Versatile precursors for multinuclear platinum(II) alkynyl assembly—synthesis, structural characterization and electrochemical studies of luminescent platinum(II) alkynyl complexes
Chi-Hang
Tao
,
Keith Man-Chung
Wong
,
Nianyong
Zhu
and
Vivian Wing-Wah
Yam
*
Centre for Carbon-Rich Molecular and Nano-Scale Metal-Based Materials Research, Department of Chemistry, and HKU-CAS Joint Laboratory on New Materials, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk; Fax: 852 2857 1586; Tel: 852 2859 2153
First published on 28th November 2002
Abstract
Newly synthesized diethynylcarbazole-bridged platinum(II) complex 3,6-{ClPt(PEt3)2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C}2-9-nBuCarb (nBuCarb
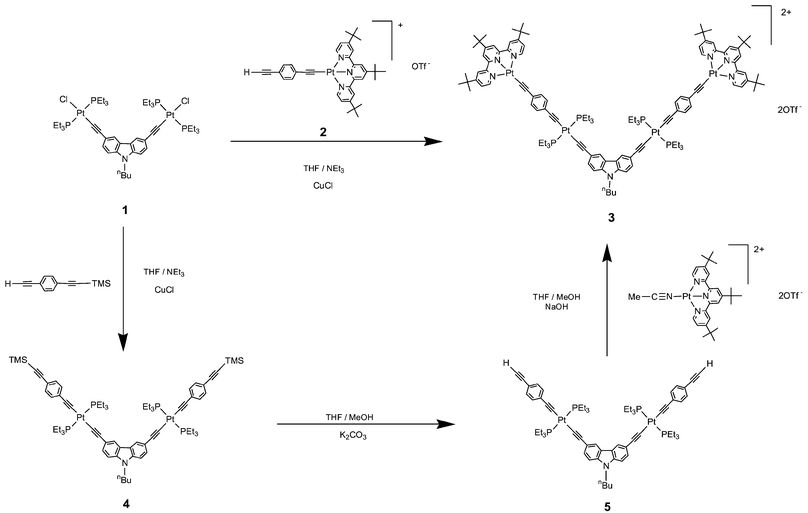
C}2-9-nBuCarb (nBuCarb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =n-butylcarbazole) and terpyridyl-containing platinum(II) alkynyl complex [Pt(tBu3trpy)(CCC6H4CCH)]OTf were shown to serve as versatile precursors for the assembly of a novel luminescent tetranuclear platinum(II) alkynyl complex [3,6-{Pt(tBu3trpy)(CCC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2. A divergent route can also be applied to synthesize the tetranuclear complex in three steps, involving the use of trimethylsilyl protecting groups on the alkynyl ligand.
=n-butylcarbazole) and terpyridyl-containing platinum(II) alkynyl complex [Pt(tBu3trpy)(CCC6H4CCH)]OTf were shown to serve as versatile precursors for the assembly of a novel luminescent tetranuclear platinum(II) alkynyl complex [3,6-{Pt(tBu3trpy)(CCC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2. A divergent route can also be applied to synthesize the tetranuclear complex in three steps, involving the use of trimethylsilyl protecting groups on the alkynyl ligand.
The construction of metal-containing branched molecules and metallodendrimers has been a growing area of research during the past decade.1 Developments based on the use of carbazole as the building unit for the synthesis of branched molecules, oligomers, polymers or even dendrimers have recently been reported, however, most of them are confined to organic systems.2,3 To the best of our knowledge, carbazole-containing metal alkynyl complexes are rare.4 These, together with our current interest in the design and synthesis of branched platinum(II) alkynyl complexes,5 have prompted us to exploit the carbazole unit as the organic backbone for the construction of branched platinum(II) alkynyl complexes. Herein we report the synthesis and structural characterization of a 3,6-diethynyl-9-butylcarbazole diplatinum(II) complex, 3,6-{ClPt(PEt3)2C
C}2-9-nBuCarb 1. This complex, with two terminal chloro ligands, may serve as versatile building blocks for the construction of multinuclear platinum(II) alkynyls. This, together with our recent efforts in platinum(II) terpyridyl complex syntheses6–9 and the rich spectroscopic and photophysical properties known of the platinum(II) polypyridine systems,10 have prompted us to synthesize a platinum(II) terpyridyl complex, [Pt(tBu3trpy)(CCC6H4CCH)]OTf 2, which together with 1 may serve as good precusors for the assembly of a novel tetranuclear platinum(II) alkynyl complex, [3,6-{Pt(tBu3trpy)(CCC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)23. The tetranuclear complex 3 can also be prepared via a divergent route from 1 in 3 steps using the trimethylsilyl-protected alkynyl ligand and [Pt(tBu3trpy)(MeCN)](OTf)2. Herein are reported their syntheses, electrochemical and photophysical studies of 1–3, and the X-ray crystal structures of 1 and 2.
Reaction of 3,6-diethynyl-9-n-butylcarbazole L2 with an excess of trans-[Pt(PEt3)2Cl2] in THF containing NEt3/CuCl afforded 1 in reasonable yield. 2 was obtained using modification of a reported procedure for terpyridyl platinum alkynyl complexes.8,9 For the convergent synthesis of the tetranuclear platinum(II) alkynyl complex, reaction of 1 with 2.2 equiv. of 2 in the presence of NEt3/CuCl in THF afforded 3
(Scheme 1), which was subsequently purified by column chromatography on activated basic alumina, followed by recrystallization from acetone–diethyl ether to give analytically pure samples of 3 as a deep red solid, while the divergent synthetic route involved the reaction of 1 and 1-ethynyl-4-trimethylsilylethynylbenzene to give 3,6-{TMSCCC6H4CCPt(PEt3)2CC}2-9-nBuCarb 4, which was subsequently deprotected to yield 3,6-{HCCC6H4CCPt(PEt3)2CC}2-9-nBuCarb 5. Finally, the reaction of the metalloligand 5 with [Pt(tBu3trpy)(MeCN)](OTf)2 gave the tetranuclear Pt(II) complex 3 in 21% overall yield (calculated from 1). All the newly synthesized complexes have been characterizaed by 1H NMR, IR and mass spectrometry, and gave satisfactory elemental analyses. Complexes 1 and 3–5 have also been characterized by 31P{1H} NMR spectroscopy. The X-ray crystal structures of 1 and 2 have also been determined.
 | ||
| Scheme 1 | ||
Figs. 1 and 2 show the perspective drawings of 1 and the complex cation of 2, respectively. All the CC bond distances of 1 and 2 were found to be in the range of 1.20–1.23 Å, which are comparable to those found in related platinum(II) alkynyl complexes.1,5,8,9 The platinum atoms in 1 showed the expected square planar coordination geometry. The coordination planes about each platinum atom are not co-planar with respect to the carbazole backbone, with interplanar angles of 86.6° and 89.7°. This kind of out-of plane twisting of coordination planes has also been observed in other related palladium(II) and platinum(II) alkynyl complexes.5 In the case of 2, the coordination geometry around the platinum atom is distorted square planar with the bond distance of the platinum to the central nitrogen atom of the terpyridine ligand slightly shorter than that to the other two nitrogen atoms and the N–Pt–N angles are deviated from the idealized values of 90° and 180°. These distortions of the coordination geometry around the platinum atom are clearly the consequences of the steric demand of the terpyridine ligand. The phenyl ring of the alkynyl ligand is not co-planar with the coordination plane around platinum, showing a dihedral angle of 9.4°, which is typical for platinum(II) terpyridyl alkynyl complexes.8,9 Both 1 and 2 did not show short intermolecular Pt⋯Pt contacts, and in the case of 2, no short π⋯π contacts were observed. It is conceivable that the presence of the bulky Et3P as well as the tBu groups on the terpyridine ligand would hinder the formation of such interactions.
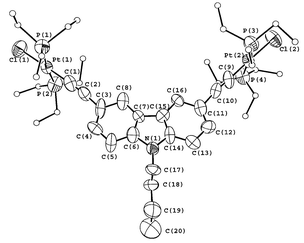 | ||
| Fig. 1 Perspective drawing of 1 with atomic numbering. Hydrogen atoms have been omitted for clarity. The thermal ellipsoids are shown at the 30% probability level. Selected bond distances (Å) and bond angles (°) of complex 1: Pt(1)–Cl(1) 2.371(6), Pt(1)–C(1) 1.97(3), Pt(1)–P(1) 2.269(6), Pt(1)–P(2) 2.261(7), Pt(2)–Cl(2) 2.343(6), Pt(2)–C(9) 1.87(3), Pt(2)–P(3) 2.288(6), Pt(2)–P(4) 2.302(6), C(1)–C(2) 1.24(3), C(9)–C(10) 1.20(3); Cl(1)–Pt(1)–C(1) 178.3(8), P(1)–Pt(1)–C(1) 89.8(7), P(2)–Pt(1)–C(1) 87.4(7), Pt(1)–C(1)–C(2) 176.0(19), C(1)–C(2)–C(3) 170(3), Cl(2)–Pt(2)–C(9) 179.0(8), P(3)–Pt(2)–C(9) 89.4(6), P(4)–Pt(2)–C(9) 88.0(6), Pt(2)–C(9)–C(10) 173(2), C(9)–C(10)–C(11) 171(3). | ||
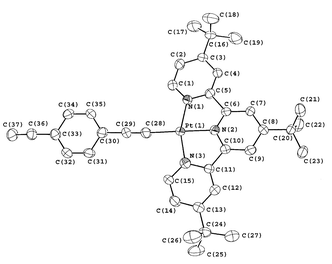 | ||
| Fig. 2 Perspective drawing of the complex cation of 2 with atomic numbering. Hydrogen atoms have been omitted for clarity. The thermal ellipsoids are shown at the 30% probability level. Selected bond distances (Å) and bond angles (°) of complex 2: Pt(1)–N(1) 2.023(10), Pt(1)–N(2) 1.980(11), Pt(1)–N(3) 2.012(12), Pt(1)–N(2) 1.992(15), C(28)–C(29) 1.192(19), C(36)–C(37) 1.20(2); N(1)–Pt(1)–N(2) 79.4(4), N(2)–Pt(1)–N(3) 80.8(4), N(1)–Pt(1)–C(28) 101.0(5), N(3)–Pt(1)–C(28) 98.9(5), N(1)–Pt(1)–N(3) 160.2(5), N(2)–Pt(1)–C(28) 178.5(5), Pt(1)–C(28)–C(29) 174.0(14). | ||
The electrochemical behaviour of 1–3 was studied by cyclic voltammetry and the electrochemical data are summarized in Table 1. The cyclic voltammogram of 1 shows two irreversible oxidative waves at ca. +0.91 and +1.12 V vs. SCE. With reference to previous electrochemical studies on related platinum(II) alkynyl complexes, in which two irreversible oxidation waves are also observable at similar reduction potentials, the two irreversible oxidation waves of 1 are tentatively assigned as Pt(II)/(III) and Pt(III)/(IV) oxidations, similar to that reported previously in the literature,11 although the possible involvement of a diethynylcarbazole ligand-centred oxidation could not be completely excluded. Complex 2, on the other hand, shows two quasi-reversible reduction couples at ca. −1.00 and −1.49 V vs. SCE, ascribed to ligand-centred reductions of the tBu3trpy ligand.8 An irreversible anodic wave at ca. +1.38 V was observed for 2. With reference to previous electrochemical studies on related platinum(II) terpyridyl alkynyl complexes,8 the irrevesible oxidation was assigned as a metal-centred oxidation from platinum(II) to platinum(III), possibly with some mixing of an alkynyl ligand character. The cyclic voltammogram of 3 shows two quasi-reversible reduction couples at ca. −1.01 and −1.53 V vs. SCE together with three irreversible anodic waves at ca. +0.78, +0.97 and +1.42 V. The two reduction couples are assigned as terpyridine-based reductions, similar to that found in 2. The two irreversible oxidative waves at ca. +0.78 and +0.97 V are similar to that observed in 1 and are assigned as metal-centred oxidations of the platinum diethynylcarbazole moiety, while the anodic wave at ca. +1.42 V which closely resembles that of 2 is likely to be attributed to the metal-centred oxidation of the [Pt(tBu3trpy)] unit.
| Complex | Oxidation Epa/V vs. SCEb | Reduction E1/2/V vs. SCEc |
|---|---|---|
|
a In acetonitrile solution with 0.1 M nBu4NPF6
(TBAH) as supporting electrolyte at room temperature: scan rate 100 mV s−1.
b
E
pa refers to the anodic peak potential for the irreversible oxidation waves.
c
E
1/2=(Epa+Epc)/2; Epa and Epc are the anodic and cathodic peak potentials, respectively.
d Not observed.
|
||
| 1 | +0.91 | —d |
| +1.12 | ||
| 2 | +1.38 | −1.00 |
| −1.49 | ||
| 3 | +0.78 | −1.01 |
| +0.97 | −1.53 | |
| +1.42 | ||
The electronic absorption spectrum of complex 1 exhibits intense absorption bands at ca. 270–290 nm and 310–390 nm, similar to that observed in other related Pt(II) alkynyl complexes of phosphines,5 and the lower energy band is tentatively assigned as an admixture of π→π*(CCR) intraligand (IL)/dπ(Pt)→π*(CCR) metal-to-ligand charge transfer (MLCT) transition with predominantly IL character. On the other hand, complexes 2 and 3 show intense low energy absorption bands at ca. 470 nm and 520 nm, respectively. The 470 nm absorption in 2 is tentatively assigned as a dπ(Pt)→π*(tBu3trpy) MLCT absorption, probably mixed with some π(CCR)→π*(tBu3trpy) ligand-to-ligand charge transfer (LLCT) character, typical of that found in related alkynylplatinum(II) terpyridyl systems.8,9 The low-energy absorption band of 3 which occurs at ca. 520 nm is found to be red-shifted with respect to that of 2. It is interesting to note that the HOMO of 3, as reflected by electrochemical studies, is higher lying than that of 2, while their LUMOs are of similar energies, giving rise to a smaller HOMO–LUMO energy gap in 3 than 2, in line with the red-shift in the lowest energy transition observed on going from 2 to 3. It is likely that the low-energy absorption at ca. 520 nm in 3 involves a charge transfer from the platinum diethynylcarbazole moiety of the molecule to the terpyridyl part of the molecule, in line with an assignment of a mixed MLCT/LLCT transition.
Upon photo-excitation at λ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 350 nm, the EtOH/MeOH (4∶1, v/v) glass of 1 at 77 K shows intense luminescence at ca. 444 nm with rich vibronic structures, which has been attributed to originate from a mixed π→π*(CCR) IL/dπ(Pt)→π*(CCR) MLCT triplet state with predominantly IL character.5 On the other hand, 2 is found to emit at ca. 586 nm in dichloromethane solution at room temperature upon photo-excitation, tentatively assigned as derived from a dπ(Pt)→π*(tBu3trpy) MLCT triplet state, probably with some mixing of an alkynyl-to-terpyridyl 3LLCT character, as is commonly observed in other related alkynylplatinum(II) terpyridyl complexes,8,9 while room-temperature dichloromethane solutions of 3 emit at ca. 627 nm, which is red-shifted relative to those of both 1 and 2. Similar to electronic absorption studies, the red-shift in emission energy of 3 relative to 2 is likely to originate from an excited state of mixed 3MLCT/3LLCT character, involving a charge transfer from the platinum diethynylcarbazole unit to the terpyridyl part of the molecule, as supported by electrochemical studies.
350 nm, the EtOH/MeOH (4∶1, v/v) glass of 1 at 77 K shows intense luminescence at ca. 444 nm with rich vibronic structures, which has been attributed to originate from a mixed π→π*(CCR) IL/dπ(Pt)→π*(CCR) MLCT triplet state with predominantly IL character.5 On the other hand, 2 is found to emit at ca. 586 nm in dichloromethane solution at room temperature upon photo-excitation, tentatively assigned as derived from a dπ(Pt)→π*(tBu3trpy) MLCT triplet state, probably with some mixing of an alkynyl-to-terpyridyl 3LLCT character, as is commonly observed in other related alkynylplatinum(II) terpyridyl complexes,8,9 while room-temperature dichloromethane solutions of 3 emit at ca. 627 nm, which is red-shifted relative to those of both 1 and 2. Similar to electronic absorption studies, the red-shift in emission energy of 3 relative to 2 is likely to originate from an excited state of mixed 3MLCT/3LLCT character, involving a charge transfer from the platinum diethynylcarbazole unit to the terpyridyl part of the molecule, as supported by electrochemical studies.
Experimental
Materials and reagents
trans-Dichlorobis(triethylphosphine)platinum(II) was obtained from Aldrich Chemical Co. [Pt(tBu3trpy)(MeCN)](OTf)28 and 3,6-diethynyl-9-n-butylcarbazole L22 were prepared according to the slight modification of reported procedures. All solvents were purified and distilled using standard procedures before use.12 All other reagents were of analytical grade and were used as received.Physical measurements and instrumentation
UV-visible spectra were obtained on a Hewlett-Packard 8452A diode array spectrophotometer, IR spectra as KBr discs on a Bio-Rad FTS-7 Fourier-transform infrared spectrophotometer (4000–400 cm−1), and steady state excitation and emission spectra on a Spex Fluorolog-2 Model F 111 fluorescence spectrophotometer equipped with a Hamamatsu R-928 photomultiplier tube. Low-temperature (77 K) spectra were recorded by using an optical Dewar sample holder. 1H and 31P{1H} NMR spectra were recorded on a Bruker DPX-400 (400 MHz) Fourier-transform NMR spectrometer. Chemical shifts (δ, ppm) of 1H NMR spectra were recorded relative to tetramethylsilane (Me4Si), while those of 31P NMR spectra were recorded relative to 85% H3PO4. Positive ion FAB mass spectra were recorded on a Finnigan MAT95 mass spectrometer. Positive ESI mass spectra were recorded on a Finnigan LCQ mass spectrometer. Elemental analyses of the new complexes were performed on a Carlo Erba 1106 elemental analyzer at the Institute of Chemistry, Chinese Academy of Sciences.Cyclic voltammetric measurements were performed by using a CH Instruments, Inc. model CHI 750A Electrochemical Analyzer. Electrochemical measurements were performed in acetonitrile solutions with 0.1 M nBu4NPF6 (TBAH) as supporting electrolyte at room temperature. The reference electrode was a Ag/AgNO3 (0.1 M in acetonitrile) electrode and the working electrode was a glassy carbon electrode (CH Instruments, Inc.) with a platinum gauze as the counter electrode. The ferrocenium/ferrocene couple (FeCp2+/0) was used as the internal reference. All solutions for electrochemical studies were deaerated with pre-purified argon gas just before measurements.
All solutions for photophysical studies were degassed on a high vacuum line in a two-compartment cell consisting of a 10 ml Pyrex bulb and a 1 cm path length quartz cuvette, and sealed from the atmosphere by a Bibby Rotaflo HP6 teflon stopper. The solution were subject to at least four freeze–pump–thaw cycles.
Synthesis
All reactions were carried out under an inert atmosphere of nitrogen using standard Schlenk techniques.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C}2-9-nBuCarb (1).
trans-[Pt(PEt3)2Cl2]
(1 g, 1.992 mmol) was dissolved in THF (50 ml) containing copper(I) chloride (1 mg, 0.01 mmol) and NEt3
(10 ml). 3,6-Diethynyl-9-n-butylcarbazole L2
(135 mg, 0.498 mmol) in THF (20 ml) was added dropwise at 0°C, and the reaction mixture was stirred overnight at room temperature. The mixture was then filtered, and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2, washed with deionized water and dried over anhydrous MgSO4. The dried organic fraction was then concentrated under reduced pressure and pass through a column of activated basic alumina (50–200 μm) using CH2Cl2–petroleum ether (bp 40–60 °C) (1∶1, v/v) as eluent. Subsequent recrystallization from CH2Cl2–n-hexane gave 1 as pale yellow crystals. Yield: 69%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2–), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2–), 2.05–2.15 (m, 24H, –CH2P), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.21 (d, JHH=8.6 Hz, 2H, Hs at 1-position of carbazole), 7.34 (dd, JHH=8.6 Hz/1.7 Hz, 2H, Hs at 2-position of carbazole), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 15.1 (JPt–P=2401 Hz); IR (KBr disk, ν/cm−1): 2117s ν(CC); positive FAB-MS: 1202 [M]+, 1084 [M−PEt3]+; anal. calcd. for 1: C, 43.9%; H, 6.24%; N, 1.16%; found: C, 44.0%; H, 6.23%; N, 1.16%.
CC6H4CCH)]OTf (2).
1,4-Diethynylbenzene (42 mg, 0.334 mmol), [Pt(tBu3trpy)(MeCN)](OTf)2
(312 mg, 0.334 mmol) and sodium hydroxide (0.2 g) in methanol (35 ml) were heated to reflux overnight. The solvent of the red reaction mixture was removed under reduced pressure. The residue was dissolved in CH2Cl2, filtered and chomatographed on silica gel (60–230 mesh) using CH2Cl2–acetone as eluent. Recrystallization from acetone–n-hexane gave 2 as orange crystals. Yield: 42%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 1.50 (s, 18H, tBu), 1.59 (s, 9H, tBu), 7.42 (dd, JHH=6 Hz/2 Hz, 4H, –C6H4–), 7.62 (dd, JHH=6 Hz/2 Hz, 2H, trpy), 8.38, (d, JHH=2 Hz, 2H, trpy), 8.45 (s, 2H, trpy), 9.10 (d, JHH=6 Hz, 4H, trpy); IR (KBr disk, ν/cm−1): 2112s ν(CC); positive ESI-MS: 720 [M−OTf]+, anal. calcd. for 2: C, 52.4%; H, 4.60%; N, 4.83%; found: C, 52.3%; H, 4.61%; N, 4.82%.
CC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2
(3)
via the convergent route.
1
(78 mg, 0.065 mmol) and 2
(100 mg, 0.142 mmol) were dissolved in THF (15 ml) containing copper(I) chloride (0.1 mg, 0.001 mmol) and triethylamine (1 ml). The mixture was stirred at room temperature overnight and the workup was similar to that of 2 using activated basic alumina (50–200 μm) for column chromatography. Subsequent recrystallization from acetone-diethyl ether gave analytically pure samples of 3 as a deep red solid. Yield: 15%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2CH3), 1.50 (s, 36H,tBu), 1.59 (s, 18H, tBu), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2CH2CH3), 2.05–2.15 (m, 24H, –CH2P), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.20–7.26 (m, 6H, aromatic Hs), 7.35–7.40 (m, 6H, aromatic Hs), 7.62 (dd, JHH=6 Hz/2 Hz, 2H, trpy), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole), 8.39, (d, JHH=2 Hz, 4H, trpy), 8.45 (s, 4H, trpy), 9.17 (d, JHH=6 Hz, 4H, trpy); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 11.4 (JPt–P=2374 Hz); IR (KBr disk, ν/cm−1): 2094s ν(CC); positive ESI-MS: 1286 [M−2OTf]2+, anal. calcd. for 3: C, 50.2%; H, 5.33%; N, 3.42%; found: C, 50.1%; H, 5.34%; N, 3.43%.
CC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2
(3)
via the divergent route.
C}2-9-nBuCarb (1).
trans-[Pt(PEt3)2Cl2]
(1 g, 1.992 mmol) was dissolved in THF (50 ml) containing copper(I) chloride (1 mg, 0.01 mmol) and NEt3
(10 ml). 3,6-Diethynyl-9-n-butylcarbazole L2
(135 mg, 0.498 mmol) in THF (20 ml) was added dropwise at 0°C, and the reaction mixture was stirred overnight at room temperature. The mixture was then filtered, and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2, washed with deionized water and dried over anhydrous MgSO4. The dried organic fraction was then concentrated under reduced pressure and pass through a column of activated basic alumina (50–200 μm) using CH2Cl2–petroleum ether (bp 40–60 °C) (1∶1, v/v) as eluent. Subsequent recrystallization from CH2Cl2–n-hexane gave 1 as pale yellow crystals. Yield: 69%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2–), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2–), 2.05–2.15 (m, 24H, –CH2P), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.21 (d, JHH=8.6 Hz, 2H, Hs at 1-position of carbazole), 7.34 (dd, JHH=8.6 Hz/1.7 Hz, 2H, Hs at 2-position of carbazole), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 15.1 (JPt–P=2401 Hz); IR (KBr disk, ν/cm−1): 2117s ν(CC); positive FAB-MS: 1202 [M]+, 1084 [M−PEt3]+; anal. calcd. for 1: C, 43.9%; H, 6.24%; N, 1.16%; found: C, 44.0%; H, 6.23%; N, 1.16%.
CC6H4CCH)]OTf (2).
1,4-Diethynylbenzene (42 mg, 0.334 mmol), [Pt(tBu3trpy)(MeCN)](OTf)2
(312 mg, 0.334 mmol) and sodium hydroxide (0.2 g) in methanol (35 ml) were heated to reflux overnight. The solvent of the red reaction mixture was removed under reduced pressure. The residue was dissolved in CH2Cl2, filtered and chomatographed on silica gel (60–230 mesh) using CH2Cl2–acetone as eluent. Recrystallization from acetone–n-hexane gave 2 as orange crystals. Yield: 42%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 1.50 (s, 18H, tBu), 1.59 (s, 9H, tBu), 7.42 (dd, JHH=6 Hz/2 Hz, 4H, –C6H4–), 7.62 (dd, JHH=6 Hz/2 Hz, 2H, trpy), 8.38, (d, JHH=2 Hz, 2H, trpy), 8.45 (s, 2H, trpy), 9.10 (d, JHH=6 Hz, 4H, trpy); IR (KBr disk, ν/cm−1): 2112s ν(CC); positive ESI-MS: 720 [M−OTf]+, anal. calcd. for 2: C, 52.4%; H, 4.60%; N, 4.83%; found: C, 52.3%; H, 4.61%; N, 4.82%.
CC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2
(3)
via the convergent route.
1
(78 mg, 0.065 mmol) and 2
(100 mg, 0.142 mmol) were dissolved in THF (15 ml) containing copper(I) chloride (0.1 mg, 0.001 mmol) and triethylamine (1 ml). The mixture was stirred at room temperature overnight and the workup was similar to that of 2 using activated basic alumina (50–200 μm) for column chromatography. Subsequent recrystallization from acetone-diethyl ether gave analytically pure samples of 3 as a deep red solid. Yield: 15%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2CH3), 1.50 (s, 36H,tBu), 1.59 (s, 18H, tBu), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2CH2CH3), 2.05–2.15 (m, 24H, –CH2P), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.20–7.26 (m, 6H, aromatic Hs), 7.35–7.40 (m, 6H, aromatic Hs), 7.62 (dd, JHH=6 Hz/2 Hz, 2H, trpy), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole), 8.39, (d, JHH=2 Hz, 4H, trpy), 8.45 (s, 4H, trpy), 9.17 (d, JHH=6 Hz, 4H, trpy); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 11.4 (JPt–P=2374 Hz); IR (KBr disk, ν/cm−1): 2094s ν(CC); positive ESI-MS: 1286 [M−2OTf]2+, anal. calcd. for 3: C, 50.2%; H, 5.33%; N, 3.42%; found: C, 50.1%; H, 5.34%; N, 3.43%.
CC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2
(3)
via the divergent route.
3,6-{TMSC
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e002.gif) CC6H4CCPt(PEt3)2CC}2-9-nBuCarb (4).
4 was synthesized according to a procedure similar to the convergent synthesis of 3, except that 1-ethynyl-4-trimethlysilylethynylbenzene was used in place of 2 and CH2Cl2 was used as the eluent instead of CH2Cl2–acetone. Yield: 78%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.24 (s, 18H, TMS), 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2CH3), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2CH2CH3), 2.05–2.15 (m, 24H, –CH2P), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.20–7.26 (m, 6H, aromatic Hs), 7.30–7.40 (m, 6H, aromatic Hs), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 11.5 (JPt–P=2374 Hz); IR (KBr disk, ν/cm−1): 2096s ν(CC); positive FAB-MS: 1525 [M]+, 1407 [M−PEt3]; anal. calcd. for 4: C, 55.1%; H, 6.62%; N, 0.92%; found: C, 55.0%; H, 6.63%; N, 0.92%.
CC6H4CCPt(PEt3)2CC}2-9-nBuCarb (4).
4 was synthesized according to a procedure similar to the convergent synthesis of 3, except that 1-ethynyl-4-trimethlysilylethynylbenzene was used in place of 2 and CH2Cl2 was used as the eluent instead of CH2Cl2–acetone. Yield: 78%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.24 (s, 18H, TMS), 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2CH3), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2CH2CH3), 2.05–2.15 (m, 24H, –CH2P), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.20–7.26 (m, 6H, aromatic Hs), 7.30–7.40 (m, 6H, aromatic Hs), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 11.5 (JPt–P=2374 Hz); IR (KBr disk, ν/cm−1): 2096s ν(CC); positive FAB-MS: 1525 [M]+, 1407 [M−PEt3]; anal. calcd. for 4: C, 55.1%; H, 6.62%; N, 0.92%; found: C, 55.0%; H, 6.63%; N, 0.92%.
3,6-{HC
CC6H4CCPt(PEt3)2CC}2-9-nBuCarb (5).
The trimethylsilyl protecting groups in 4 were removed by the standard procedure13 to give 5. Yield: 91%. 1H NMR (CDCl3, 298 K, 400 MHz): δ 0.92 (t, JHH=6.8 Hz, 3H, –NCH2CH2CH2CH3), 1.20–1.30 (m, 38H, –PCH2CH3 and –NCH2CH2CH2CH3), 1.81 (quintet, JHH=6.8 Hz, 2H, –NCH2CH2CH2CH3), 2.05–2.15 (m, 24H, –CH2P), 3.09 (s, –CCH), 4.22 (t, JHH=6.8 Hz, 2H, NCH2–), 7.20–7.26 (m, 6H, aromatic Hs), 7.30–7.40 (m, 6H, aromatic Hs), 7.92 (d, JHH=1.7 Hz, 2H, Hs at 4-position of carbazole); 31P{1H} NMR (CDCl3, 298 K, 162 MHz): δ 11.5 (JPt–P=2375 Hz); IR (KBr disk, ν/cm−1): 2096s ν(CC); positive FAB-MS: 1381 [M]+, 1263 [M−PEt3]; anal. calcd. for 5: C, 55.6%; H, 6.15%; N, 1.01%; found: C, 55.5%; H, 6.16%; N, 1.01%.
[3,6-{Pt(tBu3trpy)(C
CC6H4CC)Pt(PEt3)2CC}2-9-nBuCarb](OTf)2
(3).
5
(100 mg, 0.066 mmol), [Pt(tBu3trpy)(MeCN)](OTf)2
(153 mg, 0.164 mmol) and sodium hydroxide (0.5 g) in methanol/THF (1∶1, v/v, 35 ml) was heated to reflux overnight. The solvent of the red reaction mixture was removed under reduced pressure. The residue was dissolved in CH2Cl2, filtered and chomatographed on activated basic alumina (50–200 μm) using CH2Cl2–acetone as eluent. Recrystallization from acetone–n-hexane gave 3 as a deep red solid. Yield: 29%.
X-Ray crystallography
A single crystal of complex 1 of dimensions 0.3×0.2×0.1 mm mounted in a glass capilliary with mother liquor was used for data collection at 28°C on a MAR diffractometer with a 300 mm image plate detector using graphite monochromatized Mo-Kα radiation (λ=0.71073 Å). The images were interpreted and intensities integrated using the program DENZO.14 The structure was solved by direct methods employing the SHELXS-97 program.15 Positions of other non-hydrogen atoms were found after successful refinement by full-matrix least-squares using the program SHELXL-97.16 The ethyl groups of the PEt3 ligands have relatively high thermal parameters and a disorder model was not introduced. One crystallographic asymmetric unit consists of one formula unit, including one ethanol solvent molecule. In the final stage of least-squares refinement, atoms of the ethyl groups and the ethanol solvent molecule were refined isotropically, and other non-hydrogen atoms anisotropically. The positions of hydrogen atoms were calculated based on a riding mode with thermal parameters equal to 1.2 times those of the associated C atoms, and participated in the calculation of final R indices.
A single crystal of complex 2 of dimensions 0.6×0.2×0.1 mm mounted in a glass capillary was used for data collection at 28°C on a MAR diffractometer with a 300 mm image plate detector using graphite monochromatized Mo-Kα radiation (λ=0.71073 Å). The images were interpreted and intensities integrated using the program DENZO.14 The structure was solved by direct methods employing the SIR-97 program.17 Pt and many non-hydrogen atoms were located according to direct methods and successive least-squares Fourier cycles. Positions of other non-hydrogen atoms were found after successful refinement by full-matrix least-squares using the program SHELXL-97.16 One water, one acetone and half of n-hexane solvent molecules were also located. For convergence of least-square refinements, restraints were applied to the CF3SO3− anion, assuming similar C–F, S–O, S⋯O and C⋯O bond lengths and distances, respectively. Within the anion, due to the extremely high thermal parameter for C, restraints for the same thermal parameters were assumed for the C and three F atoms. One crystallographic asymmetric unit consists of one formula unit, including one water, one acetone and half of n-hexane solvent molecules and one anion. In the final stage of least-squares refinement, F, C and O atoms of the anion were refined isotropically, and the other non-hydrogen atoms anisotropically. Hydrogen atoms were generated by the program SHELXL-97.16 The positions of hydrogen atoms were calculated based on a riding mode with thermal parameters equal to 1.2 times those of the associated C atoms, and participated in the calculation of final R indices.
The crystallographic data and structure refinement details are summarized in Table 2, and selected bond distances and angles are listed in the captions for Fig. 1 and 2.
| Complex | 1 | 2 |
| Chemical formula | C44H75NCl2P4Pt2·EtOH | C38H40F3N3O3PtS·Me2CO·H2O·½n-hexane |
| Formula weight | 1249.08 | 990.06 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 10.420(2) | 11.259(2) |
| b/Å | 15.207(3) | 15.053(3) |
| c/Å | 19.435(4) | 15.134(3) |
| α/° | 106.31(3) | 103.65(3) |
| β/° | 101.79(3) | 111.31(3) |
| γ/° | 99.40(3) | 98.96(3) |
| U/Å3 | 2812.3(10) | 2237.9(7) |
| Z | 2 | 2 |
| T/K | 301 | 293 |
| λ/Å | 0.71073 | 0.71073 |
| μ(Mo-Kα)/cm−1 | 5.208 | 3.239 |
| Total reflections | 6049 | 13650 |
| Unique reflections | 4287 (Rint=0.0319) |
6935 (Rint=0.0461) |
| Goodness-of-fit | 0.889 | 1.018 |
|
R
1
[I>2σ(I)] |
0.0536 | 0.0820 |
CCDC reference numbers 182813 and 182814. See http://www.rsc.org/suppdata/nj/b2/b210772b/ for crystallographic data in CIF or other electronic format.
Acknowledgements
V. W. W. Y. acknowledges support from The University of Hong Kong Foundation for Educational Development and Research Limited, and the receipt of a Croucher Senior Research Fellowship (2000–2001) from the Croucher Foundation. The work described in this paper has been supported by a CERG Grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. HKU 7123/00P). C. H. T. acknowledges the receipt of a Croucher Scholarship and a Li Po Chun Scholarship, administered by the Croucher Foundation and the Li Po Chun Charitable Fund respectively. K. M. C. W. and N. Z. acknowledge the receipt of a University Postdoctoral Fellowship, administered by The University of Hong Kong.References
- N. Ohshiro, F. Takei, K. Onitsuka and S. Takahashi, J. Organomet. Chem., 1998, 569, 195 CrossRef CAS; N. Ohshiro, F. Takei, K. Onitsuka and S. Takahashi, Chem. Lett., 1996, 871 CAS; K. Onitsuka, M. Fujimoto, N. Ohshiro and S. Takahashi, Angew. Chem., Int. Ed., 1999, 38, 689 CrossRef CAS; S. Leininger and P. J. Stang, Organometallics, 1998, 17, 3981 CrossRef CAS; M. Uno and P. H. Dixneuf, Angew. Chem., Int. Ed., 1998, 37(12), 1714 CrossRef CAS.
- Y. Zhang, T. Wada, L. Wang and H. Sasabe, Chem. Mater., 1997, 9, 2798 CrossRef CAS; S. Maruyama, X. T. Tao, H. Hokari, T. Noh, Y. Zhang, T. Wada, H. Sasabe, H. Suzuki, T. Watanabe and S. Miyata, J. Mater. Chem., 1999, 9, 893 RSC.
- K. R. J. Thomas, J. T. Lin, Y. T. Tao and C. W. Ko, J. Am. Chem. Soc., 2001, 123, 9404 CrossRef; Z. Zhu and J. S. Moore, J. Org. Chem., 2000, 65, 116 CrossRef CAS.
- K. R. J. Thomas, J. T. Lin, Y. Y. Lin, C. Tsai and S. S. Sun, Organometallics, 2001, 20, 2262 CrossRef CAS.
- V. W. W. Yam, C. H. Tao, L. Zhang, K. M. C. Wong and K. K. Chueng, Organometallics, 2001, 20, 453 CrossRef CAS.
- V. W. W. Yam, R. P. L. Tang, K. M. C. Wong, C. C. Ko and K. K. Chueng, Inorg. Chem., 2001, 40, 571 CrossRef CAS.
- V. W. W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Chueng, Chem. Eur. J., 2002, 8, 4066 CrossRef CAS.
- V. W. W. Yam, R. P. L. Tang, K. M. C. Wong and K. K. Chueng, Organometallics, 2001, 20, 4476 CrossRef CAS.
- V. W. W. Yam, K. M. C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS.
- T. K. Aldridge, E. M. Stacy and D. R. McMillin, Inorg. Chem., 1994, 33, 722 CrossRef CAS; S. W. Lai, M. C. W. Chan, K. K. Cheung and C. M. Che, Inorg. Chem., 1999, 38, 4262 CrossRef CAS; R. Büchner, C. T. Cunningham, J. S. Field, R. Haines, D. R. McMillin and G. C. Summerton, J. Chem. Soc., Dalton Trans., 1999, 711 RSC; R. Büchner, J. S. Field, R. J. Haines, C. T. Cunningham and D. R. McMillin, Inorg. Chem., 1997, 36, 3952 CrossRef; J. F. Michalec, S. A. Bejune and D. R. McMillin, Inorg. Chem., 2000, 39, 2708 CrossRef CAS.
- M. Younus, A. Kohler, S. Cron, N. Chawdhury, M. R. A. Al-Mandhary, M. S. Khan, J. Lewis, N. J. Long, R. H. Friend and P. R. Raithby, Angew. Chem., Int. Ed., 1998, 37, 3036 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, 3rd edn., Pergamon, Oxford, 1988 Search PubMed.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 CrossRef CAS.
- Z. Otwinowski and W. Minor, Macromolecular Crystallography, Part A, eds. C. W. Carter and R. M. Sweet Jr., Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- SHELXS-97, G. M. Sheldrick, 1997: SHELX97, Programs for Crystal Structure Analysis (Release 97-2), University of Goettingen, Germany.
- SHELXL-97, G. M. Sheldrick, 1997: SHELX97, Programs for Crystal Structure Analysis (Release 97-2), University of Goettingen, Germany.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1998, 32, 115 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |