Lewis N.
Mander
Research School of Chemistry, The Australian National University, Canberra, ACT, 0200, Australia. E-mail: mander@rsc.anu.edu.au
Received
(in Cambridge, UK)
29th August 2002
First published on 28th November 2002
Abstract
This review covers research into the chemistry and biology of the gibberellin family of plant bioregulators carried out in the author's laboratory over the past 20 years and has 231 references.
Lew Mander
Lew Mander grew up in New Zealand and completed his B.Sc. and M.Sc. (Hons) degrees at the University of Auckland (1957–1961). He moved to Australia in 1961 and undertook a Ph.D. on steroid synthesis and alkaloid structure determination at the University of Sydney under the supervision of C. W. Shoppee, E. Ritchie and W. C. Taylor. After 2 years of post-doctoral studies with R. E. Ireland on the total synthesis of diterpenes, initially at the University of Michigan and then at the California Institute of Technology, he returned to Australia in 1966 as a lecturer in the Department of Organic Chemistry at the University of Adelaide. In 1975 he was appointed to a senior fellowship in the Research School of Chemistry (RSC) at the Australian National University, and in 1980 to his current position as Professor of Chemistry. He also served as Dean of the RSC for the periods 1981–1986 and 1992–1995. He has been a Nuffield Fellow at Cambridge University (1972)
(with A. R. Battersby), a Fulbright Senior Scholar at the California Institute of Technology (1977) and at Harvard University (1986)
(with D. A. Evans on both occasions) and an Eminent Scientist of RIKEN (1995–6, Saitama, Japan). He was elected to the Fellowship of the Australian Academy of Science in 1983 and to the Fellowship of the Royal Society in 1990. he is also an Honorary Fellow of the Royal Society of New Zealand and a Distinguished Alumnus Professor of the University of Auckland. His research interests are concerned primarily with methodology and strategies for the synthesis of complex natural products which have interesting biological properties. These activities embrace a major interest in the gibberellin family of plant bioregulators.
1 Introduction
The gibberellins (“GAs”) presently form a group of ∼130 highly functionalized diterpenoids,1–5 which are distributed widely throughout the plant Kingdom6 where they play an important role in plant growth and development. They are also produced by a number of microorganisms6 and gibberellic acid (1) is produced commercially in tonne quantities by fermentation of the fungus Gibberella fujikuroi. The quest for an understanding of the biology and biochemistry of GAs has been greatly advanced by the consequent ease of availability of this molecule.
The history of GA research7 began in the early part of the nineteenth century with reports in 1828 of a disease of rice plants that resulted in excessive elongation of the plant stem. Hori described in 1898 how the disease could be induced in healthy plants by inoculation with the “bakanae fungus”, Gibberella fujikuroi, the “perfect”, i.e. sexual, stage of Fusarium moniloforme. The first indication that a substance produced by the fungus was responsible for the effect was provided by Sawada in 1912 and confirmed by Kurosawa in 1926. Eventually, crystalline material (later shown to be a mixture of three similar compounds) was isolated in 1938 by Yabuta and Sumiki.8 This material, when applied to plants, resulted in spectacular promotion of growth, and following World War II, I.C.I. set up a screening program to search for a gibberellin-producing strain among the I.C.I. collection of the Fusaria fungus. The strain selected for fermentation studies produced mainly one gibberellin, gibberellic acid (1). Subsequently, a further 24 gibberellins based on the same ent-norgibberellane skeleton 2 or the full C20ent-gibberellane skeleton 3 have been isolated from the Gibberella fungus.
The major features of the gibberellic acid molecule had been elucidated by 1956,9 and the complete structure proposed by 1958.10 The first unequivocal assignment of structure was not achieved until 1962, however, when it could be inferred from an X-ray crystallographic study on a rearranged derivative.11 Confirmation was obtained in subsequent studies.12,13 With the availability of reasonable quantities, there was a virtual explosion in the number of studies on plant responses to the application of this compound. By 1956, groups began to report the isolation of GAs from higher plants,14,15 in which, as we now know, GAs are essential for growth and development. Of the 130 presently known naturally occurring GAs, 105 have been found exclusively in higher plants (including angiosperms, gymnosperms and ferns), 11 in the fungus only, and the rest from both sources (see Table S1† and Refs. 16–106).
Rather than assigning trivial names to naturally occurring GAs, a number has been assigned to each variant and a registry coordinated, until recently, by MacMillan and Takahashi.107 The database is now maintained by Hedden and Kamiya; for details see http://www.plant-hormones.bbsrc.ac.uk/gainplants/gibberellin_nomenclature.htm. Thus, gibberellic acid, for example, is identified as GA3. As summarised in Fig. 1, more than a third are based on the C20ent-gibberellane skeleton 3 with the variations of structure arising from different oxidation levels and hydroxylation patterns. With the sole exception of GA11, the rest are based on the 20-nor-ent-gibberellane structure and incorporate a 19,10-γ-lactone function as in GA9
(4). Six of these compounds (isolated from fern prothallia and developing apple seeds) possess an additional bond between C-9 and C-15 as in GA103
(5). Catabolic products from several GAs have been identified, namely a number of 16,17-dihydro-16,17-diols, and a handful of 1(10)-en-2-one derivatives resulting, presumably, from oxidation to 2-oxo-GAs followed by β-elimination of the lactone function.108,109 A large number of glucose conjugates are also known.2
Fig. 1
2 Bioactivity and applications
GAs appear to be involved in every aspect of plant growth and development, but their most typical (and spectacular) property is the enhancement of stem growth. It is now recognised that gibberellins were concerned in the biogenetic differences between the tall and dwarf peas used by Mendel for his classical experiments in inheritance. The phenomenon of bolting in rosette plants110
(i.e. the explosive growth which precedes flowering in plants like spinach)111 is caused naturally by endogenous GAs, while dwarfism due to a deficiency in natural GAs may be reversed by the application of exogenous GAs. The vigorous shoot growth obtained with maize hybrids has been shown to be due to the production of higher than normal levels of GAs.112 Flowering is also stimulated by GAs, although in some fruit trees, flowering may be reduced in the year following application. GAs may modify the sex expression of flowers, induce the parthenocarpic development of fruit and delay senescence. They obviate the need for exposure to red light in the germination of seeds and spores, and the need for vernalisation (winter chilling) in the growth of bulbs and tubers. They are associated with the breaking of winter dormancy and stimulate the formation of hydrolytic enzymes in germinating cereal grain.
There are several commercially valuable applications:113,114 most seedless table grapes are now grown with the application of GA3. The rind of citrus fruit typically softens at maturity, and is subject to injury by pests and environmental factors which adversely affect the appearance of otherwise marketable fruit. By inhibiting senescence, GAs maintain the rind in better condition. Control of russet, a “scabby” skin disorder in apples, especially in “golden delicious”, may be achieved by the application of GAs. A variety of ornamental plants can be induced to flower either earlier than usual, or in off-seasons. Sporadic flowering in some plants is often a problem with plant breeders, but may be ameliorated with GA applications. In the brewing of beer, malt production is a costly, time consuming step; 2–3 days may be saved by the addition of 25–500 µg of GA3 for each kg of barley.
3 Structure determination
Progress in gibberellin research in higher plants would have occurred very much more slowly without the original isolation in relatively large quantities of GAs from G. fujikuroi. Some of the richer plant sources afford milligram quantities, but concentrations in the order of µg kg−1 ranging down to ng kg−1 are more usual, and although it is still possible to accumulate sufficient material for full spectroscopic analysis, the effort can become heroic, e.g. 38 mg of GA32
(6) was isolated from one tonne of unripe peaches (it took ten students one month just to separate the seeds from the fruit),115 while 14 mg of GA19
(7) was obtained32 from 44 tonnes of bamboo shoots! With more modest quantities of plant material, however, it is only with the knowledge derived from chemical116 and metabolic117,118 studies on the fungal GAs and the availability of synthetic substrates from this source that structure determination becomes reasonably practical. Even at the nanogram level, it is often possible to arrive at quite a good estimate of molecular structure by gas-liquid chromatography–mass spectrometry (GC–MS) and then confirm tentative assignments by synthesis from one of the fungal GAs. When the guess is wrong, useful information is still gained and the deduction of the correct structure facilitated. Most importantly, comparisons may be made with an extensive database of GC–MS information held at IACR-Long Ashton, UK, a large part of which has been published in an atlas by Gaskin and MacMillan.119
4 Biosynthesis of GAs
The biosynthesis of GAs has received a great deal of attention and has been the subject of several reviews.2,120 It was established at an early stage that GAs were derived from kaurenoic acid 8 by hydroxylation at C-7 followed by ring contraction with the extrusion of C-7 to form the GA prototype, GA12aldehyde9. There are then several subsequent parallel pathways depending on the organism concerned, although the essential steps are the same, differing mainly by the position, stage, and degree of hydroxylation. Thus, in a typical sequence (Scheme 1), the 20-methyl group in GA1210 is progressively oxidised to a formyl group. This function may be oxidised further to carboxy, but this step leads to a biosynthetic dead end. On the continuing pathway, the formyl function is lost with formation of a γ-lactone bridge, affording GA94 in an as yet incompletely defined oxidative process, although it is known that the formyl group is oxidised eventually to carbon dioxide and that both oxygen atoms of the lactone function originate in the carboxy attached to C-4. The other established biosynthetic routes from GA12 involve early hydroxylation at C-3 or, more commonly, C-13.
Scheme 1
It has been long established that the biosynthesis of GA12 in both plants and G. fujikuroi is mediated by P-450 enzymes, and that subsequent steps are effected in higher plants by 2-oxoglutarate and iron-dependent non-haeme dioxygenases.121–123 However, it has been recently discovered that in the fungus, these latter steps are effected by P-450 enzymes. Thus, GA3 and other GAs are formed by multiple pathways that are entirely independent, but which converge to common GAs. An excellent summary of our current knowledge of GA biosynthesis in G. fujikuroi and in plants, as exemplified by Arabadopsis, has been provided by Hedden et al.124
The combination of a complex molecular structure with potent bioactivity made GA3
(1) a prime target for total synthesis in the mid-60's. By one estimate,125 25 groups had taken up the challenge. In an attempt to convince a University to employ him, and granting agencies to provide him with funding, the author also proposed to carry out a synthesis of this intriguing molecule. These studies, inter alia, culminated in two syntheses of gibberellic acid,126–128 several accounts of which have appeared elsewhere.129–132 These syntheses did little to increase the world's supply of these compounds, nor enhance our understanding of their biology, although we did establish that the racemate of GA1
(not surprisingly) possessed half the growth promoting potency of the naturally occurring epimer.126 A number of synthetic methods emerged from our endeavours, however, that have been of considerable value in building and manipulating GAs and other natural products. The methodology includes Birch reductions,133,134diazoketone-based chemistry,135 refinements to the Wittig reaction,136 unusual aldol and Michael reactions126 and the use of cyanoformates for the kinetically controlled C-acylation of enolate anions.137,138
A further case in point is the modification of the Takai–Nozaki methylenation of ketones developed by my post-doctoral colleague, Luciano Lombardo.139 As a prelude to extending our strategy for preparing C19 GAs to the synthesis of the C20 type,140 we needed to convert ketone11 into aldehyde13, but reactions with phosphorous or sulfur derived ylides resulted in epimerisation of the adjacent methine centre. Direct application of the Takai–Nozaki method,141 which involves adding zinc, TiCl4 and dibromomethane to the substrate, rapidly destroyed our intermediate. However, by allowing the reagent mixture to stir at 4 °C for 3 days (1–2 days may give better results), and then adding the resulting suspension to the substrate, a 90% yield of 12 was obtained (Scheme 2). The method has now been applied successfully to a wide variety of ca. 200 substrates by our group and others, but has been especially important for the preparation of deuterated GAs. Because of proton–deuteron exchange, the Wittig reaction between d2-methylene phosphorane and ketones may result in a gross mixture of d0–d4 methylenated products. The Lombardo reaction with CD2Br2 and 17-norgibberellin-16-ones is very much more specific (>96% d2) and ca. 40 different GAs have been labelled in this way. They have been invaluable for biosynthetic studies, and as standards in the determination of GA levels in plants and their various organelles.
For the author, by far the most important consequence of publishing our GA total syntheses was the response by numerous biologists who were struggling to gain access to several rare, but important GAs, preferably with incorporated isotopes (2H, 3H, 13C and 14C). New GAs were being isolated, but in miniscule quantities, and so synthesis was ultimately the only feasible option to establish their structures (after making tentative assignments from mass spectrometry and chromatographic behaviour). After some 20 years of responding to numerous requests, we have now established methodology that allows the introduction of functionality at every possible location in the GA molecule and access to any one of the 130 presently known GAs.
Most of our efforts have been focussed on (a) the conversion of C19 to C20 GAs; (b) functionalisation of the relatively inaccessible parts of the GA skeleton, notably C-11, C-12, C-14 and C-18; and (c) combinations of (a) and (b). Modification of the skeleton has also been pursued, especially for the conversion of C19 GAs into C20 GAs and kaurenoids. As noted earlier, we are fortunate in having a cheap and limitless supply of GA3, so this molecule has been the starting point for most of our studies. There has been a certain notoriety associated with the chemistry of this molecule. Corey, in his introduction125 to his total synthesis of this compound, noted “a singularly diabolical placement and density of functionality…”. With the right kind of handling, however, GA3 is well behaved. Even so, it is unstable in non-buffered aqueous solvents142 and rearrangement may occur in either or both of the A- and D-rings when exposed to acid.143,144 With 3-hydroxy GAs, bases may induce epimerisation at C-3 (through a retro-aldol mechanism)126,145,146 and in the case of the allylic systems represented by GA3 methyl ester 14 and its 13-desoxy analogue 15, dilute NaOH induces rapid isomerisation to their isolactones 16 and 17, respectively (Scheme 3).147,148 Nevertheless, these transformations may be put to good use in the preparation, inter alia, of 2-hydroxy and 9,15-cyclo GAs (see below).
Scheme 3
6.1 Dismantling and reconstituting the A-ring functionality in GA3
When converting GA3 into other GAs, one of the first problems to be encountered is the task of discriminating between the two alkene bonds. Fortunately, because of the flanking oxygen functions, the A-ring double bond is quite electron deficient and unreactive towards many electrophiles. Selective ozonolysis149 or epoxidation150 of the 16-ene function, for instance, is feasible, allowing the A-ring double bond to be reduced, and then the 16-ene function to be reconstituted as desired. Direct and selective hydrogenation of the A-ring double bond is also possible with amine-deactivated Pd catalysts.151 GA1 methyl ester (18) may be prepared in this way, but the major products are formed by hydrogenolysis to afford Δ1(9)-ene 4-carboxylic acids such as 19
(Scheme 4a). This type of GA, however, may be used in a variety of ways to prepare C20 GAs and to introduce functionality into the C-ring, as described in subsequent sections of this review. Reduction with lithium in liquid ammonia also gives access to this type of GA, e.g.20
→
21
(Scheme 4b), but with loss of the 3-substituent.152 If removal of the 13-oxy substituent is also required, then this outcome may be achieved by performing the reduction on the 3,13-bismesylate 22
(Scheme 4c), thereby furnishing 23.153
Scheme 4
An alternative approach to the removal of the A-ring double bond is the 1,4-addition of metal hydrides to 1-en-3-one derivatives, which may be achieved with combinations of NaBH4 with lithium154 or copper salts,155 or with L- or K-selectrides.156 If reduction at C-3 also occurs, the products have the 3α-configuration, which is rare among naturally occurring GAs. The 3β- configuration may be obtained, however, by SN2-displacement of a 3α-mesylate substituent with CsOAc,157 or by K-selectride reduction of the 3-one function, provided that the GA possesses a free 7-carboxy group.156 Reconstitution of the 1-en-3β-ol array is less straightforward, but has been achieved by a variety of approaches as summarised in Scheme 5. The original method, established by Corey, involved hydrolysis of the iso-lactone function in 16, in situ iodolactonisation, then reductive elimination of the vicinal 1,2-iodohydrin (via the 2-trifluoroacetate).158 A much more direct approach, accidentally discovered by Furber, is the treatment of the 1(10)-ene 19,2-lactones with Me2BBr or Ph2BBr.159 This process, given the ease with which the 19,2-lactones are formed from the 19–10-lactones with base or acid in the first place, is clearly contrathermodynamic and so we presume that the equilibrium is driven by selective complexation between the reagent and the isomeric lactones. As part of our approach to the synthesis of GA3, we had envisaged the stereoelectronically controlled opening of a 2β,3β-epoxide by HBr followed by elimination of HBr to form the Δ1-ene function.126 Employing peroxycarboxylic acids for the epoxidation, however, required forcing conditions and resulted in a 2 ∶ 3 mixture of β to α isomers. This difficulty has recently been resolved by the use of the dioxirane derived from trifluoroacetone, but at the time we employed a benzylacetal as a surrogate for the epoxide as shown.
Scheme 5
6.2 Introduction of the 9(11)-ene functionality
The methyl ester of GA9
(4) was isolated by the Takahashi group160 from prothallia of the fern Lygodium japonicum and shown to induce antheridia formation at 10−10 molar concentrations. This was the first example of bioactivity associated with a gibberellin ester, rather than the free acid. However, the amount of compound present in the culture medium was too low to account for the total activity of the extract and the presence of a second, more potent, substance was suspected. Careful fractionation of 12 litres of culture filtrate yielded 40 nanograms of a second antheridiogen and mass spectrometry established a molecular formula which corresponded to two fewer hydrogen atoms than those possessed by GA9 methyl ester, the incorporation of an additional double bond. The 1,2- and 2,3-didehydro derivatives were prepared, but neither of these corresponded to the natural material. The next candidate, 9(11)-ene 28 was synthesised, and proved to be identical with the natural antheridiogen72 and to have extraordinary potent bioactivity:161 the synthetic material induced antheridia in prothalli of L. japonicum at dilutions approaching the femtomolar level (32% at 10−14 M; 98% at 10−13 M). In addition, inhibition of archegonia formation was observed at picomolar concentrations. The key sequence in the preparation of 28
(Scheme 6) involved iodolactonisation of alkenoic acid 25 followed by DBU-induced elimination of the iodo group to form ketone27. Alkenoic acids such as 25 may be prepared from gibberellenic acid142 methyl ester 24 or from 2,19-lactones like 17.162 Since the isolation of 28, four further Δ9(11)-GAs have been isolated, either from Lygodium species or from apple seeds, and their structures confirmed by synthesis.83,89,96,101
Scheme 6
6.3 Introduction of the 11β-hydroxy
The 11-hydroxy gibberellin, GA35
(30), was isolated from loquat fruit many years ago, as well as several other unidentified GAs, but in insufficient amounts to determine their structures. Nevertheless, it appeared likely that they too were hydroxylated at C-11. With access to Δ9(11)-GAs established, hydroxylation at C-11 was expected to be routine, but because of steric hindrance, the 9(11)-ene bond is difficult to functionalise. Our solution to the problem163 was the addition of diborane to a 9(11),16-diene followed by oxidation to an 11β,17-diol, a process that was precedented by a similar addition to an equivalent kaurene derived diene.164 An important aspect of this approach is the re-establishment of the normal 9β-stereochemistry, although reconstitution of the 16-ene functionality was somewhat tedious. If the 17-hydroxy group is simply converted to a leaving group in anticipation of an elimination step, nucleophilic displacement by the 11β-hydroxy group occurs with ether formation. Hence, it is vital to mask the 11-hydroxy group first, which then requires a protection–deprotection cycle to temporarily mask the 17-hydroxy group. Two further 11β-hydroxy GA methyl esters77 were prepared in this way as illustrated for GA35 methyl ester (29)
(Scheme 7).
Scheme 7
6.4 12-Hydroxylation of GAs
The 12-hydroxylation of GAs has been effected via allylic bromination of Δ9(11)-GAs,101 but there is a more direct method,165 that may be carried out without disturbing the A-ring functionality. It is based on the trans-annular oxidation of 16α-bromo-17-ols with Pb(OAc)4 or PhI(OAc)2, which induce hydrogen abstraction from C-12 and ether formation. The C-ring in GAs normally adopts a boat-like conformation so the 12β-proton is at the optimal distance for reaction with the intermediate 17-oxy radical. Subsequent reaction with zinc metal induces reductive elimination to reveal a 12β-hydroxy with re-establishment of the 16-ene function (Scheme 8). Approximately 30 GAs are functionalised at C-12, and so this protocol has been invaluable for gaining access to a large number of new GAs.81,84,166
Scheme 8
To convert the 12β-alcohols into their 12α-epimers is not simple. Oxidation followed by borohydride reduction simply returns the 12β-epimer. Mitsunobu inversion fails, but intramolecular displacement of a 12β-mesylate by the 13-acetoxy group (Scheme 9)167 is effective. Alternatively, access to the 12α-stereochemistry may be achieved by borohydride reduction of the 13-hydroxy-12-one moiety in the presence of a chelating metal ion e.g. Zn2+. Chelation induces flattening of the C-ring and opens up the top face to nucleophilic attack (Scheme 10).166
Scheme 9
Scheme 10
6.5 Oxygenation at C-17
Hydroboration of the 16-ene bond in GAs lacking a 13-substituent provides simple and efficient access to 17-hydroxy GAs. However, 13-hydroxy GAs, with and without protection, are much less reactive and yields are often poor. The bromohydrin 35 required for the transannular oxidation outlined in the preceding Section was therefore elaborated by indirect pathways (Scheme 11). Thus, hydroxylation at C-15 was effected with SeO2–t-BuOOH to give 32 and then oxidation with pyridinium chlorochromate afforded a mixture of aldehyde33 and its 15,16-epoxide. Reduction of the mixture with Cr(II)Cl2 then afforded aldehyde34 which could be brominated at C-16 prior to borohydride reduction.81 In an alternative approach, epoxide36 was converted to aldehyde34 by treatment with Cp2Ti(III)Cl (Scheme 12).166 Two equivalents of reagent were required for complete reaction which, we assume, proceeds via free radical 37 and elimination of hydride from a subsequent titanium complex. In principle, the aldehyde could be formed from the epoxide by Lewis acid-catalysis, but such treatment engenders a Wagner–Meewein rearrangement with migration of C-12 to C-16. In very recent studies we have revisited the hydroboration of 13-substituted GAs and established that satisfactory yields can be obtained with 13-acetates168 and free 13-ols.105
Scheme 11
Scheme 12
6.6 15-Hydroxylation of GAs – synthesis of GA32
GA32
(6), isolated from the immature seeds of peaches, apricots and other Prunus species is the most biologically potent of the 130 gibberellins obtained to date from natural sources. It is, unfortunately, one of the least accessible.46 It was of special interest to us to attempt a synthesis since, not only had it been isolated and its structure determined in a sister department at the University of Adelaide,45 it was also an important GA to colleagues at the CSIRO Division of Plant Science in connection with flowering studies on Lolium temulentum.169,170 A possible route was first checked out on methyl ester 38, even though we knew that hydrolysis to a free acid at the end of the sequence was likely to prove very difficult. Beginning with the protected GA87 derivative 38
(Scheme 13), we applied the MacMillan procedure for the introduction of the 15β-ol function,67,69i.e.allylic oxidation with selenium dioxide followed by oxidation to the enone39
(for this conversion we found Dess–Martin reagent to be more reliable than the Swern procedure reported by MacMillan), then reduction with zinc and acetic acid. However, mainly 1,4-reduction occurred and only a very modest yield (20%) of the target 15β-ol 40 was obtained, an outcome that was similar to that observed for other polyhydroxylated GAs.74 We therefore explored the option of temporarily masking the 17-methylene group during the reduction of the C-15 carbonyl function. Conjugate addition of various nucleophiles to 39 was effected without difficulty, but the epoxide41 proved to be the most useful intermediate; hydride reduction followed by acetylation, then deoxygenation171 gave a good yield of 42. We then duplicated the sequence with the more labile 7-methoxymethyl (“MOM”) ester analogue of 38, but could not hydrolyse the sterically hindered 15-acetate function without isomerisation of the A-ring allylic functionality. We decided, therefore, to test the possibility that the free 7-carboxy group might be enlisted in directing172 the reduction of the 15-oxo function. In the event, hydrolysis with trifluoroacetic acid of the MOM ester corresponding to 39 afforded acid 43, and then treatment with either borane–dimethyl sulfide complex or with sodium triacetoxyborohydride173 afforded triacetate 44
(Scheme 14). This product could be hydrolysed under sufficiently mild conditions so as to afford a reasonable yield of GA326. The assisted reduction by a carboxy group is unusual and it is of interest that with the borane complex, reduction of the ketone carbonyl group occurs in preference to reduction of the carboxy function.174
Scheme 13
Scheme 14
6.7 14-Hydroxylation of GAs
Although a 14-hydroxylated GA has yet to be identified from natural sources, the mass spectra of a number of recently isolated GAs appeared to be consistent with a 13,14-dihydroxylation pattern. Accordingly, we sought to establish a general protocol for the preparation of such GAs with a view to providing a set of reference structures. Hydroxylation of C-14 had been achieved by means of an ingenious route175 involving a double rearrangement of the C/D-ring system, as outlined in Scheme 15, but did not appear suitable for retention of the 13-hydroxy group.
Scheme 15
Our approach, which is illustrated by the preparation of 14β-hydroxy-GA1 methyl ester (49)
(Scheme 16),176 broadly followed the strategy encompassed by Scheme 15, but acyloin rearrangements were used to achieve both the initial “stereochemical inversion” of the D-ring and the re-establishment of the normal skeleton. As noted above, the C-ring in GAs tends to adopt a boat-like conformation, but migration of C-12 to C-16 allows the C-ring to take up the energetically favoured chair conformation. It appeared likely, therefore, that on exposure to base, ketol 45 would rearrange to 46. A highly characteristic feature of C19-GA 1H-NMRspectra is an isolated AB spin system derived from H-5 and H-6 in the range 2.5–3.5 ppm, with a coupling constant of J
=
∼10 Hz [e.g. for 45, J
= 10.6 Hz]. Because of a smaller torsion angle between H–C-5 and H–C-6 in 8α,13α-GAs, however, J is reduced to ∼7 Hz, so any rearrangement to this structural type is readily evident. Thus, the rearrangement to 46 was confirmed by the observation of an AB system at δ 3.24 and 2.65, (J
= 6.7 Hz) for H-5 and H-6, respectively. Hydroxylation at C-15 was effected by treatment of the derived enol TBDMS ether with dimethyldioxirane and then treatment of the product with sodium methoxide resulted in hydrolysis of the 13-acetate function and rearrangement to dihydroxy ketone 48, as indicated by the vicinal coupling (J
= 9.4 Hz) of H-5 and H-6, showing that the regular GA stereochemistry had been re-established. Since the equilibrium between ketols 45 and 46 had strongly favoured 46, the high yield of 48 came as an agreeable surprise. Presumably, the alignment of the dipoles associated with the hydroxy and carbonyl groups in ring-D of 47 destabilises this isomer. Ab initio molecular orbital calculations indicate that the cisoid conformer of glyco-aldehyde, for example, is of significantly higher energy (ΔE
= 24 kJ) than the transoid form.177
Scheme 16
6.8 18-Hydroxylation of GAs
GAs in which the 18-methyl group has undergone oxidation have been isolated from immature seeds of sword bean (Canavalia gladiata), e.g. GA21
(50)35 and GA22
(51),36 and from germinating barley grain (Hordeum vulgare), e.g. 18-hydroxy-GA4
(52).70 For this last example, the structure was determined by converting 7β,18-dihydroxy kaurenolide 53 into 18-hydroxy GA1255 by means of a pinacolic rearrangement of the 6α-hydroxy tosylate derived from lactone54
(Scheme 17) and then carrying out the metabolic transformation of the resulting material to a series of 18-hydroxy C19 GAs with the fungus, Gibberella fujikuroi
(B1-41a mutant).178 In order to confirm these assignments and, more importantly, provide sufficient quantities of this type of GA for more extensive biological studies, we have developed a more general procedure for the synthesis of these compounds.179
Scheme 17
The synthesis of 18-hydroxy-GA4
(2) is outlined in Scheme 18 and pivots on the tandem transformation 56
→
57 which is based on the well established aldol process that has been shown to be quite general for forming the C-3–C-4 bond of both C19126 and C20140gibberellins. Hydroxide ion itself failed to add, but several primary alcohols did so, including methyl, allyl, benzyl, trichloroethyl and propargyl. Treatment of aldehyde56 with allyl alcohol and DBU afforded a 2 ∶ 3 mixture of 3α and 3β-OH allyl ethers 57 in good yield. Following separation, treatment of the desired 3β-OH allyl ether with RhCl(PPh3)3 and DABCO, followed by acidic workup resulted in liberation of the free hydroxy group at C-18, thereby affording the methyl ester of the target diol52.
Scheme 18
7 Conversion of C19 GAs to C20 GAs
Approximately one third of naturally occurring GAs possess the full C20 skeleton, but only one, GA13
(58), is available in reasonable quantities. It has therefore been a useful substrate for making other C20 GAs, but lacks the 13-hydroxy that is a characteristic feature of many plant GAs, especially the biosynthetic intermediates, GA53
(59), GA44
(60) and GA19
(7) which have been obtained in only minute amounts from natural sources. The only readily available 13-hydroxylated gibberellin that might serve as a synthetic precursor to these derivatives is the fungal GA, gibberellic acid (1). The introduction of the 10α-substituent into a compound of this type is not trivial, however, given the sterically congested nature of this position. Moreover, to obtain the correct stereochemistry at C-10, reagents must approach the more hindered concave α-face of the molecule. Previous work by Beale, for example, showed that conjugate addition of Li2Cu3Me5 to gibberell-1(10)-en-2-one dimethyl ester 61 resulted only in the production of the 10-epi-GA53 derivative 62
(Scheme 19).180 It was apparent, therefore, that an intramolecular approach was required and an effective solution is outlined in the next Section.
Scheme 19
7.1 Synthesis of the 13-hydroxy C20 GAs: GA19, GA44 and GA53
GA3 methyl ester was partly dismantled by means of a Li–NH3 reduction (c.f.Scheme 4b) and the resulting acid converted into diazo ketone63
(Scheme 20), treatment of which with a copper catalyst, then afforded cyclopropyl ketone64.152 A second Li–NH3reduction resulted in selective fission of the C-1–C-20 bond (better overlap with the π-orbitals of the C-19 carbonyl group) and then the potassium enolate of the resulting ketone65 cleaved with molecular oxygen to afford 66.181 To complete the synthesis of GA19
(7), all that was required was alkaline hydrolysis of the 7-methyl ester function and removal of the methoxymethyl (MOM) group from the 13-hydroxy. Preparation of GA44
(60) was easily achieved by reduction with borohydride, but access to GA53
(59) required rather more heroic efforts.182
Scheme 20
Of all the possible options for the deoxygenation of the 10α-formyl group that we explored, Wolff–Kishner methodology, appeared to be the most promising. Application of the procedure developed for hindered ketones by Barton et al.183 to the diacid 67, afforded the GA53 derivative 68 mixed with the isomeric 15-ene 69
(Scheme 21). Better yields were obtained at higher temperatures, but at the expense of greater alkene bond migration. Although the yield of the Wolff–Kishner reduction is modest, the method is general and has allowed comprehensive access to 10α-Me C20 GAs for the first time.
Scheme 21
7.2 Preparation of 2β-hydroxy C20 GAs – synthesis of GA97, GA98, GA99 and GA110
Several 2β-hydroxy C20gibberellins had been tentatively identified from a number of plant sources, including spinach, tomato, barley, and maize. In order to gain access to workable amounts of these gibberellins, to confirm their structural identity, and to explore their biosynthetic origins, we adapted the methodology described in the previous Section to the synthesis of 2β-hydroxy GA19 and its analogues. This aim proved to be deceptively difficult. We proposed to make the precursor 71 to diazoketone 72 from lactone70, which we expected to be readily prepared by deoxygenation of the known lactone16
(Scheme 22). This last compound was more conveniently prepared by brief dissolution of GA3 methyl ester (14) in trifluoroacetic acid (TFA), a procedure that is especially preferable on a large scale. However, application of the well-established Barton–McComie deoxygenation184 to this substrate, very much to our surprise, afforded the bislactone 73 by means of a complex free radical cyclisation sequence (Scheme 23).185 To circumvent this problem, GA3 methyl ester was treated with LiOtBu to effect epimerisation at C-3 (i.e. to the equatorial 3α-isomer) and the product treated with TFA to afford the 3α-epimer of 16 that could then be deoxygenated smoothly.
Scheme 22
Scheme 23
After hydrolysis and protection of the resulting carboxylic acid, the 2β-stereochemistry was established by means of an oxidation–reduction cycle using Luche conditions186 for the reduction to ensure that approach by the borohydride was blocked on the upper face by complexation with the cerium(III) cation, thereby ensuring approach to the lower face of the enone. After protection of the resulting 2β-hydroxy and formation of diazoketone 72
(R = MOM), the cyclopropanation process was initiated. Not surprisingly, this was complicated by the presence of the 2β-oxy function, and in addition to the desired cyclopropyl ketone 77, products from CH insertion (ketone76) as well as hydride transfer were formed (Scheme 24). The transfer of hydride implicit in the formation of 74 has precedent in the work of Doyle187 and Lee188 and has been observed by us with a number of other substrates where stabilisation of an incipient cation by neighbouring functionality could occur.189 A continuation of the synthesis as described in Section 7.1, led to the 2β,13-dihydroxy C20 GAs: GA97, GA98, and GA99.190,90 As well, C-13 deoxygenation of GA97 afforded 2β-hydroxy-GA12
(GA110) which has been found in spinach leaves and oil palm inflorescences.97
Scheme 24
7.3 Preparation of 13,15β-dihydroxy C20 GAs: synthesis of GA100, GA101 and GA102
Thirteen 15β-hydroxygibberellins have been isolated from seeds of the sunflower (Helianthus annuus L.) and their structures determined. Three of these gibberellins, GA6478, GA6580 and GA6682, were identified as C20 derivatives and their structures established by metabolic transformation of 15β-hydroxykaur-16-en-19-oic acid by cultures of Gibberella fujikuroi
(B1-41a mutant).68 The putative structures, 15β-hydroxy GA53
(79), 15β-hydroxy GA44
(81), 15β-hydroxy-GA19
(83), and 15β-hydroxy GA17
(84), were assigned to four further C-20 gibberellins, but attempts to confirm these structures by similar metabolism of 13,15β-dihydroxykaur-16-en-19-oic acid were unsuccessful. We therefore undertook the syntheses of the methyl esters of 79, 81, and 83 from the now readily available GA19 derivative 66, following the protocol developed by the MacMillan group, although with some minor modifications (Scheme 25).91 Thus, after methylation of 66 to give 85, allylic oxidation with selenium dioxide and tert-butyl hydroperoxide under sonication readily afforded the 15α-hydroxy derivative. Oxidation to the enone86 by the Dess–Martin periodinane and reduction with Zn in acetic acid gave an excellent yield of 87. Although the yield is often poor when applied to GAs containing 13-oxygen substituents,74enone86 was reduced to the desired 15β-hydroxy compound 87 in 91% yield. Transformation into the target compounds was then effected as described earlier (cf. Section 6.5).
Scheme 25
7.4 Preparation of 12-hydroxy C20 GAs: synthesis of GA111, GA112, GA113, GA114, GA115, GA116, GA123, GA124, GA125, GA127, GA128 and GA129
The 12β-hydroxy C20-gibberellins88, 90 and 92 and their 12α-epimers were believed to occur in a number of cruciferous plants, while the 12β,13-dihydroxy C20 GAs 89, 91 and 93 had all been tentatively identified as endogenous gibberellins in strawberries. The latter isolations were of particular interest from a biosynthetic perspective, given the occurrence of several 12α,13-dihydroxy C19-GAs in the same plant. With the availability of the epimeric 12-alcohols 94 and 95
(Section 6.4), it appeared to be a relatively simple, although lengthy task to confirm the putative assignments by preparing these 12-hydroxy C20 GAs, using the Birch reduction on the derived trismethoxymethyl ethers (Section 6.1) to dismantle the A-ring, and then applying the standard cyclopropanation methodology (Section 7.1) as summarised in Scheme 26.191Diazoketone96, however, failed to react under conditions (copper–bronze, reflux in cyclohexane–THF) that had induced highly efficient intramolecular cyclopropanation in the related system lacking the 12β-methoxymethyl ether function, while catalysis by Cu(acac)2, the preferred catalyst for the 2β,13-bismethoxymethyl analogue, afforded only a low yield of the desired cyclopropyl ketone 98, the main product arising from insertion into the 2α-CH bond. Fortunately, use of bis(N-tert-butylsalicylaldiminato)copper(II)192 gave a reasonable yield and subsequent transformation through to the 12β-methoxymethoxy analogues of GA19, GA44 and GA53 proceeded without incident. Deprotection of the 12β-hydroxy in the GA19 analogue could be effected under the standard conditions of reflux in methanol over Dowex resin, developed earlier193 for such a purpose, but under these conditions, both the GA44 and GA53 analogues underwent a Wagner–Meerwein rearrangement. However, we have subsequently gained access to 12β-hydroxy-GA44 by an alternative route, whereby the cyclopropanation sequence preceded introduction of the 12β-hydroxy.167 When deoxygenation of C-20 in 12β-hydroxy GA19 failed to afford the GA53 analogue, this last derivative was made by applying the 12-hydroxylation protocol (Section 6.4) to GA53 itself.105
Scheme 26
The 12α-series was generally better behaved, and superior outcomes were obtained.191 A further (unanticipated) advantage of the 12α-epimeric series was the option of inducing hydrogenolysis of the 13-substituent during the Li–NH3 reduction of 99. 12α-Hydroxy GA3
(GA87) methyl ester was converted into its trismethoxymethyl ether as for the 12β-epimer, and thence into the derived diazomethyl ketone 97. For cyclopropanation with this epimer, however, Cu(acac)2 was the preferred catalyst. Li–NH3reduction of 99 afforded not only the expected ketone101, but the 13-desoxy product 104 as well. A high yield of the bismethoxymethyl ether 101 could be obtained if the reaction was stopped after the addition of 2 equivalents of lithium, while reaction with an excess of lithium afforded mainly 104. It had been established that acetoxy groups attached to C-13 can be removed during Birch reductions of cyclopropyl ketones,181 and the mechanism for this process can be understood by transfer of an electron from the lithium metal into the π* molecular orbital of the carbonyl group followed by fragmentation to a tertiary radical that then undergoes further reduction and protonation.194 For the deoxygenation of 101, however, a different deoxygenation mechanism must apply, since the 13-methoxymethoxy group does not possess a sufficiently low-lying unoccupied molecular orbital to accommodate an electron in the initial step. It is proposed, therefore, that a solvated electron adds to the terminal olefin function, resulting in ejection of the methoxymethoxide anion, this loss being assisted by chelation of the (almost) coplanar 12α and 13-methoxymethoxy substituents to lithium cations formed during the reduction. This process would form an allylic radical delocalised over C13, C16 and C17, thereby violating Bredt's rule, but examination of molecular models shows a torsion angle of 65° between the singly occupied orbital at C-13 and the 16,17-π orbital, allowing overlap to occur to a limited extent. Analogous anti-Bredt structures have been invoked as possible intermediates in the formation of 13-selenides in work reported by Harrison et al.195
With both ketones101 and 104 accessible in this way, elaboration of all of the remaining 12-hydroxy GAs was straightforward. To obtain the 12β-epimers from 104, it was necessary to oxidise the intermediates to 12-oxo derivatives and reduce with borohydride. Thus, the structures for the cruciferous GAs (GA111, GA112, GA113, GA114, GA115 and GA116)100 were confirmed as well as the set of 12,13-dihydroxy C20 GAs (GA123, GA124, GA125, GA127, GA128 and GA129) recently isolated from strawberry fruitlets.104,106
7.5 Preparation of Δ9(11) GA24, a probable biosynthetic precursor to GA73
GA9
(4) would appear to be the obvious biosynthetic precursor of the potent antheridiogen, GA73 methyl ester (28) isolated from prothallia of several Lygodium fern species, but attempts to demonstrate incorporation of isotopically labelled material by prothallia of L. japonicum were unsuccessful.196 However, the 17,17-d2-derivative of GA24105 was converted into d2-GA7328 raising the possibility that an intermediate in the formation of this compound might be 9,11-didehydro-GA24106
(Scheme 27).197
Scheme 27
Our previous syntheses of C-ring functionalised C20gibberellins (Section 7.4) were an amalgam of earlier procedures that addressed separately the problems of introducing a functional group into the C-ring and of incorporating a formyl group at C(10). The resulting syntheses required ca. 18 steps, however, and in the hope of coalescing the two transformations, we examined the Lewis acid-catalysed cyclisation of diazoketone107
(readily obtained from 23) and were pleased to find that 108 was obtained in good yield (Scheme 28). After reconstitution of the 17-methylene group by a selective Lombardo methylenation, oxidative cleavage, as had been applied to similar compounds, gave access to 109 and thence 9,11-didehydro-GA24106.198 The overall sequence required only 13 steps and was readily modified to allow preparation of 17,17-d2-106 which was added to 5-week-old prothallia of L. circinnatum and L. flexuosum, respectively, and the cultures maintained for a further 10 days. d2-GA73 methyl ester was identified as a major metabolite in both L. flexuosum and L. circinnatum by full-scan GC–MS, no other metabolites being detected in either species. Thus, these results, together with the earlier experiments, provided strong circumstantial evidence that GA73 methyl ester is formed from GA24via 9,11-didehydro-GA24 in these Lygodium ferns.
Scheme 28
8 Antheridiogens from prothallia of the fern genus, Anemia
The major antheridiogen isolated from prothallia of the fern Anemia phyllitidis was shown to have structure 110. This antheridiogen was also discovered in gametophytes of the related species A. hirsuta, A. rotundifolia and A. flexuosa. The derivation in 1971 by Nakanishi et al. of such a novel structure by spectroscopic and traditional methods without reference to X-ray crystallography was a tour-de-force,199 although the stereochemistry at C(3) was incorrectly determined by them to be 3β. After completing the total synthesis of the racemate of the corresponding methyl ester200 and finding that it was different from the natural product, however, Corey and Myers concluded that the correct structure should be the 3α-epimer 110, confirmed this assignment by synthesis, and coined the name “antheridic acid”.201 In 1987, Nester et al. reported the discovery of another gibberellin-like antheridiogen, this time from A. mexicana.93 The new compound (now identified as GA104) was subsequently assigned structure 111 following spectroscopic and synthetic studies.94
8.1 Conversion of GA7 into antheridic acid
It had been suggested that antheridic acid could well have been formed biogenetically by rearrangement of a 9,10-epoxide.199 Irrespective of whether this hypothesis had any foundation, the equivalent chemical transformation appeared to be an attractive prospect for gaining access to these rare compounds, and the epoxide113 was prepared by an intramolecular transfer of oxygen from a 4α-peroxycarbonyl function to the more hindered face of the Δ9-olefinic bond in 112, which had been obtained from the 7-methyl ester 24 of gibberellenic acid202
(Scheme 29). However, treatment with Lewis acids did not induce rearrangement.203
Scheme 29
Given the speculative nature of the epoxide initiated rearrangement and the highly functionalised nature of the substrate, this outcome was hardly surprising. An alternative strategy based on an intramolecular alkylation to form a 9,15-cyclogibberellin followed by fragmentation of the C-8–C-15 bond was therefore explored. The successful sequence, beginning with GA7 methyl ester (15) and culminating with 110, proceeded smoothly as summarised in Scheme 30.203 The stereochemistry at C-3 was inverted by means of an oxidation–reduction cycle and then, after protection of the 3-hydroxy group, the diene acid 114 was formed by treatment with hydrazine in an analogous way to that of gibberellenic acid 7-methyl ester (24). Iodolactonisation followed by ozonolysis of the 17-methylene group, afforded the desired substrate 115 for the intramolecular alkylation to 116, which was effected with potassium hydride. After reconstruction of the 19,10-lactone function and reduction of the A-ring double bond, the 17-norantheridane skeleton 118 was obtained by heating ester117 with DBU (retrograde Michael reaction). The 17-methylene group was restored by a Wittig reaction and then deconjugation of the Δ6(8)-alkene bond effected by kinetically controlled protonation of the derived ester enolate. The upper face of this molecule is the more accessible one, but stereoelectronic control was expected to favor protonation along an axial-like trajectory and afford the desired 6β-isomer. In the event, only the desired diastereomer 119 was formed, along with recovered 118. The remaining stereochemical issue of concern in the sequence centered on allylic hydroxylation at C-15, but reaction on the more accessible face of the system was expected, and selenium dioxide–tert-butyl hydroperoxide treatment indeed afforded the desired 15β diastereomer as a 9 ∶ 1 mixture with its 15α-epimer. Hydrolysis of the methyl ester to the target acid 110 occurred very much more readily than for regular GAs and may be assumed to be assisted by the 15β-hydroxy.
Scheme 30
8.2 Structure determination and synthesis of the A. mexicana antheridiogen, GA104
Joan Nester, at the time a graduate student in botany at Iowa State University, Ames, Iowa, had painstakingly gathered together a few micrograms of the A. mexicana and had obtained a rough 1H-NMR spectrum. The antheridiogen could be methylated to give a product with the same molecular formula as GA7 methyl ester (15), while the NMR spectrum displayed an AB system at δ 2.70 and δ 2.80 which is typical for H-5 and H-6 in many GAs and a methine resonance at δ 4.30 associated with a secondary alcohol function. However, apart from the signals from a presumed 17-methylene group, there were no olefinic resonances. It appeared, therefore, that the degree of unsaturation, over and above the standard gibberellin skeleton as represented by GA9
(4), might be accounted for by an extra ring. It was also observed that the methyleneprotons were slightly (but significantly) upfield of the normal range for GAs, i.e.δ 4.74 and 4.77 compared with δ 4.88 and 4.99 for GA9
(4). On a visit to Canberra in 1987, Isao Yamaguchi of the University of Tokyo relayed a speculation by his colleague, Hisakazu Yamane that the antheridiogen might be based on a 9,15-cyclogibberellin structure with the cyclopropyl ring being responsible for the observed shift in frequencies.
Quite fortuitously, the structural studies coincided with our synthetic efforts on the conversion of GA7 into antheridic acid, and we had in hand a reasonable supply of the 3β-epimer of 116. Cyclo-GA 122 was therefore prepared from this intermediate (Scheme 31) with a view to determining the location of the hydroxy group before attempting the synthesis of the new antheridiogen itself. Hydrogenolysis of the derived mesylate 121 produced 122 which was subjected to the “usual” Wittig reaction to re-introduce the 17-methylene group, but when this process gave mainly water soluble material, resort was made to the Lombardo methylenation to obtain the target 123.
Scheme 31
1H-NMR comparisons with the natural product indicated that both H-5 and H-15 were deshielded relative to the parent system 123. It was therefore concluded that the hydroxy group must be located in the 1β-position, i.e. that the new antheridiogen should be formulated as 111.204 The mass spectrum of the trimethylsilyl methyl ester derivative showed a peak at m/z 116 (CH2CHO+SiMe3) a result that was also consistent with this assignment. The structure was then confirmed by synthesis, beginning with mesylate 121 as outlined in Scheme 32.94 Thus, reaction of 121 with NaOAc in HMPA afforded a 4 ∶ 5 mixture of acetates 124 and 125. Hydrolysis of the acetate function in the latter product followed by hydrogenation, Lombardo methylenation and thiolate demethylation afforded 111, the 1H NMR and mass spectra of which, were identical to those of the natural material.
Scheme 32
8.3 Second generation synthesis of cycloGAs: preparation of GA104 and GA108
A serious deficiency in the preparations outlined above stemmed from the low yields of the diene acid 114 and its analogues. In an attempt to bypass these intermediates, lactone126
(which may be derived from careful ozonolysis of the acetate derived from isolactone 17) was examined as an alternative substrate. It was envisaged that allylic bromination of this substrate with N-bromosuccinimide would take place with rearrangement of the olefinic bond to afford the 1-bromo-9-ene 127 which could then serve as an alternative substrate to 115
(Scheme 33).203 In the event, it was difficult to prevent further bromination to the 1,11-dibromide 128 and the sequence was continued with this product. Intramolecular alkylation with KH gave 130 and the 11-bromo substituent could be removed at a later stage by base-induced elimination of HBr followed by hydrogenation. Alternatively, treatment of 128 with CsOAc effected both cyclopropane formation and substitution at C-11 by acetate ion to furnish 129. This product could then be used to prepare the methyl ester of the 11-hydroxy-cycloGA, GA108
(131).205
Scheme 33
8.4 Biosynthesis of antheridic acid
With the discovery of the antheridiogen 111, it appeared possible that the biogenetic precursor to antheridic acid 110 might be based on the same cyclogibberellin skeleton. Strong support for the hypothesis was obtained when both the 17,17-d2-acid corresponding to 123 and its 3α-hydroxy derivative 132 were converted by prothallia of the fern Anemia phyllitidis into 17,17-d2-antheridic acid.95
8.5 Third generation synthesis of cycloGAs: preparation of 12-hydroxy-GA103
In order to probe the biosynthetic origins of antheridic acid 110 and related antheridiogens more thoroughly, the fate of [17,17-d2]-GA103 in metabolic studies conducted with the related ferns, Lygodium japonicum and L. circinnatum was also explored. These experiments resulted in the formation of [17,17-d2]-GA108
(131), its 11-epimer, and 2α-hydroxy-GA103, as well as two further products that were tentatively identified by GC-MS as 12β- and 12α-hydroxy-GA103.197 This last GA was also formed during the biosynthetic studies with Anemia phyllitidis. To confirm these assignments, the syntheses of these 12-hydroxy derivatives was undertaken as outlined in Scheme 34.206 The 11-acetoxy-cycloGA 129 was readily converted into enone134 and thence diene 135. We expected that the addition of borane to the exocyclic double bond in 135 would occur predominantly on the exo face followed by the intramolecular addition of the resulting alkylborane to the Δ11-alkene bond, thereby affording a 12,17-cycloborane. After oxidation to diol136, functional group manipulation led to the target structure 137. We also explored an alternative route to 134 based on the intramolecular alkylation 133
→
134. This conversion was highly successful and, given the more direct accessibility of enone27
(10 steps from GA3), this became our preferred approach. Most recently, the alkylation strategy has also been applied to the preparation of 9,15-cyclokaurenoids207 for which the earlier methods of cyclisation would not have been feasible.
Scheme 34
The syntheses of 137 and its 12-epimer were then completed as outlined in Scheme 35. Unlike the earlier examples involving 11,17-diols (cf.Scheme 7), it was essential to effect greater spatial separation between the 12β- and 17-substituents in order to avoid interference from the 12β-hydroxy during the restoration of the 17-methylene group. This objective was achieved (Scheme 35) by selectively acetylating the hydroxymethyl group to give 138, masking the 12-hydroxy (MOM ether), removing the 17-acetate function, oxidising the resulting 17-carbinol, and isomerising the product aldehyde139 with base to achieve the thermodynamically favoured exo-aldehyde 140. Reduction, formation of the 17-iodide 141, displacement with o-nitrophenyl selenide and removal of the MOM protecting group then gave 142, the selenoxide of which smoothly underwent elimination at 50 °C to afford the target alkene137. A sample of 137 was oxidised to the 12-oxo derivative and this product reduced with NaBH4, returning mainly 137 and accompanied by only a trace of the 12α-epimer, but sufficient to carry out GC–MS comparison of the trimethylsilyl derivatives. These comparisons showed that both 137 and its 12-epimer corresponded to two of the metabolites of [17,17-d2]-GA103
(123) isolated from L. japonicum and L. circinnatum,197 while 12-epi-137 corresponded to the unidentified product formed from [17,17-d2]-123 during the biosynthetic studies conducted with A. phyllitidis.95
Scheme 35
8.6 Total syntheses of antheridiogens
Just as the cyclisation of diazoketones 143 and 144 to the cyclohexadienones 145 and 146
(Scheme 36) had proven to be so effective in the efficient total synthesis of regular GAs,129 we wondered whether it would be possible, using carbenoid based processes to prepare cyclopropylketones such as 148
(Scheme 37), geometric constraints being expected to inhibit the normally facile electrocyclic rearrangement to the tautomeric cycloheptatriene. There was also the issue of whether the cyclopropyl motif could be preserved throughout the remainder of the synthesis.
Scheme 36
Scheme 37
If the highly strained ring system could be preserved, then a particularly direct and efficient total synthesis of GA103 and its congeners would be possible. We were encouraged to find that the reaction of 147
(R = 6-OMe) with Rh2(OAc)4 afforded ketone148
(R = 6-OMe) in excellent yield, and after exploring the scope of this type of transformation,189 proceeded to extend the protocol to the conversion of diazoketone149 into ketone150
(Scheme 38), although in this case, Cu(II)(acac)2 was the preferred catalyst. The A-ring was added by means of a high yielding regio- and diastereoselective [4 + 2] cycloaddition with citraconic anhydride, thereby affording the hexacycle 151 in 75% overall yield for the two steps. Functional group adjustment, Wolff ring-contraction of diazoketone153 followed by deletion of the superfluous substituent attached to C-3, then led smoothly to racemic GA103123.208 It was also possible to gain access to the antheridane and standard GA skeletons from intermediates of this kind. Photolysis of ketone154 in benzene–methanol, for example, afforded a 92% yield of 155, from which ketone156 could be obtained as indicated in Scheme 39, thereby achieving a formal synthesis of antheridic acid (110).209
Considerable effort has been invested in transforming kaurene derivatives into GAs either by incubation with the fungus, Gibberella fujikuroi,118 or by chemical means.119 Most kaurenoids, however, are not as easily obtained as the more common GAs. It was therefore of interest to explore the possibility of utilising GA3
(1) as a source of semi-synthetic kaurenoids. A particularly attractive aspect of such a conversion would be the opportunity to draw on the wealth of experience gained from the transformation of GA3
(1) into other GAs. The densely functionalised nature of these substrates was also expected to facilitate the preparation of the more complex kaurenoids that show interesting therapeutic potential. Of special interest was the Rabdosia family of seco-B-ring diterpenes, many of which have antibacterial and antineoplastic properties, e.g.oridonin157210 and enmein158.211
Our efforts directed towards the expansion of the B-ring (Scheme 40) were initially focussed on the pinacolic rearrangement of mesylate 159, but in spite of a number of encouraging precedents,212–215 it failed to undergo rearrangement, furnishing instead the corresponding 6,7-epoxide. We turned, therefore, to the acyloin rearrangement of the hydroxy aldehyde 160. Several precedents existed in the steroid literature, the ring expansions being attributed to the reduction of ring strain.216–219 The relief of additional strain in the GA skeleton was expected to favour the desired outcome even more strongly and in the event, treatment of 160 with KH furnished an excellent yield of ketol 161
(Scheme 40).220 Compounds of this type are readily transformed into 7,20-hemiacetals and thence seco-B-ring derivatives analogous to 157 and 158, respectively.
Scheme 40
10 The quest for selective bioactivity in gibberellins
In their seminal work with maize mutants, MacMillan and Phinney221 demonstrated that GA1
(162) had the primary role in promoting stem elongation; the same conclusion has since been made for rice, while GA4
(163) plays an equivalent role in cucumber.222 Although many other GAs are clearly biosynthetic intermediates or catabolites, it seems reasonable to assume that much of the structural diversity is related to a corresponding diversity of function.
10.1 Flowering studies on Lolium temulentum
When applied externally, GAs induce or promote flowering in many species, and are especially effective on “long day (LD) plants”, i.e. those plants that will not flower unless exposed to a sufficiently long period of light. Endogenous GA levels are typically higher in LD plants exposed to LD, whereas inhibitors of GA biosynthesis inhibit flowering in some LD requiring plants. In a study on the LD plant Lolium temulentum, it was discovered that the level of endogenous polyhydroxylated GAs increased on exposure to one LD.223 The general nature of these GAs was apparent from their chromatographic behaviour and the external application of GA32
(6) was especially efficacious in substituting for the LD exposure. Following the GA32 lead, a series of analogous GAs were prepared and tested for flowering efficacy on L. temulentum with very encouraging results. It was discovered that hydroxylation at either C-12 or C-15β promoted flowering.169 The balance in favour of flowering could also be shifted by inverting the stereochemistry of the 3β-OH, or removing it altogether,224 changes that flow logically from the knowledge that the 3β-hydroxy group in GA1 is essential for promoting growth in maize and other plants.
10.2 16,17-Dihydrogibberellins
At one stage in our research into flowering, the supply of GA5
(164) that we had been using as a key reference material, was fully consumed. It was a relatively simple task to prepare another batch, but then we discovered that the fresh material promoted vegetative growth more strongly than the original material. On recovering the “empty” vial, we analysed the trace of residual sample by GCMS and discovered that it was a mixture of GA5 and two dihydro derivatives. The fragmentation patterns of the latter were clearly consistent with the extra hydrogens being added to the Δ16-ene bond, the two isomers arising from differences in the configuration of C-16. When pure samples of these two compounds were prepared and screened, it was found that they both inhibited vegetative growth, with the derived 13-acetates showing enhanced potency. Within the prevailing dogma for GA properties, this effect came as a complete surprise. Moreover, these changes in activity associated with the 16,17-dihydro moiety also occurred with a wide range of GA types.170,225 The most promising aspect of this discovery, however, was the prospect that this kind of modified gibberellin should be relatively cheap to make, unlike the hydroxylated derivatives like GA32 that had been the subject of the initial study. We also had a product that would almost certainly be environmentally benign and applications of this class of GAs as an anti-lodging agent for wheat and barley crops have been especially encouraging.
10.3 16,17-Methanogibberellins
In a search for more potent inhibitors, we then embarked upon the synthesis of a series of analogues, aiming to answer the following questions: Would potency be enhanced with two 16-methyl groups. Would potency be enhanced by replacing the methyl groups with larger alkyl groups? What was the molecular basis of the inhibition?
To answer the first of these questions, we designed a synthesis based on the Simmons–Smith procedure to provide a cyclopropyl group as a precursor to the desired 16,16-gem-dimethyl. The protected GA3 molecule did not withstand the conditions of the reaction, however, undergoing a rearrangement of the lactone functionality (c.f.Scheme 3) catalysed by some of the inorganic by-products from the reagents involved. We turned therefore to dibromocarbene for the introduction of the three membered ring and the sequence through to the GA5 analogue 165 is summarised in Scheme 41.226 Although the dimethyl analogue proved to be more inhibitory of vegetative growth than either of the simple methyl derivatives, this result was eclipsed by the discovery that dichloromethano GA5166 was an even more potent inhibitor and also inhibited flowering. It was successfully tested on turf grasses227
(once we had fenced out the local kangaroos that consumed the first field test), slowing growth to as little as 30% of normal with monthly applications, but the cost of converting GA3 into 166 is likely to prohibit any commercial development.
Scheme 41
10.4 17-Alkylgibberellins
We turned, next, to the second question that we had posed, attempting to answer it by systematically replacing the methyl group with ethyl, propyl and butyl groups in both configurations at C-16 as outlined in Scheme 42.228 We found that stereochemistry and steric bulk were both important and that the growth inhibitory activity peaked with the exo-propyl derivative (167, R = Me). Although we have made gram-quantities of this GA, the prospects of making it on a commercial scale are again not encouraging.
Scheme 42
10.5 Molecular basis of growth inhibition
The final question, regarding the molecular basis of activity, was addressed by studying the fate of exogenously applied, isotopically labelled GA20 to dwarf rice (Oryza sativa L. cv. Tan-ginbozu).229 Normally, the GA20 would be converted by the plant into the growth-active GA1. In the presence of exo-dihydro GA5, however, the amount of labelled GA1 that was formed, was substantially decreased. It was concluded, therefore, that the dihydro GA5 is a competitive substrate for the enzyme that effects 3β-hydroxylation in rice. Similar results have been obtained with sorghum230 and Lolium.231
11 Conclusion
Gibberellins have fascinated chemists and biologists alike for half a century. The appeal stems partly from their complex structures, chemistry and biology, but mainly from the possibilities that they appear to hold out for the manipulation of plant growth and development. Their early promise is yet to be fully realised, however, and the original enthusiasm of industry has waned. The modified GAs described in the previous section, promise to lead to new agricultural applications, however.
The remote effects that we have observed with the chemistry of gibberellins, e.g. the cyclopropanation reactions and the zinc–acetic reductions of 15-oxo GAs, are especially puzzling. Despite the enormous progress achieved in organic synthesis over the past few decades, it still appears that more art than science is too frequently involved. The challenge here as elsewhere continues to be greater predictability and reliability. For the author, personally, he still rejoices in the challenges, complexities and subtleties presented by these molecules.
Acknowledgements
It has been a privilege and an enormous pleasure for this chemist to work with numerous biologists and biochemists scattered around the globe. This activity has only been possible, however, through the major contributions from the students, postdoctoral fellows and technicians who have worked in my research group – too many to acknowledge individually, but their names appear in the list of publications. Among all of these individuals, however, Bruce Twitchin deserves a special mention. Some 40-odd groups involved globally in GA research have benefited from his truly exceptional experimental skills. He has been a technical officer in my research group since 1980 and has prepared almost all of the unlabelled, deuterated and 14C-labelled GAs that we have provided to the GA community over the past two decades. The introduction of the isotopes has been a major undertaking in itself, but even more effort has gone into preparing the rare plant GAs from fungal GAs prior to labelling.
References
Phytohormones and Related Compounds – A Comprehensive Treatise, eds. D. S. Letham, P. B. Goodwin and T. J. V. Higgins, Elsevier, Amsterdam, 1978, vol. 1 and 2 Search PubMed.
The Biochemistry and Physiology of Gibberellins, ed. A. Crozier, Praeger, New York, 1983, vol. 1 and 2 Search PubMed.
Plant Hormones and Their Role in Plant Growth and Development, ed. P. J. Davies,Martinus Nijhoff, Dordrecht, 1987 Search PubMed.
N. Takahashi, I. Yamaguchi and H. Yamane, in Chemistry of Plant Hormones, ed. N. Takahashi, CRC Press, Boca Raton, Florida, 1986, pp. 57–151 Search PubMed.
M. H. Beale and C. L. Willis, in Methods in Plant Biochemistry Vol 4, eds. C. Banthorpe and B. V. Charlewood, Academic Press, London, 1991, pp. 289–330 Search PubMed.
J. MacMillan, J. Plant Growth Regul., 2002, 20, 387 Search PubMed.
B. O. Phinney, in The Biochemistry and Physiology of Gibberellins, ed. A. Crozier, Praeger, New York, 1983, vol. 1, pp. 19–52 Search PubMed.
T. Yabuta and T. Sumiki, J. Agric. Chem. Soc. Jpn., 1938, 14, 1526 Search PubMed.
P. W. Brian, J. F. Grove and J. MacMillan, Fortschr. Chem. Org. Naturst., 1960, 18, 350 Search PubMed.
B. E. Cross, J. F. Grove, J. MacMillan, J. S. Moffatt, T. P. C. Mulholland and J. C. Seaton, Proc. Chem. Soc., London, 1959, 302 RSC.
F. McCapra, A. I. Scott, G. A. Sim and D. W. Young, Proc. Chem. Soc., London, 1962, 185 RSC.
J. A. Hartsuck and W. N. Lipscomb, J. Am. Chem. Soc., 1963, 85, 3414 CrossRefCAS.
L. Kutschabsky and G. Adam, J. Chem. Soc., Perkin Trans. 1, 1983, 1653 RSC.
(a) I. Yamaguchi, T. Yokota, N. Murofushi, N. Takahashi and Y. Ogawa, Agric. Biol. Chem., 1975, 39, 2399 Search PubMed;
(b) I. Yamaguchi, T. Yokota, N. Murofushi and N. Takahashi, Agric. Biol. Chem., 1975, 39, 2405 Search PubMed.
H. Yamane, I. Yamaguchi, N. Murofushi and N. Takahashi, Agric. Biol. Chem., 1974, 38, 649 Search PubMed.
J. R. Bearder and J. MacMillan, Agric. Biol. Chem., 1972, 36, 342 Search PubMed.
(a) K. Hiraga, T. Yokota, N. Murofushi and N. Takahashi, Agric. Biol. Chem., 1972, 36, 345 Search PubMed;
(b) K. Hiraga, T. Yokota, N. Murofushi and N. Takahashi, Agric. Biol. Chem., 1974, 38, 2511 Search PubMed.
(a) H. Fukui, K. Koshimizu, S. Usuda and Y. Yamazaki, Agric. Biol. Chem., 1977, 41, 175 Search PubMed;
(b) H. Fukui, R. Nemori, K. Koshimizu and Y. Yamazaki, Agric. Biol. Chem., 1977, 41, 181 Search PubMed.
(a) I. Yamaguchi, M. Miyamoto, H. Yamane, N. Takahashi, K. Fujita and M. Imanari, Agric. Biol. Chem., 1973, 37, 2453 Search PubMed;
(b) I. Yamaguchi, M. Miyamoto, H. Yamane, N. Murofushi, N. Takahashi and K. Fujita, J. Chem. Soc., Perkin Trans. 1, 1975, 996 RSC.
J. R. Bearder and J. MacMillan, J. Chem., Soc., Perkin Trans. 1, 1973, 2824 RSC.
L. Beeley, P. Gaskin and J. MacMillan, Phytochemistry, 1975, 14, 779 CrossRefCAS.
V. M. Frydman, P. Gaskin and J. MacMillan, Planta, 1974, 118, 123 CrossRefCAS.
G. C. Martin, F. G. Dennis, Jr., P. Gaskin and J. MacMillan, Phytochemistry, 1977, 16, 605 CrossRefCAS.
L. J. Beeley and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1976, 1022 RSC.
A. G. McInnes, D. G. Smith, R. C. Durley, R. P. Pharis, G. P. Arsenault, J. MacMillan, P. Gaskin and L. C. Vining, Can. J. Biochem., 1977, 55, 728 Search PubMed.
H. Fukui, K. Koshimizu and R. Nemori, Agric. Biol. Chem., 1978, 42, 1571 Search PubMed.
V. M. Sponsel and J. MacMillan, Planta, 1977, 135, 129 CrossRefCAS.
V. M. Sponsel, P. Gaskin and J. MacMillan, Planta, 1979, 146, 101 CrossRefCAS.
N. Murofushi, M. Sugimoto, K. Itoh and N. Takahashi, Agric. Biol. Chem., 1979, 43, 2179 Search PubMed.
P. Gaskin, P. S. Kirkwood, J. R. Lenson, J. MacMillan and M. E. Radley, Agric. Biol. Chem., 1980, 44, 1589 Search PubMed.
N. Murofushi, M. Sugimoto, K. Itoh and N. Takahashi, Agric. Biol. Chem., 1980, 44, 1583 Search PubMed.
J. E. Graebe, P. Hedden and W. Rademacher, Br. Plant Growth Group Monogr., 1980, 5, 31 Search PubMed.
T. Yokota and N. Takahashi, Agric. Biol. Chem., 1981, 45, 1251 Search PubMed.
P. S. Kirkwood and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1982, 689 RSC.
S. C. Dolan, D. W. Holdup, M. Hutchison and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1985, 651 RSC.
M. Hutchison, P. Gaskin, J. MacMillan and B. O. Phinney, Phytochemistry, 1988, 27, 2695 CrossRefCAS.
S. C. Dolan and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1985, 2741 RSC.
P. Gaskin, S. J. Gilmour, J. R. Lenton, J. MacMillan and V. J. Sponsel, J. Plant Growth Regul., 1984, 2, 229 Search PubMed.
H. Yamane, I. Yamaguchi, M. Takahashi, Y. Sato, N. Takahashi, K. Iwatsuki, B. O. Phinney, C. R. Spray, P. Gaskin and J. MacMillan, Plant Physiol., 1985, 78, 899 CrossRefCAS.
H. Yamane, Y. Satoh, K. Nohara, M. Nakayama, N. Murofushi, N. Takahashi, K. Takeno, M. Furuya, M. Furber and L. N. Mander, Tetrahedron Lett., 1988, 29, 3959 CrossRefCAS.
N. Murofushi, M. Nakayama, N. Takahashi, P. Gaskin and J. MacMillan, Agric. Biol. Chem., 1988, 52, 1825 Search PubMed.
S. J. Castellaro, J. MacMillan, A. K. Singh and C. L. Willis, J. Chem. Soc., Perkin Trans. 1, 1990, 145 RSC.
M. Nakayama, H. Yamane, T. Yokota, I. Yamaguchi, N. Murofushi, N. Takahashi, T. Nishijima, N. Katura, M. Nonaka, P. Gaskin and J. MacMillan, Agric. Biol. Chem., 1990, 54, 837 Search PubMed.
C. L. Willis, Tetrahedron Lett., 1990, 31, 6437 CrossRefCAS.
E. Yuda, S. Nakagawa, N. Murofushi, N. Takahashi, T. Yokota, M. Koshioka, Y. Murakami, D. Pearce, R. P. Pharis, P. Kraft-Klaunzer, L. N. Mander and G. L. Patrick, Biosci. Biotechnol. Biochem., 1992, 56, 17 Search PubMed.
V. M. Sponsel, Planta, 1986, 168, 119 CrossRefCAS.
P. Gaskin, G. V. Hoad, J. MacMillan, I. K. Makinson and J. E. Readman, Phytochemistry, 1992, 31, 1869 CrossRefCAS.
C. Sheng, V. K. Bhaskar, W.-L. A. Chu, L. N. Mander, D. W. Pearce and R. P. Pharis, Biosci. Biotechnol. Biochem., 1992, 56, 564 Search PubMed.
V. K. Bhaskar, W.-L. A. Chu, P. A. Gaskin, L. N. Mander, N. Murofushi, D. W. Pearce, R. P. Pharis, N. Takahashi and I. Yamaguchi, Tetrahedron Lett., 1991, 32, 6203 CrossRef.
P. S. Blake, G. Browning, A. W. L. Chu and L. N. Mander, Phytochemistry, 1993, 32, 781 CrossRefCAS.
P. Hedden, G. V. Hoad, P. Gaskin, M. J. Lewis, J. R. Green, M. Furber and L. N. Mander, Phytochemistry, 1993, 32, 231 CrossRefCAS.
C. Sheng, K. V. Bhaskar, L. N. Mander, D. W. Pearce, R. P. Pharis and S. Young, Phytochemistry, 1992, 31, 4055 CrossRefCAS.
M. Penny, C. L. Willis, P. Gaskin and J. R. Lenton, Phytochemistry, 1993, 33, 951 CrossRefCAS.
M. Penny, C. L. Willis, P. Gaskin and J. R. Lenton, Phytochemistry, 1994, 37, 1063 CrossRefCAS.
S. Findlow, P. Gaskin, P. A. Harrison, J. R. Lenton, M. Penny and C. L. Willis, J. Chem. Soc., Perkin Trans. 1, 1997, 751 RSC.
M. Nakayama, T. Yokota, R. Sohma, L. N. Mander, B. Twitchin, H. Komatsu, H. Matsui and M. J. Bukovac, Phytochemistry, 1996, 42, 913 CrossRefCAS.
T. Yamauchi, N. Oyama, H. Yamane, N. Murofushi, H. Schraudolf, M. Pour, M. Furber and L. N. Mander, Plant Physiol., 1996, 111, 741 CAS.
L. N. Mander, D. J. Owen, S. J. Croker, P. Gaskin, P. Hedden, M. J. Lewis, M. Talon, D. A. Gage, J. A. D. Zeevaart, M. L. Brenner and C. Sheng, Phytochemistry, 1996, 43, 23 CrossRefCAS.
D. J. Owen, L. N. Mander, P. Gaskin and J. MacMillan, Phytochemistry, 1996, 42, 921 CrossRefCAS.
N. Oyama, T. Yamauchi, H. Yamane, N. Murofushi, M. Agatsuma, M. Pour and L. N. Mander, Biosci. Biotechnol. Biochem., 1996, 60, 305 Search PubMed.
J. E. Nester, S. Veysey and R. C. Coolbaugh, Planta, 1987, 170, 26 CrossRefCAS.
M. Furber and L. N. Mander, J. Am. Chem. Soc., 1988, 110, 4084 CrossRefCAS.
T. Yamauchi, N. Oyama, H. Yamane, N. Murofushi, N. Takahashi, H. Schraudolf, M. Furber, L. N. Mander, G. L. Patrick and B. Twitchin, Phytochemistry, 1991, 30, 3247 CrossRefCAS.
G. W. Wynne, L. N. Mander, N. Oyama, N. Murofushi and H. Yamane, Phytochemistry, 1998, 47, 1177 CrossRefCAS.
D. J. Owen, L. N. Mander, J. M. D. Storey, R. P. Huntley, P. Gaskin, J. R. Lenton, D. A. Gage and J. A. D. Zeevaart, Phytochemistry, 1998, 47, 331 CrossRefCAS.
S. Blechschmidt, U. Castel, P. Gaskin, P. Hedden, J. E. Graebe and J. MacMillan, Phytochemistry, 1984, 23, 553 CrossRefCAS.
H. Yamane, S. Fujioka, C. R. Spray, B. O. Phinney, J. MacMillan, P. Gaskin and N. Takahashi, Plant Physiol., 1988, 86, 857 CAS.
M. Nakayama, T. Nishijima, M. Koshioka, H. Yamane, D. J. Owen and L. N. Mander, Phytochemistry, 1998, 48, 587 CrossRefCAS.
G. W. Wynne, L. N. Mander, N. Goto, H. Yamane and T. Omori, Phytochemistry, 1998, 49, 1837 CrossRefCAS.
M. Nakayama, M. Koshioka, H. Matsuib, H. Oharab, L. N. Mander, S. K. Leitch, B. Twitchin, P. Kraft-Klaunzer, R. P. Pharis and T. Yokota, Phytochemistry, 2001, 57, 749 CrossRefCAS.
M. Koshioka, M. Roh, M. Nakayama, T. Hisamatsu and L. N. Mander, J. Jpn. Soc. Hortic. Sci., 1999, 68, 1158 Search PubMed.
P. S. Blake, D. R. Taylor, C. M. Crisp, L. N. Mander and D. J. Owen, Phytochemistry, 2000, 55, 887 CrossRefCAS.
D. W. Pearce, O. E. Hutt, S. B. Rood and L. N. Mander, Phytochemistry, 2002, 59, 679 CrossRefCAS.
P. S. Blake, unpublished results.
J. MacMillan and N. Takahashi, Nature (London), 1968, 217, 170 CAS.
(a) V. M. Sponsel and J. MacMillan, Planta, 1978, 144, 69 CrossRefCAS;
(b) V. M. Sponsel and J. MacMillan, Planta, 1980, 150, 46 CrossRefCAS.
P. Gaskin, P. S. Kirkwood and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1981, 1083 RSC.
T. Nishijima, M. Koshioka, H. Yamazaki and L. N. Mander, Acta Hortic., 1995, 394, 199 Search PubMed.
J. A. D. Zeevaart, Plant Physiol., 1971, 47, 821 CAS.
S. B. Rood, R. I. Buzzell, L. N. Mander, D. Pearce and R. P. Pharis, Science, 1988, 241, 1216 CAS.
G. C. Martin, in The Biochemistry and Physiology of Gibberellins, ed. A. Crozier, Praeger, New
York, 1983, vol. 2, pp. 395–444 Search PubMed.
R. D. Carlson and A. J. Crovetti, in Plant Growth Substances 1988, eds. R. P. Pharis and S. B. Rood, Springer Verlag, Berlin, 1990, pp. 604–610 Search PubMed.
I. Yamaguchi, T. Yokota, N. Murofushi, Y. Ogawa and N. Takahashi, Agric. Biol. Chem., 1970, 35, 1439 Search PubMed.
J. R. Bearder, J. MacMillan, C. M. Wels, M. B. Chaffey and B. O. Phinney, Phytochemistry, 1974, 13, 911 CrossRefCAS.
P. Gaskin and J. MacMillan, GC-MS of Gibberellins and Related Compounds: Methodology and a Library of Reference Spectra, Cantocks Enterprises, Bristol, 1991 Search PubMed.
T. P. C. Mulholland, J. Chem. Soc., 1958, 2693 RSC.
J. MacMillan and R. J. Pryce, J. Chem. Soc., 1967, 740 Search PubMed.
P. S. Kirkwood, J. MacMillan and M. Hutchison, J. Chem. Soc., Perkin Trans. 1, 1982, 707 RSC.
B. E. Cross, J. F. Grove and A. Morrison, J. Chem. Soc., 1961, 2498 RSC.
P. S. Kirkwood, J. MacMillan and M. L. Sinnott, J. Chem. Soc., Perkin Trans. 1, 1980, 2117 RSC.
L. Lombardo, L. N. Mander and J. V. Turner, Aust. J. Chem., 1981, 34, 745 CAS.
A. G. Avent, M. K. Baynham and J. R. Hanson, J. Chem. Soc., Perkin Trans. 1, 1989, 627 RSC.
D. F. Jones and P. McCloskey, J. Appl. Chem., 1963, 13, 324 Search PubMed.
R. D. Dawe, L. N. Mander and J. V. Turner, Tetrahedron Lett., 1985, 26, 363 CrossRefCAS.
M. Furber, P. Kraft-Klaunzer, L. N. Mander, M. Pour, T. Yamauchi, N. Murofushi, H. Yamane and H. Schraudolf, Aust. J. Chem., 1995, 48, 427 CAS.
M. H. Beale and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1980, 877 RSC.
Z. J. Duri, B. M. Fraga and J. R. Hanson, J. Chem. Soc., Perkin Trans. 1, 1981, 1612 Search PubMed.
R. A. Bell and J. V. Turner, Tetrahedron Lett., 1981, 22, 4871 CrossRefCAS.
C. L. Willis, Tetrahedron Lett., 1987, 28, 6705 CrossRefCAS.
E. J. Corey, T. M. Brennan and R. L. Carney, J. Am. Chem. Soc., 1971, 93, 7316 CrossRefCAS.
M. Furber, L. N. Mander and G. L. Patrick, J. Org. Chem., 1990, 55, 4860 CrossRefCAS.
H. Yamane, N. Takahashi, K. Takeno and M. Furuya, Planta, 1979, 147, 251 CrossRefCAS.
K. Takeno, H. Yamane, T. Yamaguchi, N. Takahashi, M. Furber and L. N. Mander, Plant Cell Physiol., 1989, 30, 201 Search PubMed.
P. Kraft-Klaunzer, M. Furber, L. N. Mander and B. Twitchin, Tetrahedron Lett., 1990, 31, 6235 CrossRefCAS.
L. N. Mander and G. L. Patrick, Tetrahedron Lett., 1990, 31, 423 CrossRefCAS.
N. J. Lewis and J. MacMillan, J. Chem. Soc., Perkin Trans. 1, 1980, 1270 RSC.
A. Chu and L. N. Mander, Tetrahedron Lett., 1988, 29, 2727 CrossRefCAS.
K. V. Bhaskar and L. N. Mander, Tetrahedron Lett., 1996, 37, 719 CrossRefCAS.
S. K. Leitch, PhD thesis, Australian National University, 2000.
L.-T. Phuoc, unpublished results.
L. T. Evans, R. W. King, A. Chu, L. N. Mander and R. P. Pharis, Planta, 1990, 182, 97 CrossRefCAS.
L. N. Mander, D. Camp, L. T. Evans, R. W. King, R. P. Pharis, M. Sherburn and B. W. Twitchin, Acta Hortic., 1995, 394, 45 Search PubMed.
V. Calo, L. Lopez, A. Mincuzzi and G. Pesce, Synthesis, 1976, 200 CrossRefCAS.
A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307 CrossRefCAS.
D. A. Evans, V. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1993, 48, 4155.
Cf.
A. Fadel, J.-L. Canet and J. Salaun, Tetrahedron Lett., 1989, 30, 6687 Search PubMed.
B. M. Fraga, J. R. Hanson, M. G. Hernández and F. G. Tellado, Tetrahedron Lett., 1989, 30, 6899 CrossRefCAS.
J. Liu, L. N. Mander and A. C. Willis, Tetrahedron, 1998, 54, 11637 CAS.
S. Vishveshwara and J. A. Pople, J. Am. Chem. Soc., 1997, 99, 2422.
L. J. Beeley, PhD Thesis, University of Bristol, 1975.
L. N. Mander and R. J. Thomson, J. Chem, Soc., Perkin Trans. 1, 2000, 2893 RSC.
M. H. Beale, J. Chem. Soc., Perkin Trans. 1, 1985, 1147 RSC.
R. D. Dawe, L. N. Mander, J. V. Turner and X. F. Pan, Tetrahedron Lett., 1985, 26, 5725 CrossRefCAS.
L. N. Mander, D. J. Owen and B. Twitchin, Aust. J. Chem., 1996, 49, 249 CAS.
D. H. R. Barton, D. A. J. Ives and B. R. Thomas, J. Chem. Soc., 1955, 2056 Search PubMed.
D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574 RSC.
L. N. Mander and M. S. Sherburn, Tetrahedron Lett., 1996, 37, 4255 CrossRefCAS.
A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454 CrossRefCAS.
M. P. Doyle, A. B. Dyatkin and C. L. Autry, J. Chem. Soc., Perkin Trans. 1, 1995, 619 RSC.
E. Lee, I. Choi and S. Y. Song, J. Chem. Soc., Chem. Commun., 1995, 321 RSC.
J. C. Morris, L. N. Mander and D. C. R. Hockless, Synthesis, 1998, 455 CrossRefCAS.
L. N. Mander and D. J. Owen, Tetrahedron Lett., 1996, 37, 723 CrossRefCAS.
L. N. Mander and D. J. Owen, Tetrahedron, 1997, 53, 2137 CrossRefCAS.
E. J. Corey and A. G. Myers, Tetrahedron Lett., 1984, 25, 3559 CrossRefCAS.
H. Seto and L. N. Mander, Synth. Commun., 1992, 22, 2823 CAS.
A. G. M. Barrett, P. A. Prokopiou, D. H. R. Barton, R. B. Boar and J. F. McGhie, J. Chem. Soc., Chem. Commun., 1979, 1173 RSC.
P. A. Harrison, L. Murtagh and C. L. Willis, J. Chem. Soc., Perkin Trans. 1, 1993, 3047 RSC.
Y. Sato, H. Yamane, M. Kobayashi, I. Yamaguchi and N. Takahashi, Agric. Biol. Chem., 1985, 49, 255 Search PubMed.
T. Yamauchi, N. Oyama, H. Yamane, N. Murofushi, H. Schraudolf, M. Pour, L. N. Mander and H. Seto, Plant Physiol., 1997, 113, 773 CAS.
L. N. Mander, G. M. Wynne, N. Goto, H. Yamane and T. Omori, Tetrahedron Lett., 1998, 39, 3877 CrossRefCAS.
K. Nakanishi, M. Endo, U. Näf and L. F. Johnson, J. Am. Chem. Soc., 1971, 93, 5579 CrossRefCAS.
E. J. Corey and A. G. Myers, J. Am. Chem. Soc., 1985, 107, 5574 CrossRefCAS.
E. J. Corey, A. G. Myers, N. Takahashi, H. Yamane and H. Schraudolf, Tetrahedron Lett., 1986, 27, 5083 CrossRefCAS.
J. F. Grove and T. P. C. Mulholland, J.
Chem. Soc., 1960, 3007 RSC.
M. Furber and L. N. Mander, J. Am. Chem. Soc., 1987, 109, 6389 CrossRefCAS.
M. Furber, L. N. Mander, N. Takahashi, H. Yamane and J. E. Nester, Phytochemistry, 1989, 28, 63 CrossRefCAS.
M. Pour, P. Kraft-Klaunzer, M. Furber, L. N. Mander, B. Twitchin, N. Oyama, N. Murofushi, H. Yamane and T. Yamauchi, Aust. J. Chem., 1997, 50, 289 CrossRefCAS.
M. Pour, G. M. Wynne, L. N. Mander and A. C. Willis, Tetrahedron Lett., 1998, 39, 1991 CrossRefCAS.
H. Zhang, G. M. Wynne and L. N. Mander, ARKIVOC, 2001, 2, 40 Search PubMed.
G. R. King, L. N. Mander, N. J. T. Monck, J. C. Morris and H. Zhang, J. Am. Chem. Soc., 1997, 119, 3828 CrossRefCAS.
H. Zhang, PhD thesis, Australian National University, 1998.
E. Fujita, T. Fujita, H. Katayama, M. Shibuya and T. Shingu, J. Chem. Soc. (C), 1970, 1674 Search PubMed.
E. Fujita, T. Fujita and M. Shibuya, J. Chem. Soc., Chem. Commun., 1966, 297 RSC.
E. J. Corey, M. Ohno, R. B. Mitra and P. A. Vatakencherry, J. Am. Chem. Soc., 1964, 86, 478 CrossRefCAS.
W. Tubiana and B. Waegell, Angew. Chem., Int. Ed. Engl., 1972, 11, 640 CrossRefCAS.
T. Nakata, S. Nomura and H. Matsukara, Tetrahedron Lett., 1996, 37, 213 CrossRefCAS.
C. W. Shoppee and D. A. Prins, Helv. Chim. Acta, 1943, 26, 201 CrossRefCAS.
T. C. Miller, J. Org. Chem., 1969, 34, 3829 CrossRefCAS.
R. B. Turner, J. Am. Chem. Soc., 1953, 75, 3484 CrossRefCAS.
D. N. Kirk and A. Mudd, J. Chem. Soc. (C), 1970, 2045 RSC.
L. J. Benjamin, G. Adamson and L. N. Mander, Heterocycles, 1999, 50, 365 Search PubMed.
J. MacMillan and B. O. Phinney, in Physiology of Cell Expansion during Cell Growth, eds. D. J. Cosgrove and D. P. Knievel, American Society of Plant Physiology, Rockville, MD, 1987, p. 156 Search PubMed.
M. Nakayama, H. Yamane, N. Murofushi, N. Takahashi, L. N. Mander and H. Seto, J. Plant Growth Regul., 1991, 10, 115 Search PubMed.
R. P. Pharis, L. T. Evans, R. W. King and L. N. Mander, Plant Physiol., 1987, 84, 1132 CAS.
L. T. Evans, R. W. King, L. N. Mander and R. P. Pharis, Planta, 1994, 192, 130 CAS.
L. T. Evans, R. W. King, L. N. Mander, R. P. Pharis and K. A. Duncan, Planta, 1994, 193, 107 CrossRefCAS.
L. N. Mander, M. Sherburn, D. Camp, L. T. Evans, R. W. King and R. P. Pharis, Phytochemistry, 1998, 49, 2195 CrossRefCAS.
R. W. King, C. Blundell, L. T. Evans, L. N. Mander and J. T. Wood, Crop Sci., 1997, 37, 1878 Search PubMed.
L. N. Mander, G. Adamson, B. Twitchin, D. Camp, L. T. Evans and R. W. King, Phytochemistry, 1998, 49, 1509 CrossRefCAS.
M. Takagi, D. W. Pearce, L. M. Janzen and R. P. Pharis, Plant Growth Regul., 1994, 15, 207 Search PubMed.
K. F. Foster, I. Lee, R. P. Pharis and P. W. J. Morgan, J. Plant Growth Regul., 1997, 16, 79 Search PubMed.
O. Junttila, R. W. King, A. Poole, G. Kretschmer, R. P. Pharis and L. T. Evans, Aust. J. Plant Physiol., 1997, 24, 359 Search PubMed.

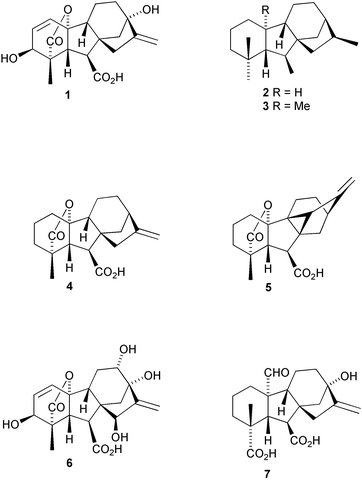
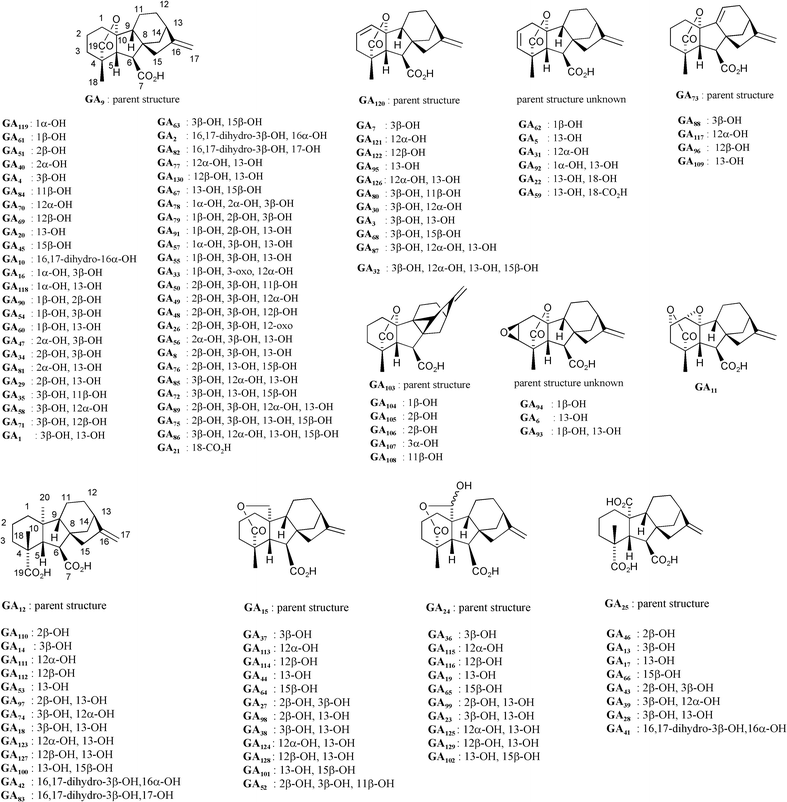
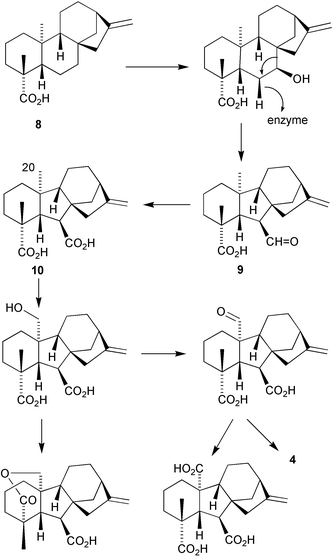
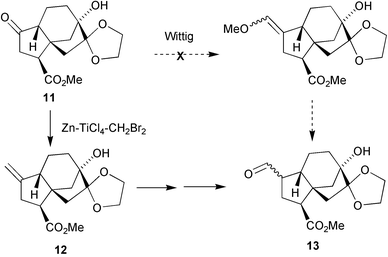
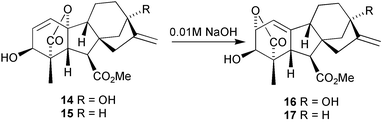
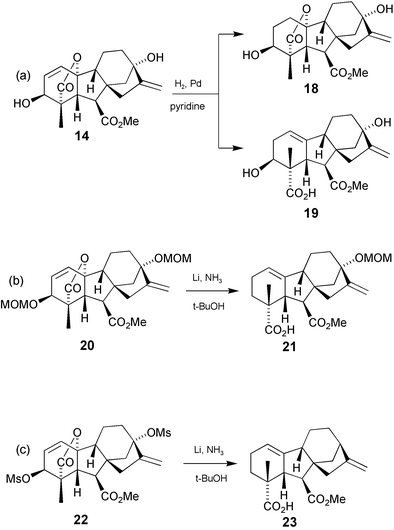
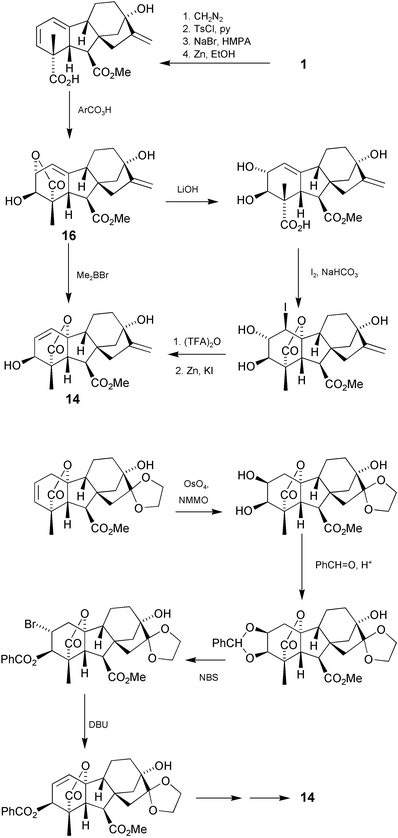
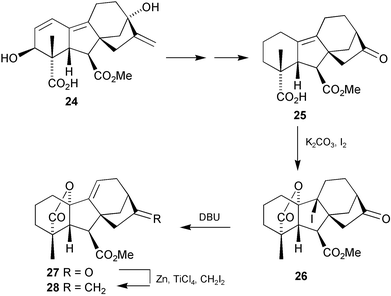
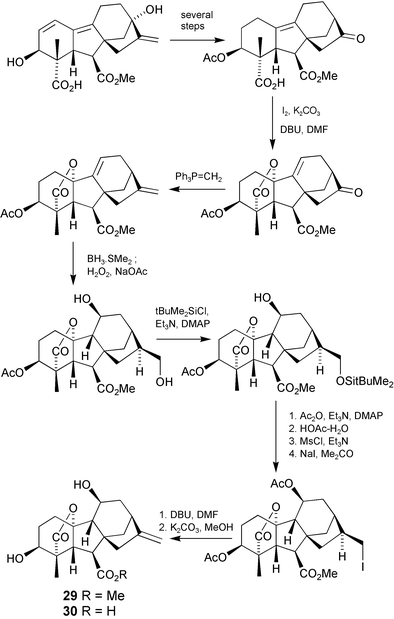
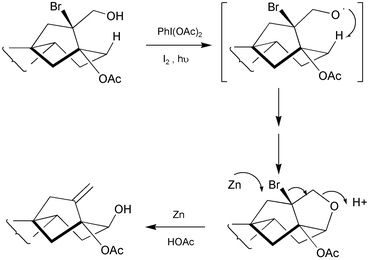
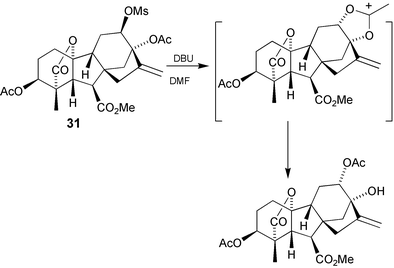
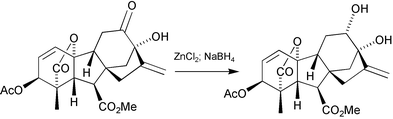
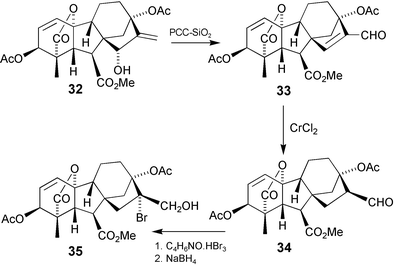
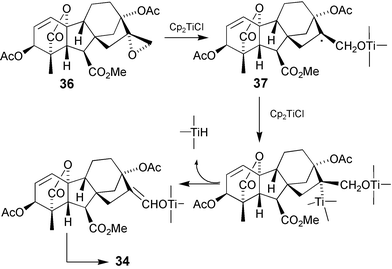
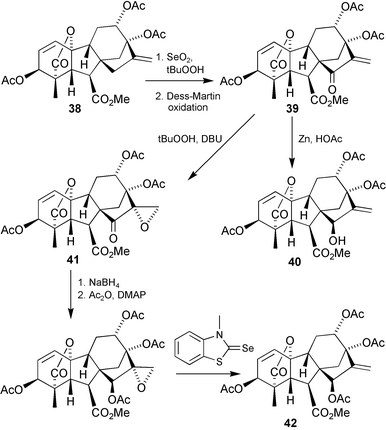
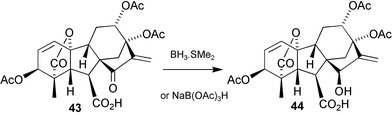
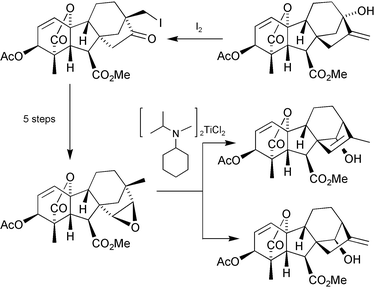
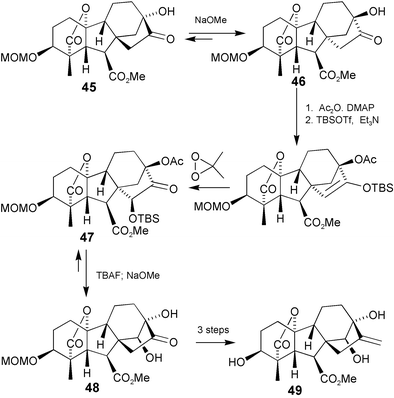
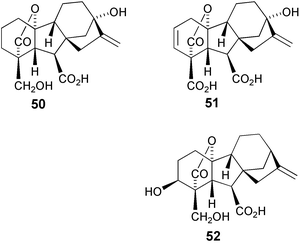
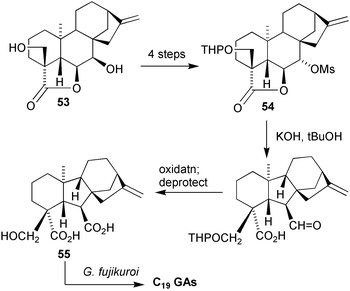
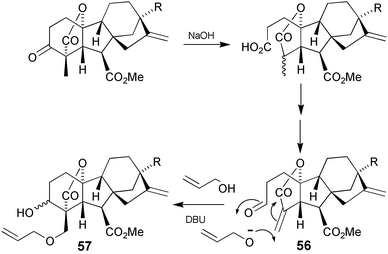
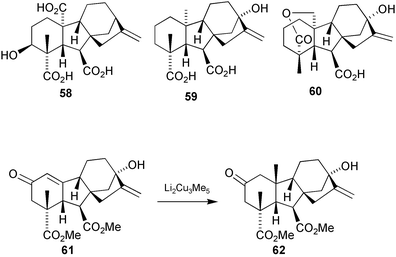
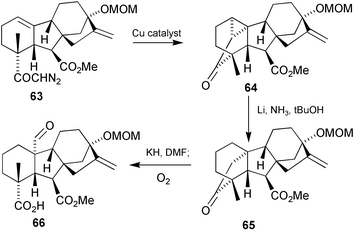
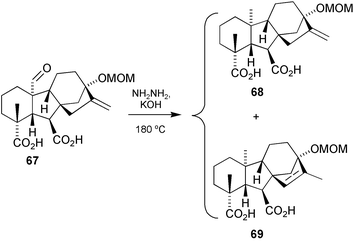
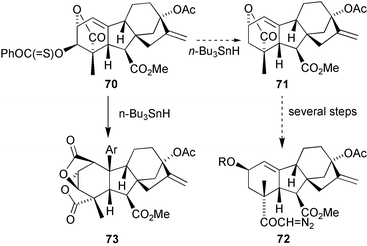
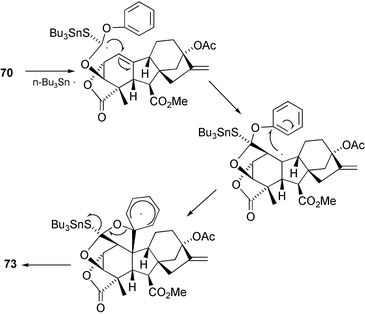
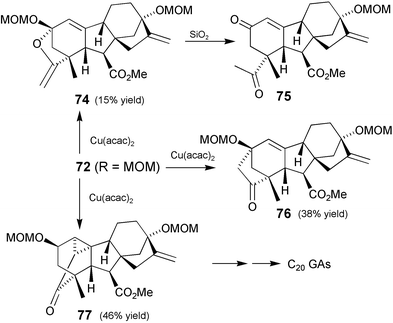
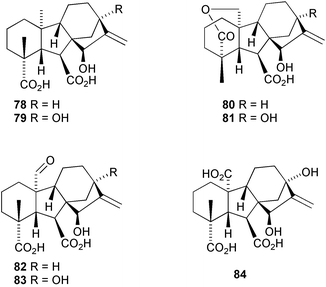
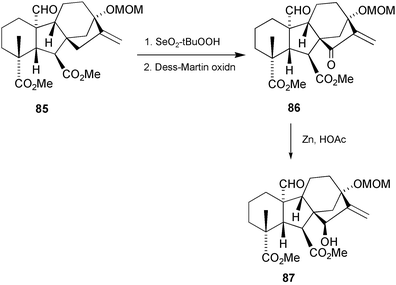
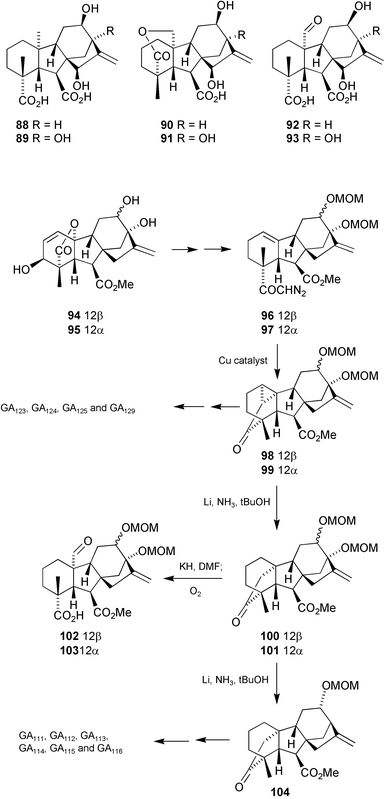
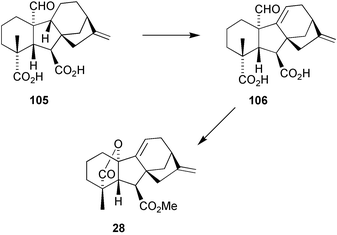
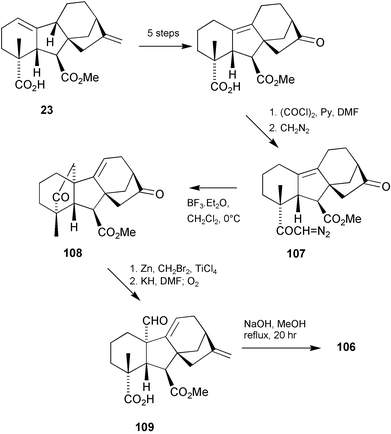
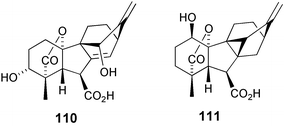
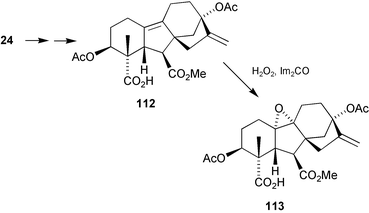
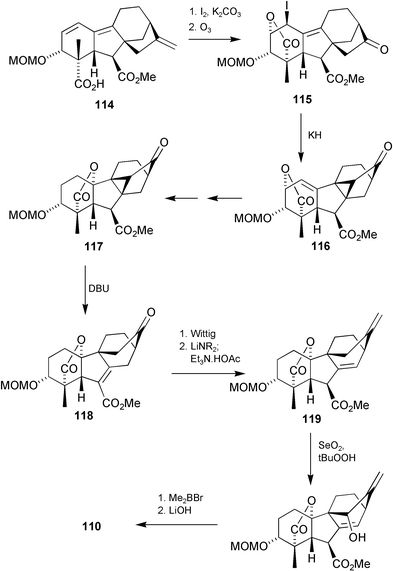
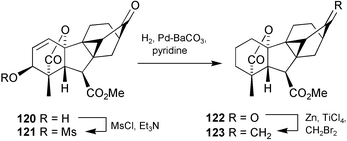
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2CH
CH2CH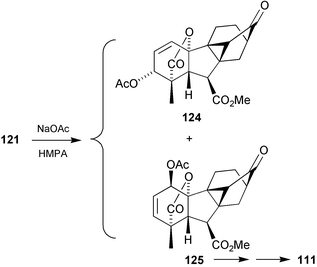
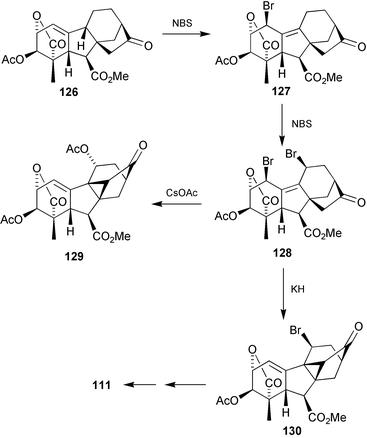
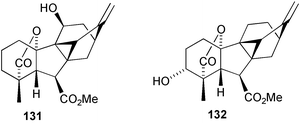
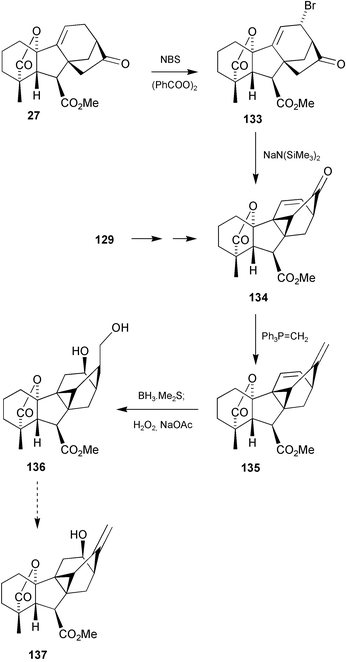
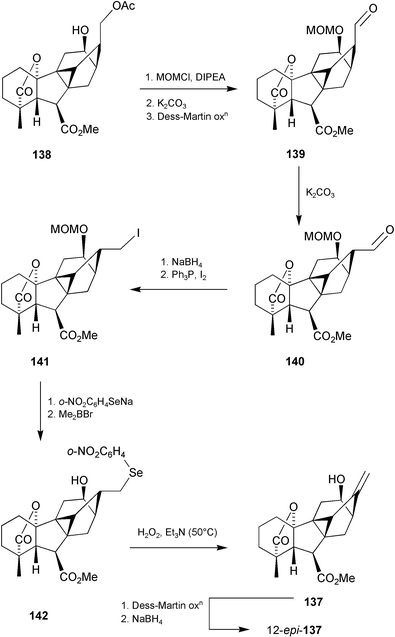
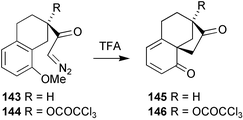
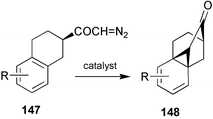
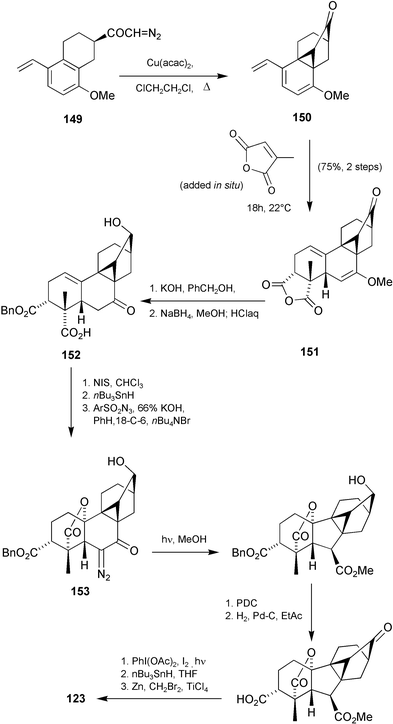
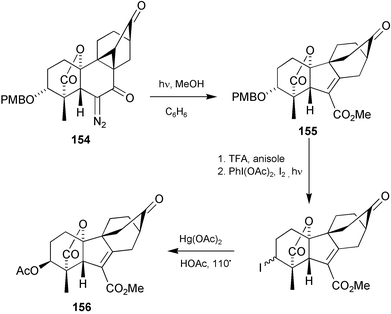
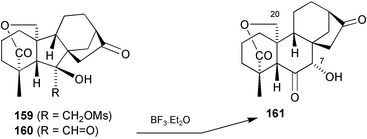
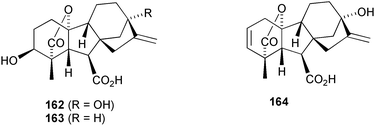
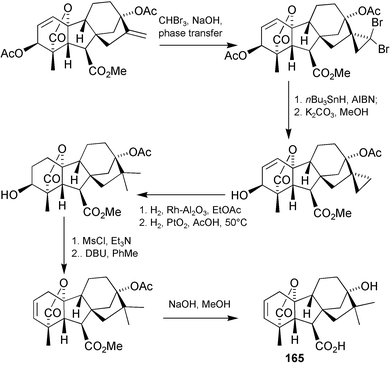
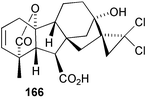
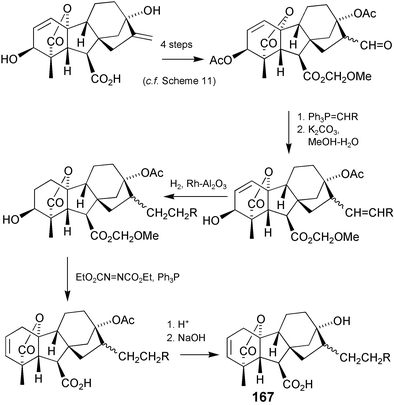
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 637 CAS.
637 CAS.