amylostaticus FERM-P 2499 has also been reported in the patent literature.43–46 Amylostatin A was reported as a neutral oligosaccharide, whereas oligostatins B–F and J–N were claimed to be weakly basic oligosaccharides showing an amylase inhibitory activity. Except for the amylostatins A (neutral oligosaccharide)44 and Y (42),43 the chemical structures of the amylostatins seem to be very close to those of acarbose analogues. In fact, some of them are identical with compounds reported by Truscheit et al.3 Amylostatin XG (35), XGG (36) and GXGG (39), for example, are identical with “component 2”, “component 3” or acarbose (26) and “component 4” described in Truscheit's report, respectively.
2.3 Adiposins
Adiposin-1 (44) and −2 (45) were isolated from Streptomyces calvus TM-521, a soil bacterium collected in Hoya City, Tokyo, by Õmura and co-workers.47,48 The molecular formulas of 44 and 45 were defined as C19H33NO14 and C25H43NO19, respectively. Structurally, they are very similar to acarbose and the amylostatins, except that they have a 4-amino-4-deoxyglucose in place of the 4-amino-4,6-dideoxyglucose moiety of acarbose. This difference was confirmed through hydrolysis of the adiposin complex with 0.6% sulfuric acid at 100 °C for 1 h, which gave adiposin-D (46),49 a tricyclic compound similar to the degradation product 27 of acarbose. Adiposin-1 and −2 and six other related compounds represented by general formula 47 have also been isolated from another soil bacterium, Streptomyces sp. strain A2396.50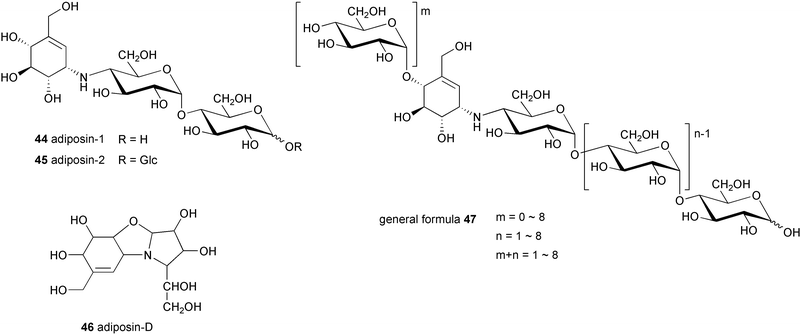
Adiposin-1 and −2 showed potent inhibitory activities against α-amylase, human α-amylase and disaccharidases isolated from porcine small intestine. They also exhibit antibacterial activity against some Gram positive bacteria (e.g., Bacillus licheniformis, Staphylococcus aureus), Gram negative bacteria belonging to Enterobacteriaceae, some anaerobic bacteria, and phytopathogenic fungi.49
2.4 Oligostatins (SF-1130 X)
Using antibacterial activity-guided separation and purification, scientists at Meiji Seika Kaisha, Ltd. isolated five compounds from the culture filtrate of Streptomyces myxogenes nov. sp. SF1130, three of which, namely oligostatins C (48), D (49), and E (50), were reported as basic oligosaccharide antibiotics.51 Oligostatins C (48), D (49), and E (50) were defined as pseudopenta, -hexa, and -heptasaccharides, respectively, with a nitrogen containing glycosidic bond.52 In contrast to the previously described α-glucosidase inhibitors, the oligostatins lack the valienamine (1) moiety, the unsaturated C7N aminocyclitol core unit, in their structures. Neither olefinic protons (in 1H NMR) nor sp2 carbons (in 13C NMR) were observed in their NMR spectra. Interestingly, methanolysis of oligostatin C gave methyl acarviosine (29) instead of the expected core structure, methyl oligobiosaminide (51). Likewise, acid hydrolysis of oligostatin C produced 3 moles of D-glucose and the tricyclic compound 27, which is known to be a hydrolytic product of acarbose. It was proposed that the core C7N aminocyclitol unit in oligostatin C contains an axially oriented C-6 hydroxy group, which undergoes a β-elimination (dehydration) during methanolysis or acid hydrolysis to give an unsaturated product. However, attempted acid-catalyzed dehydration of synthetically prepared methyl oligobiosaminide, under conditions similar to those used for the degradation of oligostatin, was unsuccessful.53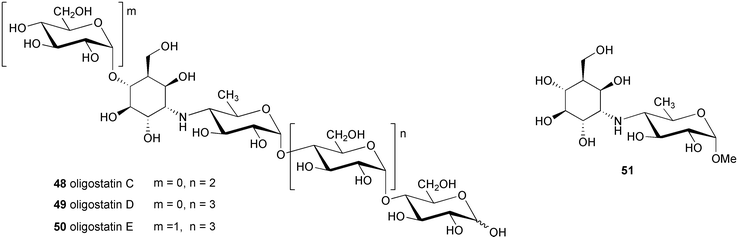
2.5 Trestatins
Trestatins A (52), B (53), and C (54) are the major components of a complex of oligosaccharides isolated from fermentation cultures of S. dimorphogenes nov. sp. NR-320-OM7HB,54 and its close relative, strain NR-320-OM7HBS (spirales).55 The latter microorganism, which is different from NR-320-OM7HB in its spore chain morphology, reportedly produced trestatin A in a higher yield than the former one.55 As in the case of other pseudooligosaccharides, their chemical structures were elucidated mainly based on combinations of their physico-chemical properties and chemical degradations. Upon a complete acid hydrolysis with 4M hydrochloric acid, trestatins gave D-glucose and a basic product identical to the tricyclic compound 27, which suggests they possess a similar core structure as acarbose and the amylostatins.56 However, trestatins are non-reducing oligosaccharides as indicated by negative color reaction with tetrazolium red. Under mild acid hydrolysis conditions, trestatins were partially hydrolyzed to a number of products, which were separated into neutral and basic fragments. HPLC analyses of the neutral fragments from each trestatin showed identical chromatograms with peaks corresponding to glucose, maltose, α,α-D-trehalose, glucotriose (58), and glucotetraose (59). On the other hand, the basic fragments each contained a certain combination of partially degraded products, which reflected the full structure of the parent compound. The non-reducing property of the trestatins as well as the chemical degradation results indicate the location of the trehalose moiety to be at the anomeric end of the molecules. This was in good agreement with the NMR experimental data that demonstrated that the anomeric carbon of the trehalose residue in trestatin has longer 13C spin–lattice relaxation times (T1s) than the other anomeric carbons in the molecule, suggesting its location is at the terminus of the oligosaccharide.56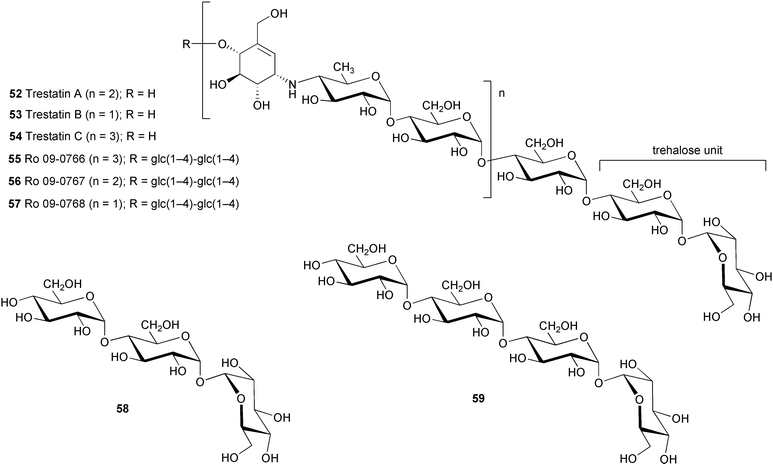
Three minor components of the trestatin complex, all possessing potent α-amylase inhibitory activity, were also isolated from the same bacteria and were characterized as Ro 09–0766 (55), Ro 09–0767 (56) and Ro 09–0768 (57).57 Their structures were confirmed by enzymatic degradation using β-amylase in acetate buffer followed by separation of the reaction mixtures on Sephadex G-10 which gave maltose and trestatin C, maltose and trestatin A, and maltose and trestatin B from 55, 56, and 57, respectively.
2.6 Oxirane pseudooligosaccharides
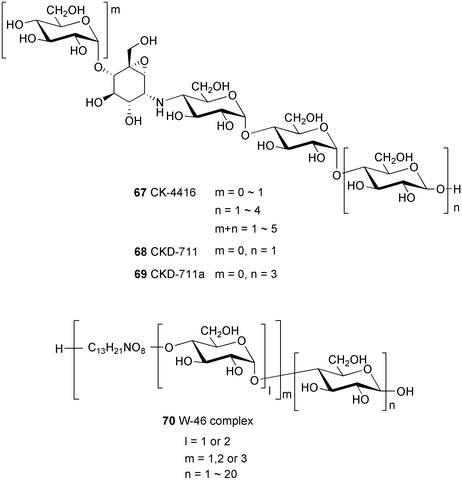
Oxirane pseudooligosaccharides refer to aminooligosaccharides carrying one or more epoxide-containing C7N aminocyclitol in their core structure connected to glucose or other sugar residues. NS complex (60),58 W-46 (70),59 and CK-4416 (67)60 are among the compounds included in this subgroup, all of which have been claimed to have potent inhibitory activity against α-amylase and saccharases.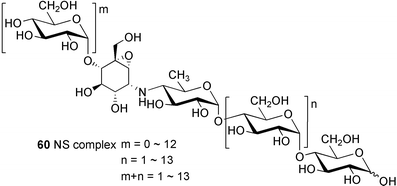
NS complex (60), which contains at least 17 related compounds designated as NS-1–NS-17, was isolated from the culture broth of Streptomyces flavochromogenes.58 The components NS-1–NS-17 all have the same core structure, differing only in the number of the glucosyl residues attached at both the anomeric and the opposite ends. Acid hydrolysis of NS complex with 2–4 M hydrochloric acid mainly gave D-glucose and the epoxidal tricyclic NS-0 (61). The latter compound is known to inhibit the aggregation of blood platelets.
W-46 complex (70) was isolated from Streptomyces galbus subsp. FH 1716 (DSM 3007)59 and mainly consists of three major components, W-46 A (62), W-46 B (63), and W-46 C (64), with molecular formulas of C44H74N2O32, C50H84N2O37, and C56H94N2O42, respectively. Their structures differ from those of the NS series particularly in the number of the active epoxide-pseudoaminosugar units, which appear twice in the W-46 active core unit. W-46 H (65) and W-46 P (66) were prepared from W-46 complex by acid hydrolysis or enzymatic elimination of glucose.61 Alternatively, these compounds can also be prepared, albeit less conveniently, by controlling the fermentation time of the producing strain.
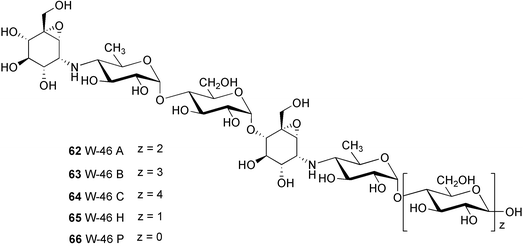
CK-4416 (67) was isolated as a complex of pseudooligosaccharides from the fermentation broth of Streptomyces sp. CK-4416 isolated from a forest soil of Jeju Island, South Korea.60 Its core structure is slightly different from that of the NS series, containing a 4-amino-4-deoxyglucose moiety instead of 4-amino-4,6-dideoxyglucose. Very recently, Chun, Han and their colleagues reported the isolation and complete structure elucidation of two of its components, CKD-711 (68) and CKD-711a (69).62–64 CKD-711 (68) (C25H43NO20) is a pseudotetrasaccharide consisting of one epoxidal C7N aminocyclitol unit connected to three molecules of glucose, whereas CDK-711a (69) (C37H63NO30) is the hexameric analog of 68 possessing two additional glucose moieties at its anomeric end. CKD-711 (68) was found to be as potent as acarbose against porcine intestinal maltase and sucrase; however, it showed about two-fold lower inhibitory activity against α-amylase compared to acarbose. The hexameric analog CDK-711a (69) showed less inhibitory activity than 68 against all enzymes tested. In contrast to acarbose, both compounds exhibited potent antimicrobial activity against Comamonas terrigena.63
2.7 Salbostatin and suidatrestin
Salbostatin (71) is a basic, non-reducing pseudodisaccharide which consists of valienamine linked to 2-amino-1,5-anhydro-2-deoxyglucitol.65 It was isolated from the fermentation culture of the polyether antibiotic salinomycin producer Streptomyces albus ATCC 21838. Despite having a similar chemical structure as acarviosine and other pseudodisaccharides, salbostatin does not inhibit the enzymes saccharase (EC 3.2.1.26) and maltase (EC 3.2.1.20) from the mucus of porcine small intestine, and only weakly (IC50 = 0.68 mM L−1) inhibits murine liver aldose reductase (EC 1.1.1.21). Salbostatin also shows no antibiotic activity; however, it does effectively inhibit trehalase from porcine kidney (EC 3.2.1.28) with a Ki of 1.8 × 10−7 M.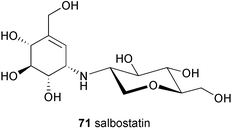
Another pseudodisaccharide that demonstrates potent trehalase inhibition is called suidatrestin (molecular weight 335).66 This compound was isolated from the culture broth of Streptomyces sp. strain Yo-23. It was proposed that suidatrestin is structurally similar to validoxylamine A, a degradation product of validamycin A, as judged by a direct comparison of their 13C NMR spectra; however, neither comprehensive molecular characterization nor the chemical structure of suidatrestin are available.
2.8 Pyralomicins
The pyralomicins are a group of antibiotics, reported in 1996 by Takeuchi and co-workers, with unique chemical structures, a benzopyranopyrrole chromophore containing a nitrogen atom which is glycosylated by C7 cyclitols [pyralomicins 1a–1d (72–75)] or by glucose [pyralomicins 2a–2c (76–78)].67 The cyclitol structure in pyralomicins 1a–1d (72–75) is similar to the valienamine moiety of acarbose (26) and other related compounds, the only difference being the opposite stereochemistry at the C-1′ position. These antibiotics were isolated from a soil bacterium, Actinomadura spiralis MI178–34F18,67 which was later renamed Microtetraspora spiralis MI178–34F18,68 and recently was again changed to Nonomuraea sp. according to a new classification.69 Their chemical structures were deduced mainly from NMR spectroscopy including various 2D NMR experiments, e.g., 1H–15N HMBC, decoupled HMBC, and 13C{1H} NOE.70 The absolute configurations of pyralomicins 1a (72) and 2a (76) were determined by X-ray crystallographic analysis of crystalline 7′-O-p-bromobenzopyralomicin 1a and 2′-O-p-bromobenzopyralomicin 2a, whereas absolute configurations of pyralomicins 1b–1d (73–75) and 2b–2c (77–78) were suggested to be similar to those of 72 and 76, respectively, based on comparison of CD spectra.71 The cyclitol moiety of pyralomicins 1a–1d (72–75) is proposed to be in the same half-chair conformation that, in the case of acarbose and the validamycins, is known to be critical for biological activity.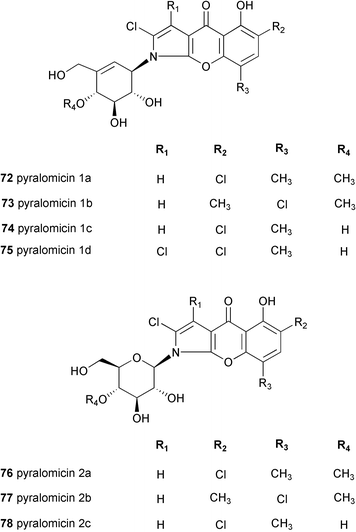
Pyralomicins 1a–1d (72–75) are the only examples of natural products reported thus far having a C7N aminocyclitol act as the glycone attached to a polyketide-derived core structure.
2.9 Epoxyquinomicins and cetoniacytones
A number of structurally unusual C7N aminocyclitols have been reported. The epoxyquinomicins, which were isolated from the culture broth of Amycolatopsis sp. strain MK 299–95 F4, are structurally quite different from other types of C7N aminocyclitols.72,73 They consist of a unique C7N aminocyclitol unit having an epoxide at C-5, C-6 as in the NS complex,56 W-4659 and CK-4416,60 but with different degrees of oxidation on other carbon positions. In contrast to most of the secondary metabolites belonging to this family, which commonly entail an N-ether linkage at the C-1 position, the nitrogen in the epoxyquinomicins is located at C-2 and is linked to a chloro-substituted or an unsubstituted salicyloyl moiety via an amide bond. Four epoxyquinomicins (79–82) have been isolated so far and their chemical structures were determined based on various NMR and X-ray analyses.74 Epoxyquinomicins B (80) and C (81) were revealed to be the deschloro analogs of epoxyquinomicins A (79) and D (82), respectively, while epoxyquinomicins A (79) and B (80) possess a carbonyl group at the C-1 position of their core units instead of the hydroxyl group seen in the epoxyquinomicins C (81) and D (82).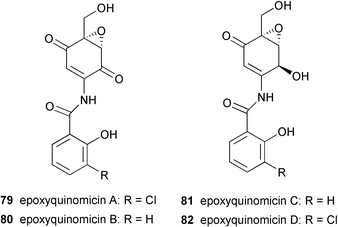
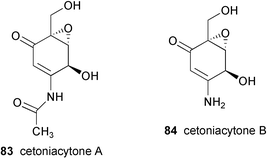
Very recently, Zeeck and co-workers75 reported the isolation of cetoniacytones A (83) and B (84), two new metabolites having the same core unit as epoxyquinomicins C (81) and D (82), from an endosymbiotic Actinomyces sp. strain Lu 9419. This microorganism was isolated from the intestines of an insect Cetonia aureata. Their chemical structures were elucidated by detailed spectroscopic analysis, and the absolute configuration of the major component, cetoniacytone A (83), was determined by X-ray analysis as well as through derivatization with chiral acids. Despite their unusual chemical structures, the core units of the cetoniacytones appear to be derived via the pentose phosphate pathway as has also been shown for acarbose, the validamycins and the pyralomicins.
3 Biosynthesis of the C7N aminocyclitols
3.1 The origin of the core C7N units
The C7N aminocyclitol moiety present in validamycin, acarbose and related compounds may be viewed as an aliphatic version of the mC7N units found in a variety of secondary metabolites, particularly the ansamycin (rifamycin, geldanamycin, and maytansine) and mitomycin antibiotics.76,77 The mC7N units in the latter compounds are exclusively formed from an unusual aromatic amino acid, 3-amino-5-hydroxybenzoic acid (AHBA), which is derived from 4-amino-3,4-dideoxy-D-arabino-heptulosonic acid 7-phosphate (aminoDAHP), through a branch of the shikimate pathway.76,78 Another type of aliphatic mC7N structure is found in a class of natural products known as mycosporine-like amino acids (MAAs), which are widely distributed in marine organisms, fresh water cyanobacteria and green microalgae, as well as terrestrial fungi.1 The core units in MAAs are presumably also derived from a variant of the shikimate pathway, although in contrast to that of AHBA the nitrogen atom in MAAs is derived intact from the side-chain amino acids and is introduced into the molecules at a later stage of the biosynthetic pathway. However, preliminary studies on the biosynthesis of the C7N aminocyclitol moiety present in acarbose, validamycin and a number of related compounds revealed that they are not derived from any of the above pathways nor any other branch of the shikimate pathway.79–81 Feeding experiments, carried out by the groups of Rinehart,81 Pape and Floss80 with uniformly and positionally 13C-labeled glucose, mannose and ribose,81 or [U-13C3]glycerol,80 and analysis of the labeling and coupling patterns in the products demonstrated that the carbon skeleton of the aminocyclitols in acarbose and validamycin is biosynthesized from a C2+C2+C3-derived 7-carbon sugar phosphate related to the pentose phosphate pathway (Scheme 1). A similar origin has been observed more recently for the formation of the aminocyclitol moieties in pyralomicin 1a82 and cetoniacytone A.75![Metabolic origin of acarbose and validamycin proposed by Floss80 and Rinehart81. {The heavy bonds indicate the 13C–13C coupling patterns from [U-13C3]glycerol and D-[U-13C6]glucose, respectively}.](/image/article/2003/NP/b205561a/b205561a-s1.gif) | ||
| Scheme 1 Metabolic origin of acarbose and validamycin proposed by Floss80 and Rinehart81. {The heavy bonds indicate the 13C–13C coupling patterns from [U-13C3]glycerol and D-[U-13C6]glucose, respectively}. | ||
Rinehart and co-workers81 suggested that either D-sedo-heptulose 7-phosphate or its epimer D-ido-heptulose 7-phosphate could be the immediate precursor of the initial cyclic intermediates. They proposed that valiolone (86) would be the first cyclitol formed, acting as the key intermediate to valiolamine (20), valienamine (1) and validamine (17), the precursors of the validamycins. Floss and co-workers83 synthesized a number of potential precursor cyclitols, including valiolone, in tritiated or stable-isotope labeled form, which were fed to the acarbose (26) producer. Rather surprisingly, while valiolone failed to act as a precursor to acarbose, its C2 and C5 epimer (87) was the only compound to be incorporated efficiently into the aminocyclitol (Scheme 2). As neither the 2-epimer nor the 5-epimer of valiolone was incorporated into 26, it would appear that 2-epi-5-epi-valiolone (87) is indeed the initial cyclitol precursor of the core moiety. The same compound was also efficiently incorporated into validamycin84 and pyralomicin 1a85 giving an overall enrichment of 12.2% and 23%, respectively. In the case of validamycin both cyclitol moieties were enriched, suggesting that 2-epi-5-epi-valiolone is the precursor of all the aminocyclitol moieties in these compounds.84
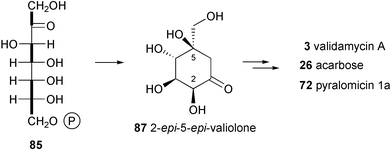 | ||
| Scheme 2 Formation of 2-epi-5-epi-valiolone, the precursor of the cyclitol moieties in validamycin, acarbose and pyralomicin. | ||
The involvement of 2-epi-5-epi-valiolone (87) in the biosynthesis of acarbose and as the immediate cyclization product of the open chain C7 sugar has been confirmed by genetic and biochemical approaches.86 Using a PCR approach with conserved sequences of known deoxythymidyl-diphospho-glucose (dTDP-glucose) 4,6-dehydratases to generate a hybridization probe, Piepersberg and co-workers cloned and sequenced part of the acarbose biosynthetic gene cluster from the acarbose producer strain Actinoplanes species SE50/110. They found three open reading frames in the region, acbA with homology to dTDP-glucose synthases, acbB with homology to dTDP-glucose 4,6-dehydratases and acbC, which showed homology to dehydroquinate synthases, enzymes that catalyze cyclization of the C7 sugar phosphate DAHP into dehydroquinate (DHQ) in the shikimate pathway. Further sequencing of the oligonucleotides in the flanking regions established the acarbose biosynthetic gene cluster (Scheme 3).87 The acbC gene was expressed heterologously in Streptomyces lividans. The recombinant protein was found to catalyze the conversion of sedo-heptulose 7-phosphate (85), but not ido-heptulose 7-phosphate, into a cyclitol which was unequivocally identified as 2-epi-5-epi-valiolone (87).86
 | ||
| Scheme 3 The organization of the acarbose biosynthetic genes of Actinoplanes sp. strain SE50/110 and their putative functions. | ||
It has been proposed that the acbC gene product operates by a mechanism similar to dehydroquinate synthases, involving transient dehydrogenation at C-5 to a ketone, followed by the elimination of phosphate to generate the enol of a 6,7-methyl ketone. The latter then undergoes intramolecular aldol condensation to give 2-epi-5-epi-valiolone (87) (Scheme 4). An experiment with D-sedo-[7-14C,7-3H]heptulose 7-phosphate supports this mechanism and argues against an alternative myo-inositol 1-phosphate synthase88,89-like mechanism.
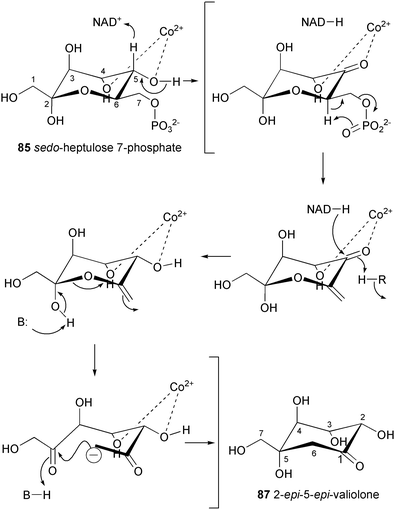 | ||
| Scheme 4 Proposed reaction mechanism of the C7-cyclitol synthase (AcbC). | ||
3.2 Validamycin formation
Validamycins A (3), C (5), D (6), E (7), F (8) and H (10) contain validoxylamine A (11) as the common core unit in their molecules, which is assembled from two C7N aminocyclitols, the unsaturated residue, valienamine (1), and the saturated one, validamine (17). Both moieties are derived from 2-epi-5-epi-valiolone (87) as the initial precursor via a number of modification steps (Scheme 5).84 The first step involves epimerization at C-2 of 2-epi-5-epi-valiolone (87) to give 5-epi-valiolone (88), followed by dehydration between C-5 and C-6 to give valienone (89). The steric course of the latter reaction has been probed by feeding experiments with stereospecifically labeled 6α- and 6β-monodeuterated 5-epi-valiolones to the validamycin A producer strain Streptomyces hygroscopicus var. limoneus. The results revealed that the dehydration of 5-epi-valiolone (88) to valienone (89) occurs by a syn elimination of water.90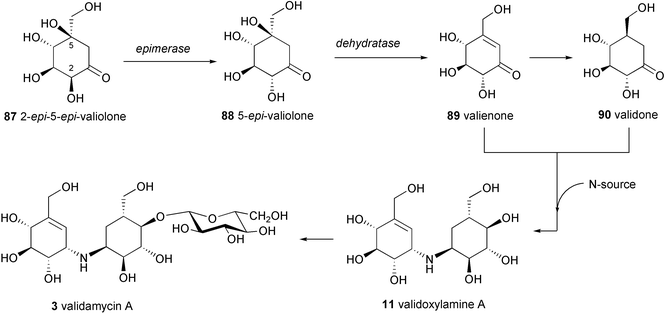 | ||
| Scheme 5 Proposed biosynthetic pathway to validamycin A. | ||
It was found that valienone (89) plays a dual role as intermediate in the biosynthesis of validamycin. Feeding experiments with radio- and stable isotope labeled valienone (89) and validone (90) showed that valienone is a specific precursor of both cyclitol moieties of validamycin, labeling the unsaturated cyclitol moiety directly and the saturated one via validone.84 The transformation of valienone (89) into validone (90) takes place by an anti addition of hydrogen. The two latter cyclitols are then connected via a bridging nitrogen to give the core structure of validamycin A (3), validoxylamine A (11). The timing and mode of introduction of the bridging nitrogen atom in validoxylamine A (3) are not completely understood. Feeding experiments with [7-3H]valienamine, [7-3H]validamine, and [7-3H]valiolamine showed no incorporation of any of these compounds into validamycin A.84 Although these experimental results could mean that none of the above compounds are involved in the biosynthesis of validamycin, it is also possible that these negative results are due to the lack of cellular uptake of the aminocyclitols. In fact, in vivo formation of [7-3H]validamine has been observed in the fermentation culture of the validamycin producer fed with the corresponding ketone, [7-3H]validone. Based on this evidence, a plausible scenario for the introduction of nitrogen and for the formation of validamycin A has then been proposed which involves as a first step transamination of the keto-cyclitol, validone (90), to its amino derivative, validamine (17), with the α-nitrogen of glutamate as probably the most efficient nitrogen source.91 The resulting validamine (17) could then undergo a reductive coupling with the second ketocyclitol, valienone (89), to give validoxylamine A (11), followed by glucosylation as the final step. The last step is in good agreement with that proposed previously by Kameda et al.,27 based on their feeding experiments with [14C]validoxylamine A to the culture of validamycin A-producing Streptomyces that demonstrated the conversion of [14C]validoxylamine A into validamycin A with a 14.25% incorporation rate. The biosynthesis of 11 from 89 and 90 could involve either the formation of an imine (92) and its subsequent reduction, or the ketocyclitol could be reduced beforehand to the alcohol (91) which could undergo SN2 displacement of the activated OH group by the amino group of 17 (Scheme 6).91
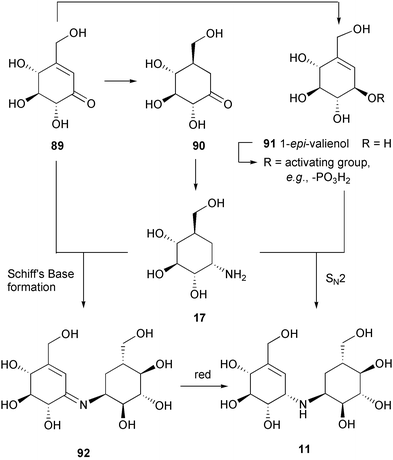 | ||
| Scheme 6 Two possible mechanisms for the formation of validoxylamine A from valienamine and validamine. | ||
While validamycins A (3), C (5), D (6), E (7), F (8) and H (10) are derived from the same core structure, validoxylamine A (11), their congeners validamycins B (4) and G (9) are assembled from different core structures, validoxylamines B (12) and G (13), respectively. Their formation would require the transamination of other ketocyclitols, such as 6-hydroxyvalidone (93), into 6-hydroxyvalidamine (18) and valiolone (86) into valiolamine (20). Condensation between 6-hydroxyvalidamine (18) and valiolamine (20) with valienone (89) would give validoxylamines B (4) and G (9), respectively (Scheme 7).84
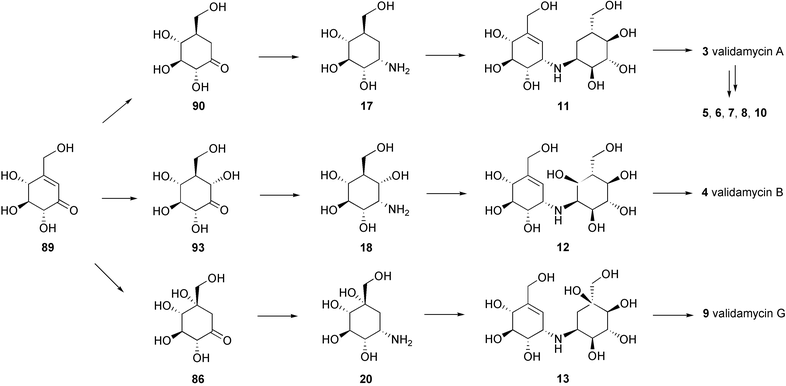 | ||
| Scheme 7 A hypothetical mode of formation of the validamycins. | ||
3.3 Acarbose formation
Despite the fact that the valienamine moiety in both acarbose and validamycin originates from the same precursors, D-sedo-heptulose 7-phosphate (85) and subsequently 2-epi-5-epi-valiolone (87), further downstream the pathways of their formation seem to be different.91 While the biosynthetic pathway to the valienamine moiety in validamycin A has been established to some extent, with a number of discrete cyclitol intermediates identified, this is not the case for the formation of the valienamine moiety of acarbose. In fact, it was found that none of the ketocyclitols involved in the formation of validamycin A, except 2-epi-5-epi-valiolone (87), was incorporated into the valienamine moiety of acarbose (26). As in the validamycin case, neither [7-3H]valiolamine nor [7-3H]valienamine were incorporated either, thus the pathway from 2-epi-5-epi-valiolone (87) to the valienamine moiety of acarbose remains purely conjectural.83Recently Piepersberg and co-workers92 reported the identification of a phosphotransferase activity modifying 2-epi-5-epi-valiolone (87) to 2-epi-5-epi-valiolone 7-phosphate (94) by ATP-dependent phosphorylation. This activity was demonstrated for the recombinant AcbM protein, which was heterologously expressed in S. lividans strain TK64. In addition, the product of acbO, which was found in the same operon as acbC and acbM (Scheme 3), was proposed to have 2-epimerase activity. This result led to a revised proposal for the biosynthetic pathway of acarbose as shown in Scheme 8. Modification of 2-epi-5-epi-valiolone 7-phosphate (94) by epimerization at C-2, dehydration at C-5 and C-6, and reduction at C-1, would give 1-epi-valienol 7-phosphate (97). A second phosphorylation and a subsequent nucleotidylation would set the stage for a transglycosylation process involving an activated amino sugar, dTDP-4-amino-4,6-dideoxy-D-glucose (100).93 This sugar moiety was shown to be assembled from two 3-carbon pieces derived intact from glycerol, most likely viaD-glucose.80 This suggests the involvement in its formation of the well-known NDP-glucose 4,6-dehydratase catalyzation, the universal entry into the biosynthesis of deoxyhexoses.94,95 The identification of the acbA (homology to dTDP-glucose synthases) and acbB (homology to dTDP-glucose 4,6-dehydratases) genes in the acarbose biosynthesis gene cluster,87 as well as evidence for the conversion of dTDP-4-keto-6-deoxy-D-glucose into dTDP-4-amino-4,6-dideoxy-D-glucose, presumably catalyzed by the acbV gene product (homology to aminotransferases),92 further support the above hypothesis.
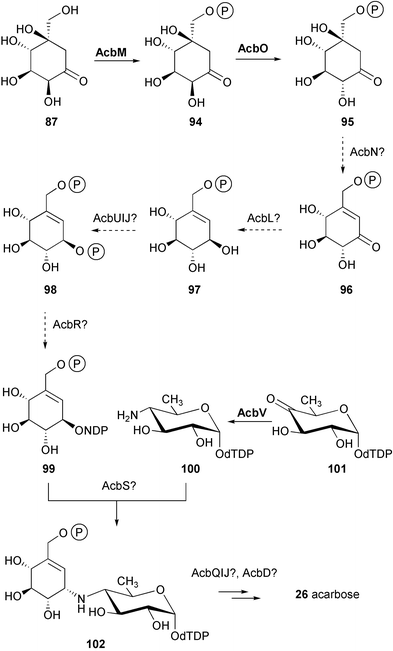
The origin of the bridge nitrogen in acarbose has been studied by Lee and co-workers through feeding experiments with a series of 15N-labeled precursors to production cultures and resting cell fermentations of the acarbose producer.96 The results identified the α-nitrogen of glutamate as the most efficient source of the acarbose nitrogen.
In contrast to the deoxysugar moiety, the two additional glucose moieties present in 26, in a ß-glycosidic linkage, were not enriched in the feeding experiment with 13C-labeled glycerol. This result suggests that they are not formed from endogenously synthesized glucose but may arise from the maltose and maltotriose which is present in abundance in the fermentation medium. It was reported that the formation of acarbose and its oligosaccharide derivatives vary broadly depending on the nature of the carbohydrates present in the production medium. The origin of the two additional glucose moieties in acarbose was confirmed experimentally by following the fate of added regiospecifically deuterated maltotriose.97 The results revealed that the maltose unit of acarbose arises from maltotriose in two ways: about 60% is formed by degradation of maltotriose to maltose through removal of the last glucose from the non-reducing end, followed by transfer of the core acarviosyl moiety to the resulting maltose, while the other 40% is formed by transfer of the core moiety to C-4 of the non-reducing glucose unit of maltotriose followed by removal of one glucose from the other end of the product.97 This result is in good agreement with the later finding of an extracellular glycosyltransferase activity in acarbose-producing cultures that recognizes a number of different sugar moieties.34
3.4 Pyralomicin formation
The antibiotic pyralomicin 1a has a unique structure, a benzopyranopyrrole that is N-glycosylated by a C7 cyclitol. The benzopyranopyrrole structure was first reported in 1992 by Funabashi et al.98 as a skeleton structure of the antibiotic TAN-876A, which is isolated together with the closely related TAN-876B. Studies on the origin of the benzopyranopyrrole core unit in pyralomicin 1a by Takeuchi and co-workers82 revealed that it is derived from two units of acetate, one unit of propionate and one molecule of proline arranged as shown in Scheme 9. A more detailed picture of the formation of the benzopyranopyrrole has been proposed by Simpson et al.,99 involving a cyclization of a simple tetraketide intermediate, which is generated from the coenzyme A thioester of pyrrole-2-carboxylate (103) as primer, followed by a rearrangement process as shown in Scheme 10. Thus, intramolecular migration of the carbonyl group from the 2-/3-position to the 3-/2-position of the pyrrole ring could take place via the key spiro-intermediate 106, with a subsequent loss of the hydroxy group and migration of the carbonyl bond to give the pyrrolinium intermediate 107. Aromatization of 107 would then lead to the core structure of pyralomicin 1a.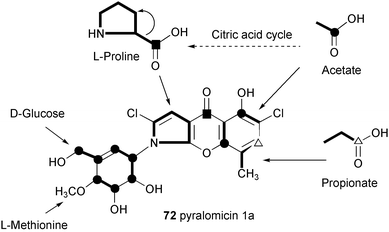 | ||
| Scheme 9 Metabolic origin of pyralomicin 1a. | ||
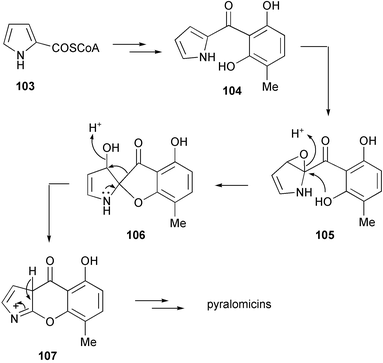 | ||
| Scheme 10 A hypothetical mode of formation of the benzopyranopyrrole moiety of the pyralomicins.99 | ||
In contrast to that of validamycin, acarbose, and other related compounds, the cyclitol structure in pyralomicin 1a was shown to be 1-epi-valienamine. Initial incorporation experiments with D-[U-13C6]glucose on pyralomicin 1a (72) suggested that it is also derived from the pentose phosphate pathway.82 However, the opposite stereochemistry at C-1′ of 72 suggests an essential biosynthetic divergence between the cyclitol in 72 and that in acarbose and validamycin, which could take place either during the formation of the cyclitol or during the condensation of the cyclitol and the core benzopyranopyrrole unit.
Very recently, Naganawa et al.85 reported the incorporation of stable-isotope labeled 2-epi-5-epi-valiolone (87) and 5-epi-valiolone (88) into pyralomicin 1a with incorporation rates of 23% and 10%, respectively, while none of the other potential intermediates, such as valiolone (86), valienone (89), 1-epi-valienol (91), valienol (108), and 5-epi-valiolol (109) were incorporated.85 To explain this surprising observation, it was proposed that 2-epi-5-epi-valiolone (87) is specifically activated (e.g., to its phosphate) and that the further transformations take place on activated intermediates (which can not be generated directly from their unactivated counterparts). A similar proposal was made recently by Piepersberg and co-workers for acarbose biosynthesis.92 While Piepersberg proposed that the phosphorylation might take place twice during the conversion of 2-epi-5-epi-valiolone (87) into the activated nucleotidyl diphospho derivative (99), Naganawa et al. suggested the involvement of an enzyme similar to phosphoglucomutase that would transfer the phosphate from C-7 to C-1 of valienol, setting the stage for the activation of the cyclitol to a nucleotidyl diphospho-valienol (NDP-valienol, 112) (Scheme 11). Alternatively, it was also suggested that the transformation of 2-epi-5-epi-valiolone (87) into 72 could involve a substrate-channeling mechanism in which enzyme-bound intermediates are directly transferred from one enzyme active site to the next in a multi-enzyme complex.85
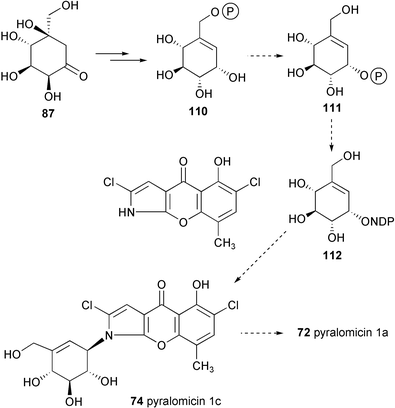 | ||
| Scheme 11 Biosynthetic pathway to pyralomicin 1a proposed by Naganawa et al.85 | ||
4 Biological properties of the C7N aminocyclitols
4.1 α-Glucosidase inhibitory activities and their related biochemistry
Many members of the C7N aminocyclitol family, e.g., acarbose, adiposins, amylostatins, oligostatins, trestatins, NS, W-46, CKD-4416, are known to display potent α-glucosidase inhibitory effects. Among these active metabolites, acarbose (26) in many respects is the best characterized and has been found to have a very pronounced inhibitory effect on a number of α-glycosidases of bacterial,3 fungal,3 and mammalian origin, including human intestinal α-glucosidases such as sucrase, maltase and glucoamylase.100,101 This strong inhibition is widely attributed to the enhanced binding of the core aminocyclitol valienamine, whose half-chair conformation mimics the substrate distortion expected in the oxocarbonium ion transition-state.102 In addition, the adjacent N-linked glycosidic bond prevents enzymatic hydrolysis. This amino linkage forms a salt link to the “acidic group” of the glycosidases, which probably contributes significantly to the unusually tight binding of the inhibitor with the enzymes.103 Its potent competitive inhibitory activity against human intestinal α-glucosidases has resulted in its use as a clinical drug for the treatment of patients with type II insulin-independent diabetes mellitus.104 Acarbose lowers postprandial serum glucose and effectively reduces postprandial hyperglycemia. Since it does not stimulate endogenous insulin secretion, it will not cause hypoglycemia when used as monotherapy.33Besides its pharmacological and dietary uses, acarbose is also of considerable enzymological interest. In addition to the earlier investigations on the mechanism of inhibition of sucrase by acarbose,105 more thorough studies on its inhibition mechanism and binding properties toward sugar hydrolyzing enzymes of various origin have been carried out rather recently at the molecular level.106–130
Interactions and binding properties of acarbose and glucoamylases of Aspergillus niger were studied using perturbation difference spectroscopy,106 stopped-flow fluorescence spectroscopy and steady-state kinetic measurements,107 NMR spectroscopy,108 and Displacement Titration Calorimetry.109 In addition, three-dimensional structures of the pseudotetrasaccharide acarbose (26) and D-gluco-dihydroacarbose (33) complexed with glucoamylase II from Aspergillus awamori var X100 have been determined to 1.7 and 2.0 Å resolution, respectively.103,110,111 Electron density for a single molecule of bound acarbose or D-gluco-dihydroacarbose defines what may be the first four subsites in the binding of extended maltooligosaccharides. It was reported that acarbose binds to the enzyme (Ki of ∼10−12 M) more tightly then D-gluco-dihydroacarbose (Ki of 10−8 M). The traditional hypothesis reasoned that this large discrepancy in affinity results from the half-chair conformation of the valienamine moiety in acarbose, constrained by a double bond between C-5 and C-6 (corresponding to the endocyclic O-5 of glucose), mimicking the putative glucopyranosyl cation intermediate at the active site for glycolysis. D-gluco-dihydroacarbose (33) lacks the double bond and hence adopts a chair conformation. In spite of this notion, both acarbose and D-gluco-dihydroacarbose have nearly identical interactions at subsites −1 and +1 (residues A and B).110,111 The two inhibitors bind in a very similar manner and each exhibits a dual binding mode with respect to the location of the last sugar residues. Aleshin et al.111 proposed that the perturbation of the catalytic water (Wat 500) from its site of optimal hydrogen bonding, caused by the shift of C-6 of the aminocyclitol residue in 33 and amplified by the addition of a hydrogen atom to it, is the most apparent cause for the reduced affinity of 33.
Structural studies and kinetics of inhibition of porcine pancreatic α-amylase isoforms (PPA I and PPA II),112–116 human pancreatic α-amylase isoforms (HPA I and HPA II),117,118 fungal α-amylase (TAKA-amylase),119 and bacterial α-amylases120–122 by acarbose and its analogs have also been reported. In all studies, acarbose was found bound to the active site with the core acarviosine moiety in the catalytic center −1 and +1 subsites. Interestingly, the observed electron densities of crystal structures of enzyme–inhibitor complex from various origins consistently suggest the presence of transglycosylated species of acarbose (Scheme 12), presumably exhibiting stronger binding to the enzymes than the original pseudotetrasaccharide. Transglycosylation is possible when O-glycosidic bonds in the initial substrate are susceptible to the first nucleophilic attack by the enzyme, giving an electrophilic intermediate in the active center, which is then transferred to the non-reducing end of another substrate molecule (Scheme 13).
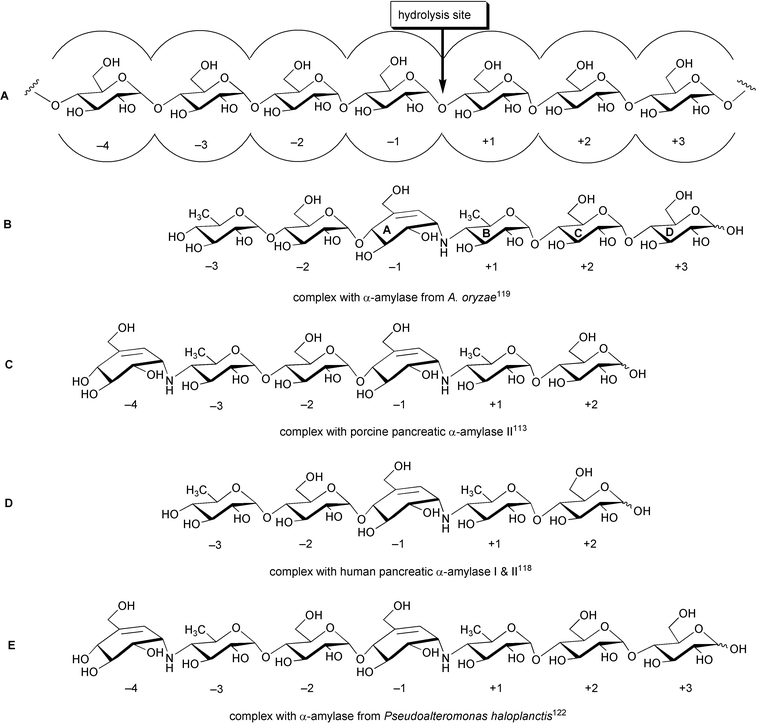 | ||
| Scheme 12 Transglycosylated species of acarbose observed in the bound complexes with α-amylases of various origins. A: An illustration of α-amylase-substrate interaction with cleavage taking place between subsites −1 and +1 (nomenclature according to Davis et al.);119 B: hexapseudosaccharide bound to α-amylase of A. oryzae; C: hexapseudosaccharide bound to porcine pancreatic α-amylase II; D: pentapseudosaccharide bound to human pancreatic α-amylases I & II; E: heptapseudosaccharide bound to α-amylase of P. haloplanctis. | ||
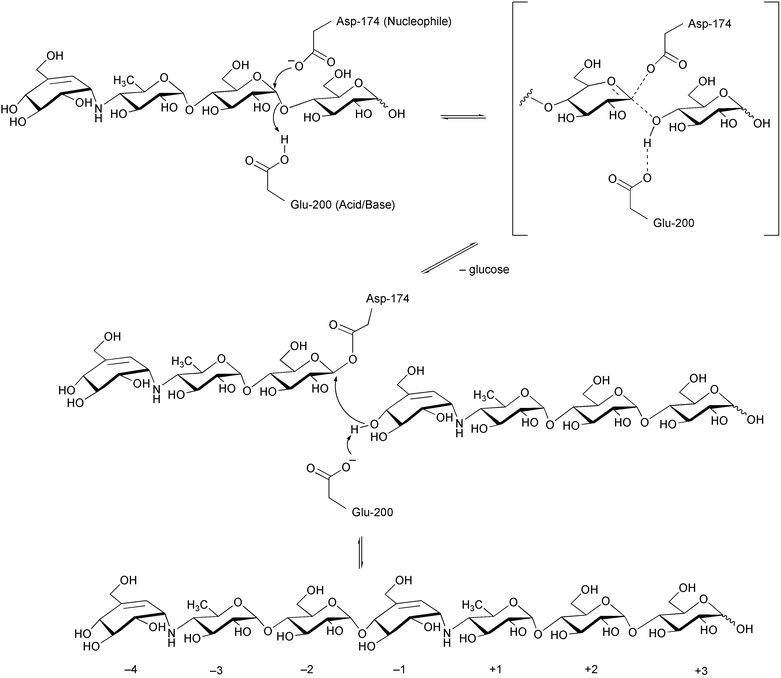 | ||
| Scheme 13 Transglycosylation mechanism at the catalytic site of α-amylase proposed by Haser et al.122 | ||
An interesting feature has been observed on maltogenic amylase of Bacillus stearothermophilus ET1 which hydrolyzes and transglycosylates acarbose into isoacarbose. Park et al.120,123 reported that a recombinant maltogenic amylase of B. stearothermophilus ET1 hydrolyzed acarbose to glucose and a pseudotrisaccharide. When the enzymatic reaction was carried out in the presence of 10% glucose, the pseudotrisaccharide molecule was then transferred to C-6 of the glucose by the same enzyme via formation of an α-1,6-glucosidic linkage to give isoacarbose.123 More interestingly, addition of a number of different carbohydrate acceptors to the incubation mixture gave the corresponding transglycosylation products, where the best acceptor was gentiobiose, followed by maltose and cellobiose.124 A similar phenomenon was also reported for a recombinant thermostable maltogenic amylase (ThMA) from a Gram negative thermophilic bacterium, Thermus strain IM6501.125 ThMA was able to transfer the acarbose-derived pseudotrisaccharide to at least 17 sugar acceptors, with transfer occurring primarily to the C-6 position of the acceptors and to a lesser extent to the C-3 and/or C-4 positions. Kinetic analysis of the acarbose transfer showed that the C-4 product formed most rapidly but was readily hydrolyzed, while the C-6 transfer product was stable and accumulated in the reaction mixture as the main product. The X-ray structure of B. stearothermophilus maltogenic α-amylase, Novamyl, complexed with acarbose and maltose showed the presence of an extended hexasaccharide species, bound in the −3 to +3 subsites.126 The valienamine moiety is bound in the −1 subsite of the enzyme in a 2C3 half-chair conformation, as expected.
The crystal structure of B. circulans cyclodextrin glycosyltransferase (CGTase) complexed with acarbose also reveals a binding mode consistent with the transition state analog character of this inhibitor.127 However, it appears that the CGTase, after hydrolyzing acarbose to glucose and a pseudotrisaccharide, couples a molecule of glucose to the non-reducing end of the pseudotrisaccharide.
The transglycosylation event was not always observed in crystal structures of sugar hydrolases complexed with acarbose. For example, in the complexes with glucoamylases,103,109–111 glycogen phosphorylase,128B. licheniformis maltogenic amylase,123E. coli maltodextrin phosphorylase (MalP),129 and amylomaltase from Thermus aquaticus,130 the unmodified molecule of acarbose seems to be bound at the catalytic site of the enzymes. In MalP- and amylomaltase-acarbose complexes, however, the acarviosine moiety apparently does not bind in the appropriate catalytic subsites (subsites −1 and +1), which suggests that acarbose is probably not a transition state analog for those enzyme-catalyzed reactions.129,130
Similar to acarbose, adiposin-1 (44) and −2 (45) exhibited potent inhibitory activity against disaccharidases, such as sucrase, maltase and isomaltase, against glucoamylase and against α-amylases, such as BSA, pancreatic α-amylase and saliva α-amylase.131 Bacterial liquefying α-amylase (BLA) and Taka-amylase were weakly inhibited, whereas α- and β-glucosidases were not affected. Further pharmacological studies reported by Õmura and co-workers revealed that adiposins suppressed increases of the blood sugar level and the secretion of insulin in mice and rats.131 Hematological and histopathological examinations of animals treated with adiposins did not reveal any remarkable change after a 3 months toxicity test. Adiposins apparently also did not show any acute toxicity in mice at dose levels (p.o.) of less than 10 g per kg of body weight.131 Adiposin-1 and −2 were found to be non-competitive inhibitors of glucoamylase, whereas acarbose and its derivatives were found to be competitive inhibitors of α-glucosidase, and mixed non-competitive inhibitors for α-amylase and cyclomaltodextrin glucanosyltransferase.120
The effect of the number of glucose residues and the position of the pseudoaminosugar residue on the inhibitory activity against glycosidases has been studied using amylostatins (XG–GXGGG).42 The results demonstrated that endo-type α-amylases, such as bacterial saccharifying α-amylase (BSA) and TAKA amylase A (TAA), were more strongly inhibited by GXG (38), GXGG (39) or GXGGG (40) than by XG (35), XGG (36) or XGGG (37); however, several pancreatic α-amylases were also inhibited strongly by XGGG (37). This observation could be due to the events taking place in the complex of α-amylase and the inhibitor where one or more residues is added to the non-reducing end of XGGG as a result of transglycosylation activity, yielding a species with stronger binding activity to α-amylase. On the other hand, glucoamylase, which is an exo-type amylase, was strongly inhibited by XGG (36) and XGGG (37), but only weakly by GXG (38), GXGG (39), and GXGGG (40). The attachment of glucose at the non-reducing end of the aminocyclitol evidently is disadvantageous for the activity against glucoamylase. A similar phenomenon has also been observed for the oligostatins.51
Despite lacking the half-chair conformation in their core structures, oligostatins (48–50) showed strong inhibitory activity against fungal glucoamylase, porcine pancreatic α-amylase, human saliva α-amylase and meilase α-amylase. They were less effective against α-amylases and invertase of microbial and fungal origin. Their inhibitory activity against α-amylase varies based on the length of the pseudosaccharides.51
Valienamine (1), the core unit of most α-glucosidase inhibitors, showed moderate inhibitory effects on various glucoside hydrolases, such as α-glucosidase, α-glucoamylase, sucrase, maltase, isomaltase, and trehalase, but weak or negligible effects on β-glucosidase, invertase, lactase and on α- and β-amylases.132,133 Its inhibitory activities against porcine sucrase and maltase were reportedly to be improved by N-alkyl or N-aralkyl derivatizations.134N-Acyl-valienamines, on the other hand, lack inhibitory activity, presumably because of the lower proton accepting ability of the N atom in these analogs, which in turn decreases the interaction of the amino linkage with the acidic group of the enzyme.134 Interestingly, valiolamine (20),25 the hydrated analog of valienamine that more likely adopts a chair conformation, as well as its N-alkyl and N-aralkyl derivatives135 showed more potent inhibitory effects on glucoside hydrolases than valienamine. This result raises a question as to the extent to which the half-chair conformation of the valienamine ring is really critical for the efficacy of the inhibitors.
4.2 Anti-fungal and insecticidal activities
A number of C7N aminocyclitol-containing compounds have been reported as potential anti-fungal agents. Validamycins, salbostatin and suidatrestin are among those classified in this category. Despite their close structural similarity to acarbose and its derivatives, validamycins, salbostatin and suidatrestin surprisingly do not show any inhibitory activity against α-amylases, glucoamylases or sucrase. The validamycins have been known as mechanistically unique anti-fungal agents, which are neither fungicidal nor fungistatic, but are able to control the spread of the pathogen by inhibiting specifically the hyphal extension without affecting the organism's specific growth rate.136,137 Therefore, it had been of great interest to investigate the mode of action for their anti-fungal activity.Validamycin A (3), the major component of the validamycin complex, is the most active compound of the complex and is widely used in the Far East and elsewhere to control sheath blight disease of rice plants caused by the fungus Rhizoctonia solani. At a concentration of 30 ppm, it was as effective as a commercial organoarsenical (Monkit dust), with no injury to the rice plant.4
Despite their strong activity against the sheath blight, the validamycins showed no antimicrobial activity against bacteria, yeast, or fungi, including P. sasakii and R. solani, in conventional in vitro tests.4 However, under certain conditions the hyphae of P. sasakii and R. solani seem to exhibit an abnormal branching at the tips and their further development appears to be repressed by the drug.5 The hyphal inhibition was more pronounced in the main hyphae than in the primary and secondary branches of R. solani.136 This inhibition was antagonized by the hyphal extension factor (HE-factor) isolated from the hyphal extract of R. solani.138 It was proposed that the formation of HE-factor in the validamycin-treated culture is inhibited by validamycin.
Extensive further studies on the mechanism of action of validamycin in controlling the hyphal extension and altering the morphology of fungi have been carried out by several groups.139–143 Shibata and co-workers reported the effects of validamycin on the production of some enzymes in R. solani, which are most likely related to the metabolism of the cell wall glycan, such as laminarinase, glucan synthetase, and β-D-glucan-degrading enzymes (β-D-glucosidase and β-1,3-glucanase).140,142,144 While validamycin had no direct effect on the activity of these enzymes, extracellular production of laminarinase and β-D-glucosidase was increased in the presence of validamycin, and the intracellular production of laminarinase, β-D-glucosidase and β-1,3-glucanase remarkably decreases. On the other hand, the intracellular activity of glucan synthetase significantly increased under the same conditions. It was proposed that the decrease in the production of the β-D-glucan-degrading enzymes and the increase of glucan synthetase activity in the mycelium may stimulate the synthesis of cell wall polymers, which in turn may cause morphological change. In addition, validamycin A (3) was also found to reduce the concentration of inositol, which is known to be associated with the cell wall, in fermentation cultures of R. solani137 and R. cerealis145 and to affect the component sugars and linkage of the cell wall glycan of the fungus.141 No effect of the validamycins has been found on the production of glucose 6-phosphate dehydrogenase, 6-phosphogluconic acid dehydrogenase, glucose phosphate isomerase, glucotransferase or glucoamylase.140
The most plausible explanation for the unique anti-fungal activity of the validamycins is probably that proposed by Matsui and co-workers.146 They found that the validamycins strongly inhibit trehalase, the trehalose-degrading enzyme, of R. solani. Trehalose (α-D-glucopyranosyl α-D-glucopyranoside) is known as a storage carbohydrate in fungi (8 to 10% of the dry cell weight of R. solani) and is recognized as a characteristic blood sugar of insects. The degrading enzyme, trehalase, appears to play a primary metabolic role in these organisms in generating glucose for energy supply or for further metabolic uses. Its inhibition might disrupt the glucose supply system of the fungus, which in turn hampers hyphal extension, altering the morphology of the fungus. When injected into young last instar larvae of the tobacco cutworm, Spodoptera litura, the validamycins evoked morphological abnormality followed by death with a 100% mortality rate.147 Matsuura and co-workers148 also suggested that the anti-fungal activity of validamycin A resulted from its anti-trehalase activity. This was based on their observation that the inhibition of growth of the mycelia of R. solani by validamycin A (3) occurred when trehalose was used as the sole carbon source in the medium, but not when the fungus was incubated in a medium containing various sugars and carbohydrate polymers. It was also found that the trehalose content increased in the mycelia treated with validamycin A.148
In vitro experiments with R. solani showed that the pseudodisaccharide, validoxylamine A (11), is the most potent inhibitor of trehalase among the validamycins.149 Attachment of a D-glucosyl residue to validoxylamine A, e.g., validamycins A (3), D (6) and G (9), as well as other synthetically prepared mono-β-D-glucosides of validoxylamine A, evidently reduced the activity against trehalase,150 whereas the pseudotetrasaccharides, such as validamycins C (5), E (7) and F (8), lost their inhibitory effect on trehalase entirely.149 Interestingly, in vivo experiments revealed that validamycin A (3), not validoxylamine A (11), is the most potent anti-fungal agent among the validamycins and the validoxylamines.149 This can be explained by a subsequent finding that validamycin A is more efficiently transported into the mycelia where it is hydrolyzed by a β-glucosidase yielding validoxylamine A with greater inhibitory activities.149,150
Salleh and Honek151 reported the time-dependent inhibition of porcine kidney trehalase by the validamycins. Without preincubation, validoxylamine A and validamycin A are competitive inhibitors against pig trehalase. However, in preincubation experiments, both compounds showed non-competitive inhibition, suggesting that the interaction is a slow binding process. Matsui and co-workers suggested that the active center of pig kidney trehalase consists of two subsites, one for recognition and one for catalysis.152 The extremely high affinity of validoxylamine A towards this enzyme may be due to the synergistic interactions of two cyclitol units in validoxylamine A with the two subsites. It was also found that validoxylamine A, which presumably mimics the reaction transition state, exhibits higher binding affinity than does D-gluco-dihydrovalidoxylamine A, a mimic of trehalose, the natural substrate.152
Salbostatin (71) and suidatrestin have reportedly shown similar inhibitory features as validoxylamine A (11) with respect to trehalase.65,66 Compared to trehalose as the substrate, suidatrestin binds about 500 000 times more strongly to the enzyme.66 This strong in vitro activity, however, is not reflected in its in vivo effects in larvae of the moth S. littoralis. In contrast to validoxylamine A, which has significant insecticidal activity, suidatrestin has almost no effect on larvae growth.66 The morphological abnormalities induced by validoxylamine A in the cabbage armyworm, Mamestra brassicae,147 and in Spodoptera litura133,153 were not observed in S. littoralis after injection of suidatrestin. Validoxylamine A (11) has also been shown to raise trehalose levels in Bombyx mori,154M. brassicae155 and the American cockroach, Periplaneta americana,156 due to inhibition of trehalase. In the latter species it also caused oocyte retardation156 and inhibition of flight.157 Although the exact reasons for the above discrepancy are not clearly understood, it was proposed that they could be due to a rapid metabolic inactivation event in the organism or due to membrane transport problems of suidatrestin to the target enzyme.66
4.3 Antibacterial activities
Although validamycin and acarbose have been shown to have no significant antibacterial activity, some of their closest relatives have evidently demonstrated notable effects on the growth of various bacteria. Adiposin, which is structurally very close to acarbose, having a glucose moiety (instead of deoxyglucose in acarbose) adjacent to the core unit valienamine, exhibits antibacterial activity against some Gram positive bacteria, e.g., Bacillus licheniformis, Staphylococcus aureus, Gram negative bacteria belonging to the Enterobacteriaceae, some anaerobic bacteria, and phytopathogenic fungi.49 Oligostatins C (48), D (49), and E (50), another structurally related group, were active against Gram negative bacteria, e.g., E. coli, Shigella sonnei, Proteus vulgaris, Salmonella typhi, and Klebsiella pneumoniae, while Gram positive bacteria such as Bacillus subtilis, Micrococcus luteus, and Staphylococcus aureus were not affected.51 The bacterial growth inhibitions of the adiposins and the oligostatins were generally enhanced by the addition of maltooligosaccharides such as maltose and maltotriose, and were inhibited by most monosaccharides and trehalose, which suggests the possibility of a close correlation between their sugar hydrolase inhibitory activity and their antimicrobial activity.In contrast to the adiposins and the oligostatins, the epoxidal aminocyclitols CKD-711 (68) and CKD-711a (69) showed no antibacterial activity against the Gram negative and Gram positive bacteria tested, including E. coli and Bacillus subtilis, fungi or yeast. However, quite interestingly, they showed a highly selective antibacterial activity against Comamonas terrigena ATCC 8461.64 Reasons for this extraordinary observation are unknown.
The benzopyranopyrrole antibiotics, the pyralomicins, are reportedly active against various bacteria, particularly strains of Micrococcus luteus.67 Their antibacterial activity appears to be dependent upon the number and the position of chlorine atoms within the molecules, and whether or not the glycon is methylated. It is also noteworthy that pyralomicin 1c (74), having 1-epi-valienamine as its glycon, exhibited more potent antibacterial activity than its glucosyl analog pyralomicin 2c (78).67
Another new type of C7N aminocyclitols, the epoxyquinomicins A (79) and B (80), exhibited moderate antimicrobial activities against Gram positive bacteria and several strains of Pasteurella piscicida.72,73 However, their C-1 hydroxy analogs epoxyquinomicins C (81) and D (82) showed almost no effect on bacterial growth.73
4.4 Other biological activities
Even though traditionally the C7N aminocyclitols, represented by acarbose and validamycin, have been known as a group of pseudoaminooligosaccharides of bacterial origin that exhibit potent sugar hydrolase inhibitory activities, more recent discoveries indicate a broader scope to their structural diversity and biological activities. For example, the epoxyquinomicins, a new type of C7N aminocyclitol having no sugar moiety in their structures, have been found to be potential drug candidates for rheumatoid arthritis.158 They exhibited potent inhibitory effects on type II collagen-induced arthritis in mice with a mode of action different from that of common non-steroidal anti-inflammatory drugs (NSAIDs). They have neither an anti-inhibitory effect on carrageenan-induced paw edema in rats nor an analgesic effect on acetic acid-induced writhing in mice. It was proposed that the anti-arthritic effect of epoxyquinomicins in vivo might be partly due to the inhibition of histidine decarboxylase, an enzyme that is involved in the biosynthesis of histamine.159Cetoniacytone A (83), the analog of epoxyquinomicins isolated recently from an endosymbiotic Actinomyces sp. living in the intestines of an insect, Cetonia aureata, has been shown to have a significant growth inhibitory effect against a number of human cancer cell lines, e.g., hepatocellular carcinoma (HEP G2) and breast adenocarcinoma (MCF 7).75
In addition, synthetically modified C7N aminocyclitols have also shown various biological activities such as antiproliferative activity against smooth muscle cells,160 anti-inflammation,161 immunomodulation,162 as well as potential for anti-cancer and HIV.163
5 Synthesis and development of the C7N aminocyclitols
5.1 Syntheses of the core C7N units
Valienamine, the core unit of many C7N aminocyclitol-containing compounds, has been synthesized in both racemic164–166 and enantiomerically pure167–180 forms. In fact, during the past twenty years more than a dozen enantiospecific syntheses have been described for valienamine alone. The first enantiospecific synthesis was reported by Paulsen and Heiker in 1980,167 using quebrachitol (2-O-methyl-L-chiroinositol) (113) as the chiral starting material (Scheme 14). Six syntheses used D-glucose in constructing the carbocyclic framework, involving either a Ferrier rearrangement,170,172,175 an aldol cyclization of a nitrofuranose,171 an intramolecular Horner–Emmon alkenylation173 or a ring-closing alkene metathesis (RCM).177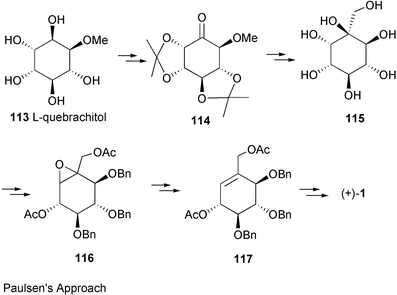 | ||
| Scheme 14 The first enantiospecific synthesis of (+)-valienamine reported by Paulsen et al.163 | ||
Schmidt and Köhn170 first reported the synthesis of valienamine from methyl-α-D-glucopyranoside via the Ferrier rearrangement product 118 (Scheme 15). Imination of 119 with chloramine T, in a two-phase process with benzyltriethylammonium chloride in dichloromethane, induces a [2,3]-sigmatropic shift of a sulfimide group to afford a diastereospecifically valienamine derivative 120 in good yield (Scheme 15). Nicotra et al.172 reported the synthesis of (+)-valienamine from enone 121via a regio- and stereoselective nucleophilic addition reaction. Compound 121 was prepared by a Ferrier cyclization from methyl 4,6-O-benzylidene-2,3-di-O-benzyl-α-D-glucopyranoside. Park and Danishefsky175 elegantly applied an intramolecular allylic displacement of the spiro-epoxide 124, derived from a Ferrier rearrangement product of readily available TIPS-glucal 123, to generate (+)-valienamine. Kitagawa and co-workers171 used the pseudo-nitro-sugar 126 to synthesize (+)-valienamine. Compound 126 was derived from D-glucose via an aldol cyclization. Fukase and Horii173 synthesized (+)-valienamine from the branched unsaturated inosose derivative 130, which was obtained by an intramolecular Horner–Emmons reaction starting from tetra-O-benzyl-D-glucono-1,5-lactone 129, which is readily available from tetra-O-benzyl-D-glucose. The amino group was introduced by taking advantage of a Mitsunobu's reaction employing phthalamide as the nitrogen source. Vasella et al.177 employed a ring-closing alkene metathesis of diene 134, generated from the ketone 133, in the presence of Grubb's catalyst 135 to give the cyclohexane 136 (Scheme 16), which was converted into (+)-valienamine in three steps in 47% yield.
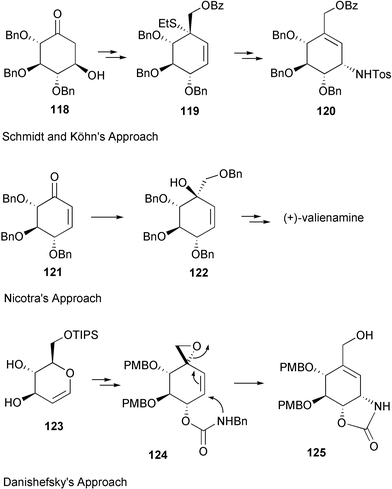 | ||
| Scheme 15 Syntheses of (+)-valienamine applying Ferrier rearrangement. | ||
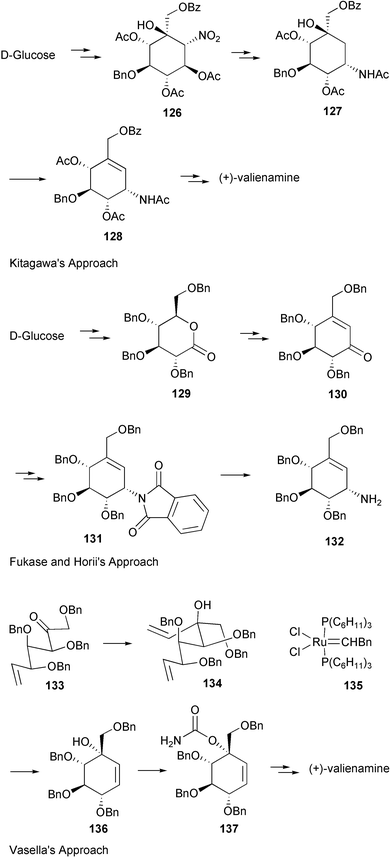 | ||
| Scheme 16 Syntheses of (+)-valienamine from D-glucose. | ||
Another synthesis of optically active valienamine was reported by Tatsuta and co-workers,179 who employed D-xylose as the chiral starting material using an aldol condensation of a sulfone derivative as the key step, which involved a one-step opening of a furanose ring containing a phenylsulfonylmethyl group (139→140) (Scheme 17).
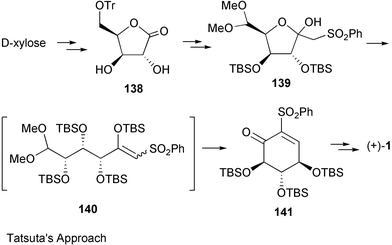 | ||
| Scheme 17 Synthesis of (+)-valienamine from D-xylose. | ||
Furthermore, three syntheses took advantage of the Diels–Alder reaction to generate the cyclohexane skeleton (Scheme 18). Ogawa et al.169 used optically active 143, derived from (–)-142, the Diels–Alder adduct of furan with acrylic acid, to synthesize (+)-valienamine, which was isolated as its pentaacetyl derivative. The approach had been applied earlier in various syntheses of racemic valienamine and its analogs.164–166 Knapp et al.174 employed an intramolecular amino delivery via the carbonimidothioate iodocyclization reaction (149→150), on material obtained from the Diels–Alder adduct 148. Oxidation of 150 to the corresponding iodoso intermediate 151 resulted in spontaneous syn elimination of HOI to give the unsaturated oxazolidinone 152. Trost and co-workers176 studied a new protocol using as co-catalyst bidentate phosphite in a palladium-based cis-hydroxyamination. With this approach, racemic valienamine was obtained in 7 steps from the Diels–Alder adduct of 153 and 154 in 10–15% overall yield. However, the asymmetric synthesis, which started from benzoate 156, required 14 steps with only 1–2% overall yield.
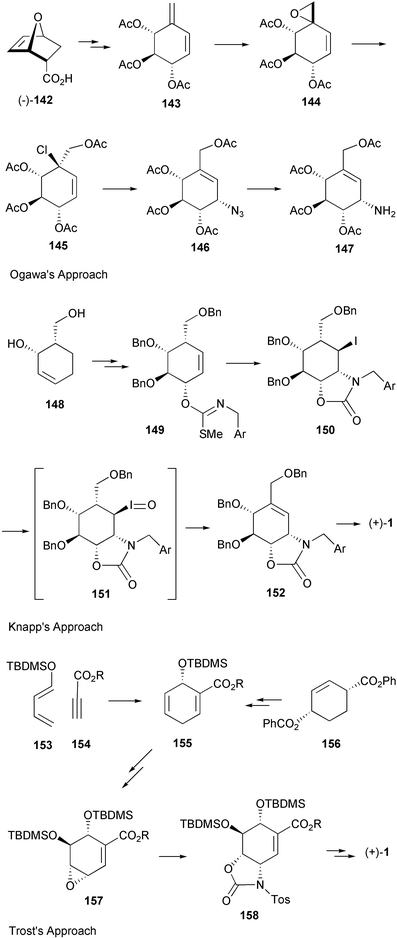 | ||
| Scheme 18 Syntheses of (+)-valienamine using the Diels–Alder reaction. | ||
Finally, two closely related syntheses were reported by Shing et al.178,180 using (–)-quinic acid (159) as a starter unit (Scheme 19). The first was based on a regio- and stereospecific ring opening of the cyclic sulfite 160 as the key step, while in the second synthesis they demonstrated the high efficiency of a palladium-catalyzed coupling strategy for the introduction of the amino group (162→163). The latter route gave pentaacetylvalienamine in 20 steps from (–)-quinic acid with overall yield of 11%.180
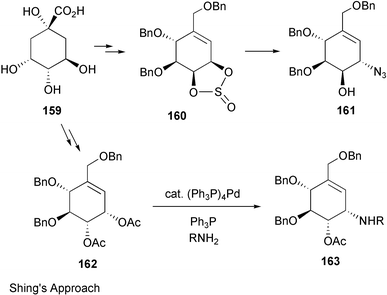 | ||
| Scheme 19 Syntheses of (+)-valienamine from (−)-quinic acid. | ||
Among the approaches described above, the two most efficient routes appear to be the ones reported by Vasella,177 who obtained valienamine in seven steps (overall yield 17%), and by Fukase and Horii,173 who obtained valienamine in nine steps (overall yield 12%), both starting from commercially available 2,3,4,6-tetra-O-benzyl-D-glucopyranose.
Syntheses of the valienamine analogs 2-aminovalienamine (164),181 2-deoxyvalienamine (165),182,183 7-nor-valienamine (166)174 and 2-epi-valienamine (167)178 have also been reported.
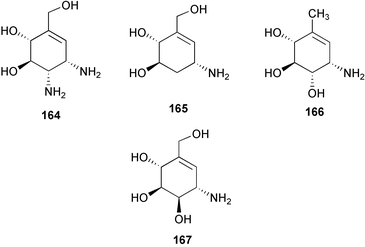
Like valienamine, validamine (17), the other component of validoxylamine A (11), has also been synthesized in both racemic and enantiomerically pure forms. The first synthesis of DL-validamine was reported by Ogawa et al. in the mid 1970s using the Diels–Alder adduct (±)-endo-7-oxabicyclo[2.2.1]hept-5-ene-2-carboxylic acid [(±)-142] as starting material.184–187 With the same approach, (+)-validamine was later prepared from chiral (–)-142 after a successful optical resolution of the racemic acid (±)-142 using resolving agent (R)-(+)- and (S)-(–)-α-methylbenzylamine.188 Similarly, hydroxyvalidamine (18),189 1-epi-validamine (168),190 2-epi-validamine (169),191 2-amino-2-deoxyvalidamine having α- and β-gluco-, and α- and β-manno configurations (171–174),192 all in racemic form, have also been synthesized in the same laboratory. Preparations of racemic validamine (17), as well as epimers 168, 169 and 170 were carried out by Aceña et al. via a stereocontrolled nucleophilic epoxidation of polyhydroxylated cyclohexenyl sulfones obtained from (phenylsulfonyl)-7-oxabicyclo[2.2.1]heptanes (Scheme 20).193 In addition, a diastereoselective synthesis of (±) 1-epi-3-epi-4-epi-validamine (179) was carried out from N-tert-butoxycarbonyl-2-[(tert-butyldimethylsilyl)oxy]pyrrole (177) and 2,3-O-isopropylidene-D-glyceraldehyde (178) via a sort of [3+3] cycloaddition involving sequential vinylogous cross aldolization–intramolecular aldolization.194 More recently, an efficient synthesis of (±)-2-epi-validamine was reported, based on a Diels–Alder cycloaddition of ethyl coumalate (180) and vinylene carbonate (181), in which the product was obtained in five steps and in 60% of overall yield.195,196
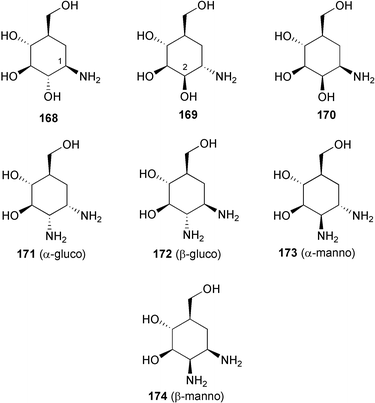
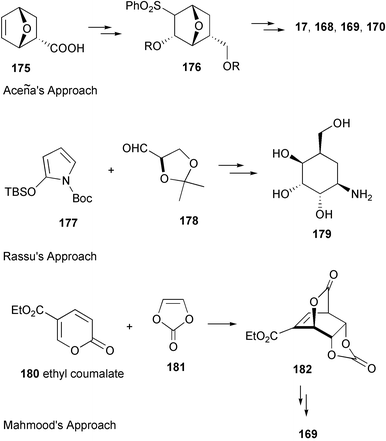 | ||
| Scheme 20 Syntheses of validamine analogs. | ||
Five enantiospecific syntheses of validamine and its epimers have been reported, including that of Ogawa, who used (–)-142 as the starting material as previously described.188 Kitagawa et al.171 employed D-glucose-derived nitrofuranose derivatives 183 and 185, which were prepared via a Michael-type addition reaction, to synthesize optically active validamine (17) and 5-epi-validamine (187) (Scheme 21). An improved route to prepare the 1-α-acetamido derivative 184 was also presented.197 A new synthetic approach to 2-epi-validamine (169) by an asymmetric Diels–Alder reaction of (S)s-3-(2-pyridylsulfinyl)acrylate 188 with furan 189 has also been reported.198 Finally, as described for the synthesis of (+)-valienamine, the groups of Shing199 and Tatsuta179 once again utilized (–)-quinic acid and D-xylose, respectively, as chiral precursors to construct optically active validamine.
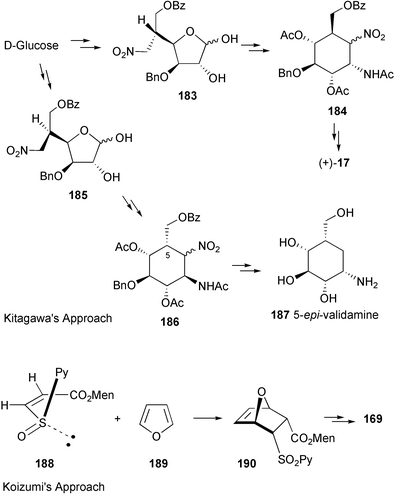 | ||
| Scheme 21 Two examples of enantiospecific syntheses of validamine and its analogs. | ||
Semi-synthetic preparations of 2-epi- and 4-epi-validamines by configurational inversion of validamine, which is prepared by the microbial degradation of validamycins, have also been reported.200 However, excessive protection and deprotection steps made this synthesis unexpectedly long.
In contrast to those of valienamine and validamine, the synthesis of valiolamine (20), the core component of validoxylamine G (13) and validamycin G (9) which is also found in fermentation cultures of S. hygroscopicus var limoneus, has been reported almost exclusively by Fukase and Horii and their collaborators.173,201–203 Optically active pure valiolamine has been synthesized in three different ways: (1) via stereoselective conversion of valienamine (1) and validamine (17),201 (2) from D-glucose via valiolone (86) involving a stereospecific intramolecular aldol cyclization,173,202 (3) from sedo-heptulose via a biomimetic approach resembling its probable biosynthesis and taking advantage of the Ferrier's intramolecular carbocyclic ring closure reaction.203 The stereoselective conversions of valienamine and validamine into valiolamine take place via transient halo cyclic carbamate intermediates 191 and 195, respectively (Scheme 22). Compound 195 was derived from an exo-methylene derivative 194. A direct dihydroxylation of 194 with osmium tetroxide gave 5-epi-valiolamine (193), which was also synthesized from valienamine via a bromo cyclic carbonate intermediate 192, followed by reductive debromination and hydrolysis.201
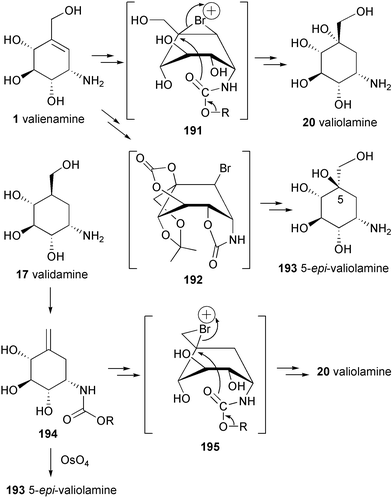 | ||
| Scheme 22 Stereoselective conversions of valienamine and validamine into valiolamine and 5-epi-valiolamine. | ||
Valienamine epoxide, the core unit of the NS complex and related compounds, was synthesized via epoxidation of N-acetyl-valienamine 196 with MCPBA in acetic acid or, in a lesser yield, the epoxide function can also be introduced via the bromohydrin 197 by treatment with potassium carbonate in methanol to give 198.204
5.2 Total syntheses of the C7N aminocyclitol-containing natural products
Validoxylamine A, the core structure of validamycins A (3), C (5), D (6), E (7), F (8) and H (10), was first synthesized in racemic form from the protected DL-validamine 199 and epoxide 200.205 Later, an enantiomerically pure form was also synthesized by selective deoxygenation of the (+)-validoxylamine B derivative 202 obtained from a coupling of the partially protected (+)-valienamine (132) and the epoxide 201.206 Deoxygenation of 202 takes place through formation of the aziridine 203, which undergoes nucleophilic displacement with toluenethiol, followed by reductive removal of the thiol with Raney nickel and deprotection to give (+)-validoxylamine A (11). Glycosylation of the protected validoxylamine A 204 with either 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl chloride (205) or 1,6-di-O-acetyl-2,3,4-tri-O-benzyl-D-glucopyranose (206) or other halides obtained from disaccharides D-mannose, D-cellobiose and D-lactose followed by deprotection, yielded validamycins A, E and their analogues.207–209 An appropriate deprotection of 202 followed by glycosylation with 205 and complete deprotection gives validamycin B.210 Alternatively, validamycin B can also be obtained through coupling of the glycosylated epoxide 207 and the partially protected valienamine derivative 132, followed by deprotection.211 Validamycins C, D, F and H have also been synthesized, mainly from appropriately protected derivatives of validoxylamine A, followed by glucosylation reactions and deprotections.212,213
Validoxylamine G (13) was synthesized via a coupling of tetra-O-benzyl-valienamine (132) and tetra-O-benzyl-valiolone (208) by a reductive N-alkylation reaction with sodium cyanoborohydride (NaBH3CN) followed by deprotection of the benzyl ethers.173 In a similar manner, validamycin G (9) was prepared from tetra-O-benzyl-valienamine and the heptabenzyl-β-D-glucopyranosyl valiolone (209), the latter of which was derived from hepta-O-benzyl-D-cellobiose.173
The total synthesis of acarbose was carried out via coupling of two synthons, the optically pure di-O-isopropylidene-(+)-valienamine (210) and the protected anhydro derivative 215 of the trisaccharide, followed by deprotection.214,215 This approach has been applied as a common strategy for the synthesis of related members of this family, e.g., the adiposins,214–217 amylostatin (XG),218 salbostatin219 and 2′-deoxyvalidamycin B.220 Thus, coupling of the protected valienamine (210) with the appropriate mono- (211, 212), di- (213, 214) or trisaccharide (216) epoxides, followed by subsequent removal of protecting groups, and in some cases also acetolysis, and O-deacetylation gave salbostatin,219 2′-deoxyvalidamycin B,220 amylostatin (XG),218 adiposin-1,216,217 and adiposin-2,214,215 respectively. Amylostatin XG was previously synthesized from the chiral cyclohexyl halide 217 and the 4′-amino-4′ -deoxy-disaccharide 218.221
A common structural unit of the antibiotic oligostatins, methyl oligobiosaminide (51), was synthesized from epoxide 200 (racemic) and methyl 4-amino-4,6-dideoxy-α-D-glucoside (219), followed by deprotection.53,222 Coupling of 200 and 219 gave only a 37% yield of a mixture of diastereomeric and regioisomeric products. An enantiospecific synthesis of 51 was later reported using epoxide 201 and aminosugar 219 followed by deprotection.223 Methyl oligobiosaminide was utilized as the precursor for the synthesis of methyl acarviosine (29), the core pseudodisaccharide of acarbose and the amylostatins, isolated as its hexaacetate (221).224 Its 6′-hydroxy analog, the core structure of adiposin, was synthesized as the hepta-acetate 222 from the corresponding 6′-hydroxy analog 220.224 An enantioselective synthesis of 220 has been reported by Paulsen and Röben225 using L-1-desoxy-1-C-(hydroxymethyl)-myo-inositol (223) as the starting material for the preparation of the enantiomerically pure hydroxyvalidamine derivative 224. Coupling of 224 with epoxide 225, followed by opening of the 1,6-anhydro ring with acetic anhydride/boron trifluoride yielded α- and β-anomers 226. Deblocking of 226 caused a spontaneous rearrangement to the tricyclic 227, consistent with the rearrangements reported in the degradation of acarbose, the adiposins, and the oligostatins.
The antibiotic pyralomicin 1c was synthesized by Tatsuta et al.226 from the aglycon, pyralomicinone (230), which was prepared from pyrrole 228, the acid chloride 229,227 and properly protected valienol (231)
(Scheme 23). The glycon 231 was prepared from L-arabinose using the same method as that developed for the preparation of (+)-valienamine from D-xylose.179 Coupling of components 230 and 231 was carried out under modified Mitsunobu's conditions using nBuP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCN to give predominantly the desired product 232 with no significant by-products resulting from the reaction of other hydroxy groups of 231.
CHCN to give predominantly the desired product 232 with no significant by-products resulting from the reaction of other hydroxy groups of 231.
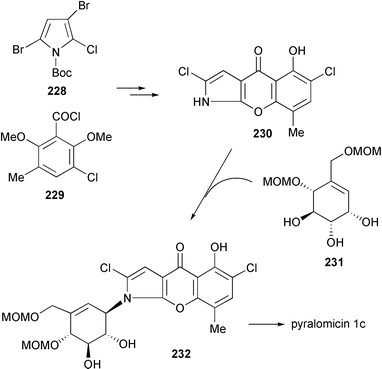 | ||
| Scheme 23 Total synthesis of pyralomicin 1c. | ||
5.3 Development of the aminocyclitols
During the past twenty years, a significant amount of work involving synthesis and biological evaluation of compounds containing C7N aminocyclitol units has been reported. This includes the synthesis of analogs of the natural products,228–231 analogs of enzyme substrates163,232–234 and analogs of enzyme inhibitors235–237 belonging to other classes of natural products, which were modified by replacing part of their structures with aminocyclitol units. In particular, tremendous contributions have been made by the group of Ogawa and Suami, who carried out syntheses and biological evaluations of analogs of validoxylamine,238 methyl acarviosine,239–241 methyl oligobiosaminide,223 analogs of kanosamine,234 trehalosamine235,236 and trehazoline,237 carba sugar versions of cell surface glycans,242,243 analogs of glycosylceramide162,244–248 and PDMP (1-phenyl-2-decanoylamino-3-morpholino-1-propanol),249 as well as an α-L-fucosidase inhibitor.163Efforts in developing more potent trehalase inhibitors, for example, have been carried out by synthesizing a number of pseudo-trehalosamines (234–236) and related compounds,235,236 as well as dicarba analogs (238–240) of trehalose (237) composed of validamine, valienamine, and valiolamine moieties.232 The latter compounds (238–240) were shown to have strong inhibitory activity against trehalase from muscidae comparable to that of validoxylamine A.
An attempt was also made to synthesize an analog of trehazoline (241),237 a trehalase inhibitor isolated from the culture broth of Micromonospora strain SANK 62390, by replacing the aminocyclitol portion of trehazolin with validamine. The product, compound 242, was apparently unstable, especially under basic conditions, and readily rearranged to isomer 243. The latter compound showed no inhibitory activity against α-amylase, β-glucosidase, α-mannosidase, maltase, sucrase, and trehalase. Furthermore, enzymatic preparation of α- and β-D-glucoside derivatives of validamycins250 and glucoside derivatives of validamine and valienamine251 using microbial glycosylation with strains of Rhodotorula spp. resulted in a number of novel entities that in general are less active against trehalase and α-glucosidases than the parent compounds.
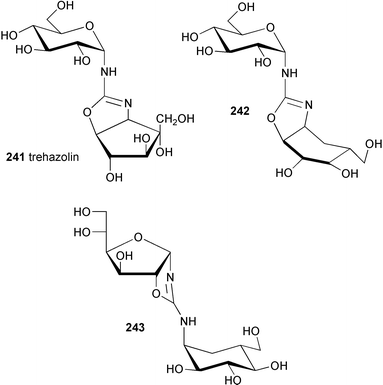
Despite the fact that acarbose has been acknowledged as a potent α-glucosidase inhibitor, and has been developed and clinically used as an anti-diabetic drug, a large amount of work aiming for better derivatives of this compound has been reported. Dihydroacarbose, which was isolated as a product of the hydrogenation of acarbose36 and later was synthesized from pullalen,252,253 was found to be a potent oligosaccharide hydrolase inhibitor,253 although its inhibitory activities were lower than those of acarbose. Syntheses of potential β-glucosidase inhibitors, 6″-hydroxy β-acarbose (β-adiposin-2) and its derivatives were also reported; however, attempts to prepare β-acarbose itself by similar approaches have been unsuccessful.229,254
It was also of great interest to many investigators to develop rather simple C7N aminocyclitols that possess the same or better biological activities as acarbose. Since it was previously proposed that the pseudodisaccharide core moiety in acarbose is responsible for its sugar hydrolase inhibitory activity, a significant numbers of analogs (244–257) of methyl oligobiosaminide and methyl acarviosine were synthesized and their biological activities evaluated.223,239–241 These have led to the discovery of strong α-D-glucosidase inhibitors, the 2-deoxy (256) and the 2,3-dideoxy derivatives (257),241 which are almost 10 × more potent than the parent methyl acarviosine (29). The latter compound itself is a 5 × more potent α-glucosidase inhibitor than acarbose.35 Surprisingly, methyl oligobiosaminide (51) has almost no inhibitory activity.223
The most successful attempt in developing new α-glucosidase inhibitors appears to have been carried out by Horii and co-workers, who synthesized and screened a series of N-substituted valiolamine derivatives.135 They found that the N-substituted valiolamine derivatives studied are more potent than the correspondent N-substituted valienamine derivatives as well as the parent valiolamine, some of which have even stronger α-D-glucosidase inhibitory activity against porcine intestinal maltase and sucrase than naturally occurring oligosaccharide α-D-glucosidase inhibitors. One of these analogs, N-[2-hydroxy-1-(hydroxymethyl)ethyl)]-valiolamine or AO-128 (voglibose, 258) has been developed as a blood glucose ameliorating agent255 due to its good activity, its easy preparation, especially in a large-scale synthesis, and the relative lack of safety concerns due to possible metabolites potentially produced from the N-substituted moiety in humans. It showed 190–3900-fold more potent inhibition of the purified rat small intestine sucrase–isomaltase (S-1) complex and 23–33-fold more potent inhibition of semi-purified porcine small intestine disaccharidases than acarbose.256,257 Kinetic studies revealed that voglibose inhibits disaccharidases competitively, suppressing the elevation of the blood glucose concentration after oral sucrose, maltose, or starch administration, but not after oral glucose, fructose or lactose. In animals and healthy volunteers, voglibose significantly reduced post prandial blood glucose concentration. As shown in the case of valienamine, validamine and other simple α-glucosidase inhibitors, the inhibitory effects of voglibose on α-amylase from porcine or rat pancreas were very weak and only about 1/3000 of those of acarbose.257 These differences, in fact, have given voglibose some advantage over acarbose with respect to the frequent undesirable side effects, e.g., diarrhea, flatulence, and abdominal distension, which are mainly due to a considerable amount of undigested starch accumulating in the large bowel. The Ki values of voglibose were found to be two or three orders of magnitude smaller than those of acarbose, which is probably one of the reasons why the clinical doses (0.2 or 0.3 mg with every meal) of voglibose are very low. Voglibose was launched as an anti-diabetic agent, known as Basen®, in September 1994.255
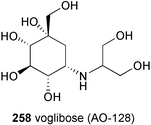
In the patent literature, scientists at Bayer AG claimed a novel biotransformation product of acarbose by Actinoplanes sp. strain SE 50/110.258 This new finding was presumably born from the understanding that the oligosaccharide residues of acarbose and its analogs originate from the sugar components most abundant in the culture media, which are transferred to the molecules via a direct transglycosylation reaction. Accordingly, they found that addition of the flavonol glycoside “rutin” to the fermentation media of the acarbose producer yields a new hybrid secondary metabolite 259, which is likely formed by a direct glycosylation of rutin by the activated acarviosine (e.g., its dTDP derivative) catalyzed by a nonspecific glycosyltransferase. Compound 259 was 6 × more potent against saccharases than acarbose. However, the production yield of this compound appears to be very low.
In a larger sense, the C7N aminocyclitols have also been envisaged to play roles in elucidating and controlling other biological events that involve sugar moieties. Ogawa and co-workers have synthesized protected derivatives of carba-sugar analogs (261–263) of the branched mannotriose structure (260) which consists of the “trimannosyl core” of the oligosaccharide chains that frequently occur as structural units in biologically important glycoconjugates.243 Those analogs were expected to be utilized as model compounds in conformational studies of complex oligosaccharides and for elucidation of the mode of action of sugar hydrolases and transferases, such as GlcNAc-transferases I–IV.
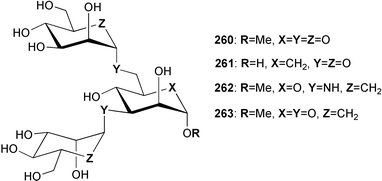
Analogs of 5a-carba glycosylceramides, structurally related to glycosphingolipids and glycoglycerolipids, have also been synthesized and their biological activities have been examined. 5a-Carba-β-glucosyl (E-264) and galactosylceramide (E-265) analogs, as well as their corresponding Z-isomers, were shown to be very potent and specific gluco- and galactocerebrosidase inhibitors.162,246N-Alkyl-β-valienamines (266–271),247 simplified versions of E-264, were also examined and all were shown to be potent and specific inhibitors of β-glucocerebrosidase. Among them, the N-octyl derivative (268) was found to be the strongest inhibitor (IC50 = 3 × 10−8 M), being almost 10-fold more potent than E-264. Biological evaluation data on the N-alkanoyl (272–275) and N-alkyl derivatives (276–282)248 of 268 revealed that the basic cationic property of the alkyl amino function is necessary for activity, while additional hydrophobic side chains seem to be less important. N-Butyl-N-octyl-β-valienamine (276) and N-decyl-N-octyl-β-valienamine (279) were shown to change the amount of GlcCer and GM3 in mouse-derived B16 melanoma cells, suggesting that they have an effect on glycolipid biosynthesis.
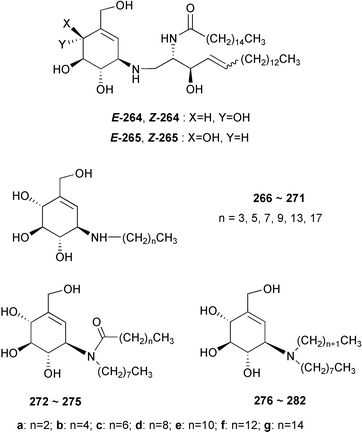
Creations of inhibitors for other classes of sugar hydrolases were also pursued based on structural similarity to substrates or inhibitors. 5a-Carba-α-L-fucopyranosamine (283),163 a validamine analog having an α-L-fucose-type structure, was found to have a specific and very strong inhibitory activity against α-L-fucosidase from bovine kidney (Ki = 1.2 × 10−8 M). α-Fucosidase inhibitors are considered to be potential candidates for cancer and HIV drugs, due to their inhibitory effect on the extracellular matrix secreted fucosidases.160,163,259
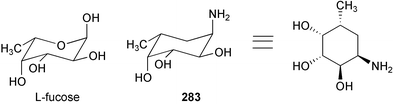
Sulfated trestatin A (284),161 which mimics the structure of heparin (285), was found to have the highest antiproliferative activity among “low-molecular weight” compounds against smooth muscle cells in vitro. In contrast to heparin, trestatin A sulfate has almost no anticoagulant properties. More recently, it was found that trestatin A sulfate strongly inhibits P-selectin and L-selectin-mediated cell adhesion in vitro and in vivo while lacking antithrombin-mediated anticoagulant activity.260 It reduced by 96% leukocytes rolling along rat mesenteric post-capillary venules and inhibited by 81% neutrophil migration into thioglycollate-inflamed peritoneum of BALB/c mice, which suggests a therapeutic potential of this compound in inflammatory disorders. Trestatin A sulfate could also be a potential drug to prevent reperfusion injury when the risk of hemorrhagic complications is high, for example in the case of thrombolytic therapy and organ transplantation.260
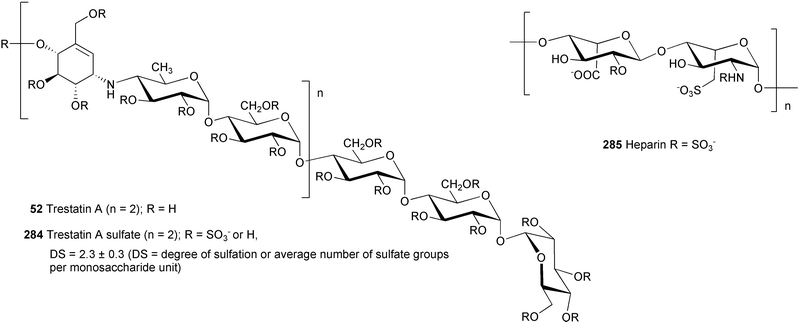
6 Summary and perspectives
First reported in the early 1970s, the C7N aminocyclitol-containing family of natural products has rapidly grown to be one of the clinically more successful groups of secondary metabolites. A number of its members have been used to combat diseases in humans and plants, while several others are still under investigation. Their excellent biological activities in combination with their relative lack of harmful side effects toward humans, animals, or plants have been particularly important from a pharmaceutical point of view and have been the most significant factors in their notable success. In addition, the C7N aminocyclitols are structurally close to sugar moieties, which are widely distributed and play physiological roles in almost all parts of living organisms, thus increasing the possibilities for these compounds to be developed for therapeutical use in numerous physiological disorders. Despite the fact that many of them exhibited inhibitory effects on some kinds of sugar-hydrolases, the spectrum of their biological activities is apparently broader than was originally thought. The isolations of a number of unusual C7N aminocyclitol-containing natural products having anti-inflammatory and anti-tumor activities were reported rather recently. In addition, synthetically designed C7N aminocyclitols have also been shown to possess a variety of biological activities, such as antiproliferative activity against smooth muscle cells, anti-inflammation, immunomodulation, as well as potential for anti-cancer and HIV applications.Studies on the biosynthesis of the C7N aminocyclitols of validamycin, acarbose, and pyralomicin have established several closely related novel metabolic pathways that involve a number of ketocyclitol intermediates that are all derived from a common open chain C7 sugar phosphate involved in the pentose phosphate pathway. The novel pathways are presumably exclusive to microorganisms, particularly of the class of Actinomycetes soil bacteria. The isolations of a variety of keto-analogues of the aminocyclitols, e.g., the glyoxalase I inhibitor 286,261,262 KD16-U1 (287)263 and the gabosines (e.g.288),264–266 in various Streptomyces species, suggest that this type of compound could be widely distributed among soil bacteria; however, neither the same biosynthetic pathway nor the same class of secondary metabolites have ever been found in plants or other organisms.
The current rapid growth in the genetic and molecular sciences has undoubtedly brought increased opportunities for the development of many secondary metabolites via genetics and combinatorial biosynthetic approaches, which should be applicable to the C7N aminocyclitols. Cloning and heterologous expression of biosynthetic genes in different hosts has become more and more common and is routinely performed. These techniques could be useful for the generation of microbial strains that can produce the secondary metabolites in an accelerated fashion with higher yields, or they could be used to generate transgenic plants harboring antibiotic or antifungal biosynthetic genes for self-protection. For instance, introduction of the biosynthetic gene cluster of the anti-fungal validamycin A into rice would likely create a transgenic rice plant resistant to R. solani or other pathogenic fungi. Also, the “mix and match” combinatorial biosynthetic approach, as popularly applied to the construction of combinatorial libraries of polyketides and non-ribosomal peptides,267–269 could be a useful tool for generating additional analogues of these aminocyclitol natural products.
Acknowledgements
The author thanks Professor Heinz G. Floss and Dr Matthew G. McDonald for proofreading this review and for their invaluable comments and suggestions.References
- J. M. Shick and W. C. Dunlap, Annu. Rev. Physiol., 2002, 64, 223 CrossRef CAS.
- T. Iwasa, H. Yamamoto and M. Shibata, J. Antibiot., 1970, 23, 595 Search PubMed.
- E. Truscheit, W. Frommer, B. Junge, L. Müller, D.D. Schmidt and W. Wingender, Angew. Chem., Int. Ed. Engl., 1981, 20, 744 CrossRef.
- T. Iwasa, E. Higashide, H. Yamamoto and M. Shibata, J. Antibiot., 1971, 24, 107 Search PubMed.
- T. Iwasa, E. Higashide and M. Shibata, J. Antibiot., 1971, 24, 114 Search PubMed.
- A. P. J. Trinci, Exp. Mycol., 1985, 9, 20 CAS.
- G. D. Robson, P. J. Kuhn and A. P. J. Trinci, J. Gen. Microbiol., 1988, 134, 3187 Search PubMed.
- T. Iwasa, Y. Kameda, M. Asai, S. Horii and K. Mizuno, J. Antibiot., 1971, 24, 119 Search PubMed.
- S. Horii, T. Iwasa and Y. Kameda, J. Antibiot., 1971, 24, 57 Search PubMed.
- S. Horii, T. Iwasa, E. Mizuta and Y. Kameda, J. Antibiot., 1971, 24, 59 Search PubMed.
- S. Ogawa, Y. Miyamoto and A. Nakajima, Chem. Lett., 1989, 725 CAS.
- S. Ogawa, A. Nakajima and Y. Miyamoto, J. Chem. Soc., Perkin Trans. 1, 1991, 3287 RSC.
- S. Horii and Y. Kameda, J. Chem. Soc., Chem. Commun., 1972, 747 RSC.
- K. Kamiya, Y. Wada, S. Horii and M. Nishikawa, J. Antibiot., 1971, 24, 317 Search PubMed.
- T. Suami, S. Ogawa and N. Chida, J. Antibiot., 1980, 33, 98 Search PubMed.
- S. Ogawa, Y. Shibata, N. Chida and T. Suami, Chem. Lett., 1980, 135 CAS.
- S. Ogawa, Y. Shibata, N. Chida and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 494 CAS.
- S. Ogawa, N. Chida and T. Suami, Chem. Lett., 1980, 139 CAS.
- S. Ogawa, N. Chida, H. Ito and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 499 CAS.
- W.-Z. Jin, K. L. Rinehart, Jr. and T. Toyokuni, J. Antibiot., 1987, 40, 329 Search PubMed.
- S. Horii, Y. Kameda and K. Kawahara, J. Antibiot., 1972, 25, 48 Search PubMed.
- Y. Kameda, N. Asano, K. Matsui, S. Horii and H. Fukase, J. Antibiot., 1988, 41, 1488 Search PubMed.
- Y. Kameda, N. Asano, T. Yamaguchi, K. Matsui, S. Horii and H. Fukase, J. Antibiot., 1986, 39, 1491 Search PubMed.
- N. Asano, Y. Kameda, K. Matsui, S. Horii and H. Fukase, J. Antibiot., 1990, 43, 1039 Search PubMed.
- Y. Kameda, N. Asano, M. Yoshikawa, M. Takeuchi, T. Yamaguchi, K. Matsui, S. Horii and H. Fukase, J. Antibiot., 1984, 37, 1301 Search PubMed.
- Y. Kameda and S. Horii, J. Chem. Soc., Chem. Commun., 1972, 746 RSC.
- Y. Kameda, S. Horii and T. Yamano, J. Antibiot., 1975, 28, 298 Search PubMed.
- Y. Kameda, N. Asano, M. Teranishi and K. Matsui, J. Antibiot., 1980, 33, 1573 Search PubMed.
- Y. Kameda, N. Asano, M. Teranishi, M. Yoshikawa and K. Matsui, J. Antibiot., 1981, 34, 1237 Search PubMed.
- N. Asano, M. Takeuchi, K. Ninomiya, Y. Kameda and K. Matsui, J. Antibiot., 1984, 37, 859 Search PubMed.
- D.D. Schmidt, W. Frommer, B. Junge, L. Müller, W. Wingender and E. Truscheit, Naturwissenschaften, 1977, 64, 535 CrossRef CAS.
- W. Puls, U. Keup, H. P. Krause, G. Thomas and F. Hoffmeister, Naturwissenschaften, 1977, 64, 536 CrossRef CAS.
- L. K. Campbell, J. R. White and R. K. Campbell, Ann. Pharmacother., 1996, 30, 1255 Search PubMed.
- M. Hemker, A. Stratmann, K. Goeke, W. Schröder, J. Lenz, W. Piepersberg and H. Pape, J. Bacteriol., 2001, 183, 4484 CrossRef CAS.
- B. Junge, F.-R. Heiker, J. Kurz, L. Müller, D.D. Schmidt and C. Wünsche, Carbohydr. Res., 1984, 128, 235 CrossRef CAS.
- K. Bock, M. Meldal and S. Refn, Carbohydr. Res., 1991, 221, 1 CrossRef CAS.
- K. Bock and H. Pedersen, Carbohydr. Res., 1984, 132, 142 CrossRef CAS.
- E. Raimbaud, A. Buléon and S. Pérez, Carbohydr. Res., 1992, 227, 351 CrossRef CAS.
- P. M. Coutinho, M. K. Dowd and P. J. Reilly, Proteins: Struct., Funct., Genet., 1997, 27, 235 Search PubMed.
- S. Murao, K. Ohyama and S. Ogura, Agric. Biol. Chem., 1977, 41, 919 Search PubMed.
- K-i. Fukuhara, H. Murai and S. Murao, Agric. Biol. Chem., 1982, 46, 2021 Search PubMed.
- K-i. Fukuhara, H. Murai and S. Murao, Agric. Biol. Chem., 1982, 46, 1941 Search PubMed.
- Jpn. Pat., JP 55-157595, 1980.
- US Pat., 4,010,258, 1977.
- Jpn. Pat., JP 55-071494, 1980.
- Jpn. Pat., JP 57-018692, 1982.
- S. Namiki, K. Kangouri, T. Nagate, H. Hara, K. Sugita and S. Õmura, J. Antibiot., 1982, 35, 1234 Search PubMed.
- S. Namiki, K. Kangouri, T. Nagate, H. Hara, K. Sugita and S. Õmura, J. Antibiot., 1982, 35, 1156 Search PubMed.
- Kangouri, S. Namiki, T. Nagate, H. Hara, K. Sugita and S. Omura, J. Antibiot., 1982, 35, 1160 Search PubMed.
- US Pat., 4, 254, 256, 1981.
- J. Itoh, S. Omoto, T. Shomura, H. Ogino, K. Iwamatsu, S. Inouye and H. Hidaka, J. Antibiot., 1981, 34, 1424 Search PubMed.
- S. Omoto, J. Itoh, H. Ogino, K. Iwamatsu, N. Nishizawa and S. Inouye, J. Antibiot., 1981, 34, 1429 Search PubMed.
- S. Ogawa, Y. Iwasawa, T. Toyokuni and T. Suami, Carbohydr. Res., 1985, 144, 155 CrossRef CAS.
- K. Yokose, K. Ogawa, T. Sano, K. Watanabe, H. B. Maruyama and Y. Suhara, J. Antibiot., 1983, 36, 1157 Search PubMed.
- K. Watanabe, T. Furumai, M. Sudoh, K. Yokose and H. B. Maruyama, J. Antibiot., 1984, 37, 479 Search PubMed.
- K Yokose, K. Ogawa, Y. Suzuki, I. Umeda and Y. Suhara, J. Antibiot., 1983, 36, 1166 Search PubMed.
- K Yokose, M. Ogawa and K. Ogawa, J. Antibiot., 1984, 37, 182 Search PubMed.
- Jpn. Pat., JP 58-172400, 1983.
- US Pat., 4,632,917, 1986.
- US Pat., 5,866,377, 1999.
- US Pat., 4, 990,500, 1991.
- J-G. Kim., H-B. Chang, Y-I. Kwon, S-K. Moon, H-S. Chun, S. K. Ahn and C. I. Hong, J. Antibiot., 2002, 55, 457 Search PubMed.
- Y-I. Kwon, H-J. Son, K. S. Moon, J. K. Kim, J-G. Kim, H-S. Chun, S. K. Ahn and C. I. Hong, J. Antibiot., 2002, 55, 462 Search PubMed.
- H-B. Chang, S-H. Kim, Y-I. Kwon, D-H. Choung, W-K. Choi, T. W. Kang, S. Lee., J-G. Kim., H-S. Chun, S. K. Ahn, C. I. Hong and K-H. Han, J. Antibiot., 2002, 55, 467 Search PubMed.
- L. Vértesy, H-W. Fehlhaber and A. Schulz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1844 CrossRef.
- I. Knuesel, S. Murao, T. Shin, T. Amachi and H. Kayser, Comp. Biochem. Phys., B, 1998, 120, 639 Search PubMed.
- N. Kawamura, R. Sawa, Y. Takahashi, K. Issiki, T. Sawa, N. Kinoshita, H. Naganawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 435 Search PubMed.
- N. Kawamura, N. Kinoshita, R. Sawa, Y. Takahashi, T. Sawa, H. Naganawa, M. Hamada and T. Takeuchi, J. Antibiot., 1996, 49, 706 Search PubMed.
- Z. Zhang, Y. Wang and J. Ruan, Int. J. Sys. Bacteriol., 1998, 48, 411 Search PubMed.
- N. Kawamura, R. Sawa, Y. Takahashi, K. Isshiki, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1996, 49, 651 Search PubMed.
- N. Kawamura, H. Nakamura, R. Sawa, Y. Takahashi, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1997, 50, 147 Search PubMed.
- T. Tsuchida, M. Umekita, N. Kinoshita, H. Iinuma, H. Nakamura, K. T. Nakamura, H. Naganawa, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1996, 49, 326 Search PubMed.
- N. Matsumoto, T. Tsuchida, M. Umekita, N. Kinoshita, H. Iinuma, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1997, 50, 900 Search PubMed.
- N. Matsumoto, T. Tsuchida, R. Sawa, H. Iinuma, H. Nakamura, H. Naganawa, T. Sawa and T. Takeuchi, J. Antibiot., 1997, 50, 912 Search PubMed.
- O. Schlörke, P. Krastel, I. Müller, I. Usón, K. Dettner and A. Zeeck, J. Antibiot., 2002, 55, 635 Search PubMed.
- H. G. Floss, Nat. Prod. Rep., 1997, 14, 433 RSC.
- H. G. Floss and J. M. Beale, Angew. Chem., Int. Ed. Engl., 1989, 28, 146 CrossRef.
- C-G. Kim, A. Kirschning, P. Bergon, P. Zhou, E. Su, B. Sauerbrei, S. Ning, Y. Ahn, M. Breuer, E. Leitsner and H. G. Floss, J. Am. Chem. Soc., 1996, 118, 7486 CrossRef CAS.
- H. G. Floss, P. J. Keller and J. M. Beale, J. Nat. Prod., 1986, 49, 957 CrossRef CAS.
- U. Degwert, R. van Hülst, H. Pape, R. E. Herrold, J. M. Beale, P. J. Keller, J. P. Lee and H. G. Floss, J. Antibiot., 1987, 40, 855 Search PubMed.
- T. Toyokuni, W-Z. Jin and K. L. Rinehart, Jr., J. Am. Chem. Soc., 1987, 109, 3481 CrossRef CAS.
- N. Kawamura, R. Sawa, Y. Takahashi, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1996, 49, 657 Search PubMed.
- T. Mahmud, I. Tornus, E. Egelkrout, E. Wolf, C. Uy, H. G. Floss and S. Lee, J. Am. Chem. Soc., 1999, 121, 6973 CrossRef CAS.
- H. Dong, T. Mahmud, I. Tornus, S. Lee and H. G. Floss, J. Am. Chem. Soc., 2001, 123, 2733 CrossRef CAS.
- H. Naganawa, H. Hashizume, Y. Kubota, R. Sawa, Y. Takahashi, K. Arakawa, S. G. Bowers and T. Mahmud, J. Antibiot., 2002, 55, 578 Search PubMed.
- A. Stratmann, T. Mahmud, S. Lee, J. Distler, H. G. Floss and W. Piepersberg, J. Biol. Chem., 1999, 274, 10889 CrossRef CAS.
- Deutsch. Pat., DE 10021667 A1, 2001.
- M. W. Loewus, F. A. Loewus, G.-U. Brillinger, H. Otsuka and H. G. Floss, J. Biol. Chem., 1980, 255, 11710 CAS.
- F. Tian, M. E. Migaud and J. W. Frost, J. Am. Chem. Soc., 1999, 121, 5795 CrossRef.
- T. Mahmud, J. Xu and Y. U. Choi, J. Org. Chem., 2001, 66, 5066 CrossRef CAS.
- T. Mahmud, S. Lee and H. G. Floss, Chem. Rec., 2001, 1, 300 Search PubMed.
- C-S. Zhang, A. Stratmann, O. Block, R. Brückner, M. Podeschwa, H-J. Altenbach, U. F. Wehmeier and W. Piepersberg, J. Biol. Chem., 2002, 277, 22853 CrossRef CAS.
- S. G. Bowers, T. Mahmud and H. G. Floss, Carbohydr. Res., 2002, 337, 297 CrossRef CAS.
- A. Trefzer, J. A. Salas and A. Bechthold, Nat. Prod. Rep., 1999, 16, 283 RSC.
- T. M. Hallis and H.-W. Liu, Acc. Chem. Res., 1999, 32, 579 CrossRef CAS.
- S. Lee and E. Egelkrout, J. Antibiot., 1998, 51, 225 Search PubMed.
- S. Lee, B. Sauerbrei, J. Niggemann and E. Egelkrout, J. Antibiot., 1997, 50, 954 Search PubMed.
- Y. Funabashi, M. Takizawa, S. Tsubotani, S. Tanida and S. Harada, J. Takeda Res. Lab., 1992, 51, 73 Search PubMed.
- M. E. Raggatt, T. J. Simpson and S. K. Wrigley, Chem. Commun., 1999, 1039 RSC.
- W. F. Caspary and S. Graf, Res. Exp. Med. (Berlin), 1979, 175, 1 Search PubMed.
- W. Puls, U. Keup, H. P. Krause, L. Müller, D. D. Schmidt, G. Thomas and E. Truscheit, Front. Hormone Res., 1980, 7, 235 Search PubMed.
- S. Cogoli and G. Semenza, J. Biol. Chem., 1975, 250, 7802.
- A. E. Aleshin, L. M. Firsov and R. B. Honzatko, J. Biol. Chem., 1994, 269, 15631.
- K. Schöffling, I. Hillebrand and P. Berchtold, Front. Hormone Res., 1980, 7, 248 Search PubMed.
- G. Hanozet, H-P. Pircher, P. Vanni, B. Oesch and G. Semenza, J. Biol. Chem., 1981, 256, 3703 CAS.
- B. Svensson and M. R. Sierks, Carbohydr. Res., 1992, 227, 29 CrossRef CAS.
- K. Olsen, U. Christensen, M. R. Sierks and B. Svensson, Biochemistry, 1993, 32, 9686 CrossRef CAS.
- L. M. Firsov, K. N. Neustroev, A. E. Aleshin, C. M. Metzler, D. E. Metzler, R. D. Scott, B. Stoffer, T. Christensen and B. Svensson, Eur. J. Biochem., 1994, 223, 293 CAS.
- B. W. Sigurskjold, C. R. Berland and B. Svensson, Biochemistry, 1994, 33, 10191 CrossRef CAS.
- B. Stoffer, A. E. Aleshin, L. M. Firsov, B. Svensson and R. B. Honzatko, FEBS Lett., 1995, 358, 57 CrossRef CAS.
- A. E. Aleshin, B. Stoffer, L. M. Firsov, B. Svensson and R. B. Honzatko, Biochemistry, 1996, 35, 8319 CrossRef CAS.
- M. Qian, R. Haser, G. Buisson, E. Duée and F. Payan, Biochemistry, 1994, 33, 6284 CrossRef CAS.
- C. Gilles, J-P. Astier, G. Marchis-Mouren, C. Cambillau and F. Payan, Eur. J. Biochem., 1996, 238, 561 CrossRef CAS.
- M. Alkazaz, V. Desseaux, G. Marchis-Mauren, F. Payan, E. Forest and M. Santimone, Eur. J. Biol., 1996, 241, 787 Search PubMed.
- M. Machius, L. Vértesy, R. Huber and G. Wiegand, J. Mol. Biol., 1996, 260, 409 CrossRef CAS.
- M. Qian, V. Nahoum, J. Bonicel, H. Bischoff, B. Henrissat and F. Payan, Biochemistry, 2001, 40, 7700 CAS.
- G. Ferey-Roux, J. Perrier, E. Forest, G. Marchis-Mouren, A. Puigserver and M. Santimone, Biochim. Biophys. Acta, 1998, 1388, 10 CrossRef CAS.
- V. Nahoum, G. Roux, V. Anton, P. Rougé, A. Puigserver, H. Bischoff, B. Henrissat and F. Payan, Biochem. J., 2000, 346, 201 CrossRef CAS.
- A. M. Brzozowski and G. J. Davies, Biochemistry, 1997, 36, 10837 CrossRef CAS.
- M-J. Kim, S-B. Lee, H-S. Lee, S-Y. Lee, J-S. Baek, D. Kim, T-W. Moon, J. F. Robyt and K-H. Park, Arch. Biochem. Biophys., 1999, 371, 277 CrossRef CAS.
- A. M. Brzozowski, D. M. Lawson, J. P. Turkenburg, H. Bisgaard-Frantzen, A. Svendsen, T. V. Borchert, Z. Dauter, K. S. Wilson and G. J. Davies, Biochemistry, 2000, 39, 9099 CrossRef CAS.
- N. Aghajari, M. Roth and R. Haser, Biochemistry, 2002, 41, 4273 CrossRef CAS.
- H-J. Cha, H-G. Yoon, Y-W. Kim, H-S. Lee, J-W. Kim, K-S. Kweon, B-H. Oh and K-H. Park, Eur. J. Biochem., 1998, 253, 251 CrossRef CAS.
- K. H. Park, M. J. Kim, H. S. Lee, N. S. Han, D. Kim and J. F. Robyt, Carbohydr. Res., 1998, 313, 235 CrossRef CAS.
- T-J. Kim, M-J. Kim, B-C. Kim, J-C. Kim, T-K. Cheong, J-W. Kim and K-H. Park, Appl. Environ. Microbiol., 1999, 65, 1644 CAS.
- Z. Dauter, M. Dauter, A. M. Brzozowski, S. Christensen, T. V. Borchert, L. Beier, K. S. Wilson and G. J. Davies, Biochemistry, 1999, 38, 8385 CrossRef CAS.
- R. Mosi, H. Sham, J. C. M. Uitdehaag, R. Ruiterkamp, B. W. Dijkstra and S. G. Withers, Biochemistry, 1998, 37, 17192 CrossRef CAS.
- E. J. Goldsmith, R. J. Fletterick and S. J. Withers, J. Biol. Chem., 1987, 262, 1449 CAS.
- M. O'Reilly, K. A. Watson and L. N. Johnson, Biochemistry, 1999, 38, 5337 CrossRef CAS.
- I. Przylas, Y. Terada, K. Fujii, T. Takaha, W. Saenger and N. Sträter, Eur. J. Biol., 2000, 267, 6903 Search PubMed.
- S. Namiki, K. Kangouri, T. Nagate, H. Hara, K. Sugita, K. Noda, Y. Tarumoto and S. Omura, J. Antibiot., 1982, 35, 1167 Search PubMed.
- Y. Kameda, N. Asano, M. Yoshikawa and K. Matsui, J. Antibiot., 1980, 33, 1575 Search PubMed.
- M. Takeuchi, N. Takai, N. Asano, Y. Kameda and K. Matsui, Chem. Pharm. Bull., 1990, 38, 1970 CAS.
- Y. Kameda, N. Asano, M. Yoshikawa, K. Matsui, S. Horii and H. Fukase, J. Antibiot., 1982, 35, 1624 Search PubMed.
- S. Horii, H. Fukase, T. Matsuo, Y. Kameda, N. Asano and K. Matsui, J. Med. Chem., 1986, 29, 1038 CrossRef CAS.
- M. Shibata, M. Uyeda and K. Mori, J. Antibiot., 1981, 34, 447 Search PubMed.
- O. Wakae, J. Pesticide Sci., 1981, 6, 455 Search PubMed.
- M. Shibata, K. Mori and M. Hamashima, J. Antibiot., 1982, 35, 1422 Search PubMed.
- T. Nioh and S. Mizushima, J. Gen. Appl. Microbiol., 1974, 20, 373 Search PubMed.
- M. Uyeda, A. Ikeda, T. Machimoto and M. Shibata, Agric. Biol. Chem., 1985, 49, 3485 Search PubMed.
- Y. Kido, T. Nagasato, K. Ono, Y. Fujimoto, M. Uyeda and M. Shibata, Agric. Biol. Chem., 1986, 50, 1519 Search PubMed.
- M. Uyeda, A. Ikeda, T. Ogata and M. Shibata, Agric. Biol. Chem., 1986, 50, 1885 Search PubMed.
- M. Ohnishi and K. Matsuura, J. Takeda Res. Lab., 1981, 40, 77 Search PubMed.
- M. Uyeda, K. Suzuki, H. Tsuruta and M. Shibata, Agric. Biol. Chem., 1988, 52, 2607 Search PubMed.
- G. D. Robson, P. L. Kuhn and A. P. J. Trinci, J. Gen. Microbiol., 1989, 135, 739 Search PubMed.
- Y. Kameda, N. Asano, T. Yamaguchi and K. Matsui, J. Antibiot., 1987, 40, 563 Search PubMed.
- N. Asano, M. Takeuchi, Y. Kameda, K. Matsui and Y. Kono, J. Antibiot., 1990, 43, 722 Search PubMed.
- R. Shigemoto, T. Okuno and K. Matsuura, Ann. Phytopath. Soc. Jpn., 1992, 58, 685 Search PubMed.
- N. Asano, T. Yamaguchi, Y. Kameda and K. Matsui, J. Antibiot., 1987, 40, 526 Search PubMed.
- N. Asano, K. Tanaka, Y. Kameda and K. Matsui, J. Antibiot., 1991, 44, 1417 Search PubMed.
- H. M. Salleh and J. F. Honek, FEBS Lett., 1990, 262, 359 CrossRef CAS.
- N. Asano, A. Kato and K. Matsui, Eur. J. Biochem., 1996, 240, 692 CrossRef CAS.
- Y. Kono, S. Takeda and Y. Kameda, J. Pestic. Sci., 1994, 19, 39 Search PubMed.
- Y. Kono, S. Takeda, Y. Kameda, M. Takahashi, K. Matsushita, M. Nishina and E. Hori, Appl. Entomol. Zool., 1993, 28, 379 Search PubMed.
- Y. Kono, S. Takeda, Y. Kameda, M. Takahashi, K. Matsushita, M. Nishina and E. Hori, J. Pestic. Sci., 1995, 20, 83 Search PubMed.
- Y. Kono, M. Takahashi, K. Matsushita, M. Nishina, Y. Kameda and E. Hori, J. Insect. Physiol., 1994, 40, 455 CrossRef CAS.
- Y. Kono, M. Takahashi, M. Mihara, K. Matsushita, M. Nishina and Y. Kameda, Appl. Entomol. Zool., 1997, 32, 293 Search PubMed.
- N. Matsumoto, H. Iinuma, T. Sawa, T. Takeuchi, S.-I. Hirano, T. Yoshioka and M. Ishizuka, J. Antibiot., 1997, 50, 906 Search PubMed.
- N. Matsumoto, N. Agata, H. Kuboki, H. Iinuma, T. Sawa, T. Takeuchi and K. Umezawa, J. Antibiot., 2000, 53, 637 Search PubMed.
- E. G.-Martin, S. G.-Seijo, C. N.-Novoa and A. F.-Briera, Int. J. Biochem. Cell Biol., 1996, 28, 651 CrossRef.
- H. P. Wessel, M. Hosang, T. B. Tschopp and B.-J. Weimann, Carbohydr. Res., 1990, 204, 131 CrossRef CAS.
- H. Tsunoda and S. Ogawa, Liebigs Ann. Chem., 1995, 267 Search PubMed.
- S. Ogawa, A. Maruyama, T. Odagiri, H. Yuasa and H. Hashimoto, Eur. J. Org. Chem., 2001, 967 CrossRef CAS.
- S. Ogawa, T. Toyokuni and T. Suami, Chem. Lett., 1980, 713 CAS.
- S. Ogawa, N. Chida and T. Suami, J. Org. Chem., 1983, 48, 1203 CrossRef CAS.
- T. Toyokuni, S. Ogawa and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 1161 CAS.
- H. Paulsen and F. R. Heiker, Angew. Chem., Int. Ed. Engl., 1980, 19, 904 CrossRef.
- H. Paulsen and F. R. Heiker, Liebigs Ann. Chem., 1981, 2180 Search PubMed.
- S. Ogawa, Y. Shibata, T. Nose and T. Suami, Bull. Chem. Soc. Jpn., 1985, 58, 3387 CAS.
- R. R. Schmidt and A. Köhn, Angew. Chem., Int. Ed. Engl., 1987, 26, 482 CrossRef.
- M. Yoshikawa, B. C. Cha, Y. Okaichi, Y. Takinami, Y. Yokokawa and I. Kitagawa, Chem. Pharm. Bull., 1988, 36, 4236 CAS.
- F. Nicotra, L. Panza, F. Ronchetti and G. Russo, Gazz. Chim. Ital., 1989, 119, 577 Search PubMed.
- H. Fukase and S. Horii, J. Org. Chem., 1992, 57, 3651 CrossRef CAS.
- S. Knapp, A. B. J. Naughton and T. G. M. Dhar, Tetrahedron Lett., 1992, 33, 1025 CrossRef CAS.
- T. K. Park and S. J. Danishefski, Tetrahedron Lett., 1994, 35, 2667 CrossRef CAS.
- B. M. Trost, L. S. Chupak and T. Lübbers, J. Am. Chem. Soc., 1998, 120, 1732 CrossRef CAS.
- P. Kapferer, F. Sarabia and A. Vasella, Helv. Chim. Acta, 1999, 82, 645 CrossRef CAS.
- T. K. M. Shing, T. Y. Li. and S. H.-L. Kok, J. Org. Chem., 1999, 64, 1941 CrossRef CAS.
- K. Tatsuta, H. Mukai and M. Takahashi, J. Antibiot., 2000, 53, 430 Search PubMed.
- S. H.-L. Kok, C. C. Lee and T. K. M. Shing, J. Org. Chem., 2001, 66, 7184 CrossRef CAS.
- S. Ogawa and H. Tsunoda, Liebigs Ann. Chem., 1992, 637 Search PubMed.
- T. M. Tagmosa and M. Bols, Chem. Eur. J., 1997, 3, 453.
- S. Ogawa, H. Ito, T. Ogawa, S. Iwasaki and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 2319 CAS.
- T. Suami, S. Ogawa, K. Nakamoto and I. Kasahara, Carbohydr. Res., 1977, 58, 240 CrossRef CAS.
- S. Ogawa, I. Kasahara and T. Suami, Bull. Chem. Soc. Jpn., 1979, 52, 118 CAS.
- S. Ogawa, K. Nakamoto, M. Takahara, Y. Tanno, N. Chida and T. Suami, Bull. Chem. Soc. Jpn., 1979, 52, 1174 CAS.
- S. Ogawa, M. Suzuki and T. Tonegawa, Bull. Chem. Soc. Jpn., 1988, 61, 1824 CAS.
- S. Ogawa, Y. Iwasawa, T. Nose, T. Suami, S. Ohba, M. Ito and Y. Saito, J. Chem. Soc., Perkin Trans. 1, 1985, 903 RSC.
- S. Ogawa, N. Chida and T. Suami, Chem. Lett., 1980, 1559 CAS.
- S. Ogawa, M. Oya, T. Toyokuni, N. Chida and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 1441 CAS.
- S. Ogawa, M. Ara, T. Kondoh, M. Saitoh, R. Masuda, T. Toyokuni and T. Suami, Bull. Chem. Soc. Jpn., 1980, 53, 1121 CAS.
- S. Ogawa and T. Tonegawa, Carbohydr. Res., 1990, 204, 51 CrossRef CAS.
- J. L. Aceña, O. Arjona, R. F. de la Pradilla, J. Plumet and A. Viso, J. Org. Chem., 1994, 59, 6419 CrossRef CAS.
- G. Rassu, L. Auzzas, L. Pinna, F. Zanardi, L. Battistini and G. Casiraghi, Org. Lett., 1999, 1, 1213 CrossRef CAS.
- K. Afarinkia and F. Mahmood, Tetrahedron. Lett., 1998, 39, 7413 CrossRef CAS.
- K. Afarinkia and F. Mahmood, Tetrahedron, 1999, 55, 3129 CrossRef CAS.
- M. Yoshikawa, N. Murakami, Y. Yokokawa, Y. Inoue, Y. Kuroda and I. Kitagawa, Tetrahedron, 1994, 50, 9619 CrossRef CAS.
- T. Takahashi, H. Kotsubo, A. Iyobe, T. Namiki and T. Koizumi, J. Chem. Soc., Perkin Trans. 1, 1990, 3065 RSC.
- K. M. Shing and V. W.-F. Tai, J. Org. Chem., 1995, 60, 5332 CrossRef.
- Y. Kameda, K. Kawashima, M. Takeuchi, K. Ikeda, N. Asano and K. Matsui, Carbohydr. Res., 1997, 300, 259 CrossRef CAS.
- S. Horii, H. Fukase and Y. Kameda, Carbohydr. Res., 1985, 140, 185 CrossRef CAS.
- H. Fukase and S. Horii, J. Org. Chem., 1992, 57, 3642 CrossRef CAS.
- S. Horii, J. Takeda Res. Lab., 1993, 52, 1 Search PubMed.
- S. Ogawa, N. Ikeda, H. Takeda and Y. Nakagawa, Carbohydr. Res., 1988, 175, 294 CrossRef CAS.
- S. Ogawa, T. Ogawa, N. Chida, T. Toyokuni and T. Suami, Chem. Lett., 1982, 749 CAS.
- S. Ogawa and Y. Miyamoto, Chem. Lett., 1988, 889 CAS.
- S. Ogawa, T. Ogawa, T. Nose, T. Toyokuni, Y. Iwasawa and T. Suami, Chem. Lett., 1983, 921 CAS.
- S. Ogawa, T. Nose, T. Ogawa, T. Toyokuni, Y. Iwasawa and T. Suami, J. Chem. Soc., Perkin Trans. 1, 1985, 2369 RSC.
- Y. Miyamoto and S. Ogawa, J. Chem. Soc., Perkin Trans. 1, 1989, 1013 RSC.
- S. Ogawa, Y. Miyamoto and T. Nose, J. Chem. Soc., Perkin Trans. 1, 1988, 2675 RSC.
- S. Ogawa and Y. Miyamoto, J. Chem. Soc., Chem. Commun., 1987, 1843 RSC.
- Y. Miyamoto and S. Ogawa, J. Chem. Soc., Perkin Trans. 1, 1991, 2121 RSC.
- Y. Miyamoto and S. Ogawa, Carbohydr. Res., 1992, 223, 299 CrossRef CAS.
- S. Ogawa and Y. Shibata, J. Chem. Soc., Chem. Commun., 1988, 605 RSC.
- Y. Shibata and S. Ogawa, Carbohydr. Res., 1989, 189, 309 CrossRef CAS.
- S. Ogawa, Y. Iwasawa, T. Toyokuni and T. Suami, Chem. Lett., 1983, 337 CAS.
- S. Ogawa, Y. Iwasawa, T. Toyokuni and T. Suami, Carbohydr. Res., 1985, 141, 29 CrossRef CAS.
- S. Ogawa, H. Sugizaki, Y. Iwasawa and T. Suami, Carbohydr. Res., 1985, 140, 325 CrossRef CAS.
- T. Yamagishi, C. Uchida and S. Ogawa, Bioorg. Med. Chem. Lett., 1995, 5, 487 CrossRef CAS.
- S. Ogawa, H. Ito and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 2326 CAS.
- N. Sakairi and H. Kuzuhara, Tetrahedron Lett., 1982, 23, 5327 CrossRef CAS.
- S. Ogawa, Y. Iwasawa, T. Toyokuni and T. Suami, Chem. Lett., 1982, 1729 CAS.
- Y. Shibata, Y. Kosuge and S. Ogawa, Carbohydr. Res., 1990, 199, 37 CrossRef CAS.
- Y. Shibata, S. Ogawa and T. Suami, Carbohydr. Res., 1990, 200, 486 CrossRef CAS.
- H. Paulsen and W. Röben, Liebigs Ann. Chem., 1985, 974 Search PubMed.
- K. Tatsuta, M. Takahashi and N. Tanaka, J. Antibiot., 2000, 53, 88 Search PubMed.
- K. Tatsuta, M. Takahashi and N. Tanaka, Tetrahedron Lett., 1999, 40, 1929 CrossRef CAS.
- S.-B. Lee, K.-H. Park and J. F. Robyt, Carbohydr. Res., 2001, 331, 13 CrossRef CAS.
- J. C. McAuliffe, R. V. Stick and B. A. Stone, Tetrahedron Lett., 1996, 37, 2479 CrossRef CAS.
- N. Asano, Y. Kameda and K. Matsui, J. Antibiot., 1991, 44, 1406 Search PubMed.
- A. Hasegawa, T. Kobayashi, H. Hibino and M. Kiso, Agric. Biol. Chem., 1980, 44, 143 Search PubMed.
- S. Ogawa, K. Sato and Y. Miyamoto, J. Chem. Soc., Perkin Trans. 1, 1993, 691 RSC.
- S. Ogawa and M. Orihara, Carbohydr. Res., 1989, 189, 323 CrossRef CAS.
- S. Ogawa, M. Ashiura and C. Uchida, Carbohydr. Res., 1998, 370, 83 CrossRef.
- S. Ogawa, K. Nishi and Y. Shibata, Carbohydr. Res., 1990, 206, 352 CrossRef CAS.
- S. Ogawa and Y. Shibata, Carbohydr. Res., 1988, 176, 309 CrossRef CAS.
- C. Uchida, H. Kitahashi, T. Yamagishi, Y. Iwaisaki and S. Ogawa, J. Chem. Soc., Perkin Trans. 1, 1994, 2775 RSC.
- T. Toyokuni, S. Ogawa and T. Suami, Bull. Chem. Soc. Jpn., 1983, 56, 2999 CAS.
- S. Ogawa, Y. Shibata, Y. Kosuge, K. Yasuda, T. Mizukoshi and C. Uchida, J. Chem. Soc., Chem. Commun., 1990, 1387 RSC.
- Y. Shibata, Y. Kosuge, T. Mizukoshi and S. Ogawa, Carbohydr. Res., 1992, 228, 377 CrossRef CAS.
- S. Ogawa and D. Aso, Carbohydr. Res., 1993, 250, 177 CrossRef CAS.
- S. Ogawa and K. Nishi, Carbohydr. Res., 1992, 229, 117 CrossRef CAS.
- S. Ogawa, Methods Enzymol., 1994, 247, 128 Search PubMed.
- S. Ogawa and H. Tsunoda, Methods Enzymol., 1994, 247, 136 Search PubMed.
- H. Tsunoda and S. Ogawa, Liebigs Ann. Chem., 1994, 103 Search PubMed.
- H. Tsunoda, J.-i. Inokuchi, K. Yamagishi and S. Ogawa, Liebigs Ann., 1995, 279 Search PubMed.
- S. Ogawa, M. Ashiura, C. Uchida and S. Watanabe, Bioorg. Med. Chem. Lett., 1996, 6, 929 CrossRef CAS.
- S. Ogawa, Y. Kobayashi, K. Kabayama, M. Jimbo and J.-i. Inokuchi, Bioorg. Med. Chem., 1998, 6, 1955 CrossRef CAS.
- S. Ogawa, T. Mito, E. Taiji, M. Jimbo, K. Yamagishi and J.-i. Inokuchi, Bioorg. Med. Chem. Lett., 1997, 7, 1915 CrossRef CAS.
- Y. Kameda, N. Asano, O. Wakae and T. Iwasa, J. Antibiot., 1980, 33, 764 Search PubMed.
- T. Furumoto, Y. Kameda and K. Matsui, Chem. Pharm. Bull., 1992, 40, 1871 CAS.
- M. Hayashida, N. Sakairi and H. Kuzuhara, Carbohydr. Res., 1986, 158, C5 CrossRef CAS.
- M. Hayashida, N. Sakairi and H. Kuzuhara, Carbohydr. Res., 1989, 194, 233 CrossRef CAS.
- R. V. Stick, D. M. G. Tilbrook and S. J. Williams, Aust. J. Chem., 1999, 52, 895 CAS and references therein.
- H. Fukase, Yuki Gosei Kagaku Kyokaishi, 1997, 55, 920 Search PubMed.
- T. Matsuo, H. Odaka and H. Ikeda, Am. J. Clin. Nutr., 1992, 55, 314S Search PubMed.
- H. Odaka and H. Ikeda, J. Takeda Res. Lab., 1995, 54, 21 Search PubMed.
- Deutsch. Pat., DE 19821038 A1, 1999.
- R. J. Bernacki, M. J. Niedbala and W. Korytnyk, Cancer Metastasis Rev., 1985, 4, 81 Search PubMed.
- X. Xie, A.-S. Rivier, A. Zakrzewicz, M. Bernimoulin, X.-L. Zeng, H. P. Wessel, M. Schapira and O. Spertini, J. Biol. Chem., 2000, 275, 34818 CrossRef CAS.
- T. Takeuchi, H. Chimura, M. Hamada, H. Umezawa, O. Yoshioka, N. Oguchi, Y. Takahashi and A. Matsuda, J. Antibiot., 1975, 28, 737 Search PubMed.
- H. Chimura, H. Nakamura, T. Takita, T. Takeuchi, H. Umezawa, K. Kato, S. Saito, T. Tomisawa and Y. Iitaka, J. Antibiot., 1975, 28, 743 Search PubMed.
- K. Tatsuta, T. Tsuchiya, N. Mikami, S. Umezawa, H. Umezawa and H. Naganawa, J. Antibiot., 1974, 27, 579 Search PubMed.
- G. Bach, S. Breiding-Mack, S. Grabley, P. Hammann, K. Hütter, R. Thiericke, H. Uhr, J. Wink and A. Zeeck, Liebigs Ann. Chem., 1993, 241 Search PubMed.
- Y.-Q. Tang, C. Maul, R. Höfs, I. Sattler, S. Grabley, X.-Z. Feng, A. Zeeck and R. Thiericke, Eur. J. Org. Chem., 2000, 149 CrossRef CAS.
- R. Höfs, S. Schoppe, R. Thiericke and A. Zeeck, Eur. J. Org. Chem., 2000, 1887.
- D. E. Cane, C. T. Walsh and C. Khosla, Science, 1998, 282, 63 CrossRef CAS.
- Q. Xue, G. Ashley, C. R. Hutchinson and D. V. Santi, Proc. Natl. Acad. Sci. USA, 1999, 96, 11740 CrossRef CAS.
- J. Staunton, Curr. Opin. Chem. Biol., 1998, 2, 339 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |