DOI:
10.1039/B206923G
(Paper)
New J. Chem., 2003,
27, 155-160
Thin films by metal organic deposition of Fe–Mo–S molecular clusters: synthesis and crystal structure of [Cp2MoFe2(μ3-S)2(CO)6]
Received
(in Montpellier, France)
11th July 2002
, Accepted 14th October 2002
First published on 28th November 2002
Introduction
The continuing interest in polynuclear and cluster complexes containing iron and molybdenum is largely due to their often unique structural features, reactivities and applications in homogeneous and heterogeneous catalysis.1 Furthermore, the presence of the Fe–Mo protein in nitrogenase, a class of enzymes able to catalyze the six-electron reduction of N2 to NH3, with concomitant reduction of protons to H2, has triggered considerable research activity in modeling the active site of this enzyme.2 A large number of Fe–Mo–S clusters have thus been prepared and structurally characterized.
The cluster [Mo2Fe2(μ3-S)2Cp2(CO)8]
(2) was found to exist in two isomeric forms. The metal core in the isomer first reported has a butterfly structure3 whereas it is planar in the second isomer.4 When considering the Mo2Fe2S2 apices, the bitetrahedral structure (with the Mo–Mo edge being shared) is particularly interesting since it differs from the octahedral structure predicted on the basis of the electron counting rules.5 In a formal sense, the isomers with a butterfly or a planar metal core can be interconverted within the bitetahedral structure by changing the orientation of the Fe–S vector. A MgO-supported bimetallic catalyst derived from this cluster showed high selectivity for C2 products in CO methanation.6 The suggestion that the cluster is not fragmenting and re-aggregating into larger crystallites was based on the fact that the selectivities of the [Mo2Fe2] catalyst differ from those of Mo/Al2O3, MoS2 or Fe/Al2O3.7
With the same heterogeneous cluster-derived [Mo
2Fe
2]
catalyst on various
oxides, activities for hydrodesulfurization of
thiophene were comparable to those of commercial HDS
catalysts while showing decreased H
2 consumption.
6,7b,8 It is interesting to note that ultrafine iron–
molybdenum oxide particles have recently been obtained from monometallic precursors by the sol–gel method. These particles were shown to be a selective
catalyst for the partial oxidation of
toluene to
benzaldehyde.
9
In view of the specific properties of the bimetallic metal particles generated from cluster 2,6–8 we sought to explore its potential for the preparation of thin films by Metal Organic Deposition (MOD) and by Metal Organic Chemical Vapour Deposition (MOCVD). Despite the potential of the latter method for the synthesis of multiphasic materials,10 only few mixed-metal molecular clusters have been used as precursors in MOCVD.11
For comparative purposes, we also considered it of interest to prepare new Fe–Mo–S clusters with a different metal ratio that would still contain ligands relatively easy to eliminate thermally, like CO and organic moieties. We thus describe in this paper the synthesis of the new cluster [Cp2MoFe2(μ3-S)2(CO)6]
(1) from [Cp2MoH2] and [Fe2(μ2-η2-S2)(CO)6]. Interestingly, the latter has been used as MOCVD single-source precursor to produce FeS films.12 We have investigated the potential of the two Fe–Mo–S clusters 1 and 2 as single-source precursors for MOCVD and MOD and compared the chemical composition of the resulting solid materials.
Results and discussion
Synthesis and structure of the Fe–Mo–S clusters
Whereas cluster 2 was prepared from the reported3 reaction between [Cp2Mo2(CO)4](Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mo) and [Fe2(μ2-η2-S2)(CO)6], the reaction of the latter complex with [Cp2MoH2] afforded the new cluster [Cp2MoFe2(μ3-S)2(CO)6]
(1) in 20% yield (eqn. 1):
Mo) and [Fe2(μ2-η2-S2)(CO)6], the reaction of the latter complex with [Cp2MoH2] afforded the new cluster [Cp2MoFe2(μ3-S)2(CO)6]
(1) in 20% yield (eqn. 1):| |  | (1) |
Cluster 1 was characterized by 1H NMR and IR (ν(CO) region) spectroscopic studies (see Experimental section). The solid state structure was established by single crystal X-ray diffraction (Fig. 1). Crystallographic data and selected bond distances and angles are given in Tables 1 and 2, respectively.
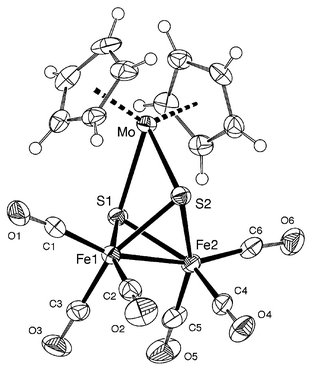 |
| | Fig. 1 ORTEP view of the molecular structure of 1. | |
| Formula |
C16H10Fe2MoO6S2 |
|
M
|
570 |
| Crystal system |
monoclinic |
| Space group |
P21/n |
|
T/K |
293 |
|
a/Å |
8.217(5) |
|
b/Å |
13.703(5) |
|
c/Å |
16.310(5) |
|
β/deg |
92.176(5) |
|
V/Å3 |
1835.1(14) |
|
Z
|
4 |
|
ρ(calcd)/g cm−3 |
2.063 |
| Radiation, λ Mo Kα/Å |
0.71069 |
|
μ/mm−1 |
2.491 |
| No. of reflcns. |
4203 |
No. of reflcns. I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) >2σ(I) >2σ(I) |
3030 |
| Residuals: R; Rw |
0.0320; 0.1018 |
| GOF |
0.940 |
Table 2 Selected bond distances (Å) and angles (deg) in 1a
|
Numbers in parentheses are estimated standard deviations in the least significant digits.
|
| Bond distances |
| Mo–S(1) |
2.540(2) |
Mo–S(2) |
2.550(2) |
| Fe(1)–S(1) |
2.294(2) |
Fe(1)–S(2) |
2.322(2) |
| Fe(2)–S(1) |
2.321(2) |
Fe(2)–S(2) |
2.288(2) |
| S(1)–S(2) |
2.761(4) |
Fe(1)–Fe(2) |
2.464(2) |
| Fe(1)–C(1) |
1.783(5) |
C(1)–O(1) |
1.157(6) |
| Fe(1)–C(2) |
1.792(5) |
C(2)–O(2) |
1.142(6) |
| Fe(1)–C(3) |
1.789(5) |
C(3)–O(3) |
1.140(6) |
| Fe(2)–C(4) |
1.786(5) |
C(4)–O(4) |
1.148(5) |
| Fe(2)–C(5) |
1.799(5) |
C(5)–O(5) |
1.133(6) |
| Fe(2)–C(6) |
1.783(5) |
C(6)–O(6) |
1.154(6) |
| Bond angles |
| Fe(1)–S(1)–Fe(2) |
64.53(4) |
Fe(1)–S(1)–Mo |
100.87(4) |
| Fe(2)–S(2)–Fe(1) |
64.60(4) |
Fe(2)–S(2)–Mo |
100.33(5) |
| S(1)–Fe(1)–S(2) |
73.47(4) |
S(1)–Fe(1)–C(1) |
107.5(2) |
| C(1)–Fe(1)–Fe(2) |
161.4(2) |
C(1)–Fe(1)–S(2) |
109.45(16) |
| S(1)–Fe(2)–S(2) |
73.62(4) |
S(1)–Fe(2)–Fe(1) |
57.21(4) |
| S(1)–Fe(2)–C(6) |
115.5(2) |
Fe(1)–Fe(2)–C(6) |
161.2(2) |
| S(2)–Fe(2)–C(6) |
103.7(2) |
Fe(1)–C(1)–O(1) |
174.2(5) |
| Fe(1)–C(2)–O(2) |
178.3(4) |
Fe(1)–C(3)–O(3) |
178.0(5) |
The molecule possesses almost a mirror plane passing through the Mo atom and the middle of the Fe–Fe bond. It contains a Fe2S2 butterfly moiety, in which the metal–metal bond constitutes the hinge whereas the S atoms form the wing tips. This contrasts with the situation in [Fe2(μ2-η2-S2)(CO)6]. The carbonyl ligands adopt a typical sawhorse arrangement. The Cp2Mo fragment forms a bridge between the two S atoms in such a way that the coordination geometry about the Mo centre is tetrahedral, when the Cp centroid is taken as an apex. Each metal centre has a 18e configuration and the total electron count of 52 is consistent with the presence of only one metal–metal bond. The “double star” structure of 1, with two opposite μ3-S atoms, is reminiscent of the structural features found in other Fe2S2Mo clusters.13 The Fe–Fe bond distance is 2.464(2)
Å. The average Fe–S (2.306(2)
Å) and Mo–S (2.545(2)
Å) bond distances are very similar to those found in [{Fe(CO)3(μ3-S)}2Mo(NNMe2)2(PPh3)](Fe–Fe).13a It is interesting to note that this structural type has often been encountered with metals other than Mo, such as Ni,14,15 Pd,15–17 Pt,15–17 Ti,15 Ge15 and Sn.15 To the best of our knowledge, the structure of 1 represents the first example where a Cp2M fragment is chelated by a [Fe2(μ2-S)2(CO)6]2− unit. The titanium analog formulated as [Cp2TiFe2(μ3-S)2(CO)6] was claimed but not fully characterized owing to its insolubility.15a
The reaction in eqn. (1) can be envisaged to proceed by a nucleophilic attack of the lone pair of electrons in an orbital situated between the two hydride ligands of [Cp2MoH2], a well known Lewis base,18 on the S–S bond of [Fe2(μ2-η2-S2)(CO)6]. The LUMO of the latter having a S–S antibonding character,19 this results in the breaking of the S–S bond and formation of the Fe2S2 butterfly core present in 1 with a Fe–Fe hinge instead of a S–S hinge in the precursor. This formal insertion of the Mo centre into the S–S bond is accompanied by loss of H2, which restores the 18e configuration of the Mo atom.
Preparation and characterization of the films obtained from clusters 1 and 2
Both precursors [Cp2MoFe2(μ3-S)2(CO)6]
(1) and [Cp2Mo2Fe2(μ3-S)2(CO)8]
(2) were initially tested in a Metal Organic Chemical Vapour Deposition (MOCVD) process. However, it was not possible to vaporize them even at high temperature (200°C/10−3 mbar) owing to their rather low vapour pressure. Consequently, a solution-coating process was envisaged to achieve metal organic deposition on silicon or borosilicate glass substrates. Both substrates were used as received. Metal Organic Deposition (MOD) is a versatile technique to produce inorganic powders and thin films as it requires neither low-pressure conditions (MOCVD) nor gel preparation (sol–gel) steps. MOD is based on the dissolution of the desired single-source molecular precursor and spin- or dip-coating the resulting solution on an appropriate substrate. The as-deposited film is dried at room temperature and then gradually heated to high temperature in order to pyrolyse the organic moieties and produce an inorganic film. In this study, compounds 1 and 2 were dissolved in a mixture of toluene and chloroform and a small amount of ethylene glycol (1% v/v) was added to obtain the coating solution, which was then spin-coated on the desired substrate. Owing to the limited solubility of the inorganic clusters, the concentrations of the solutions for deposition were kept low (1.0–0.05 mmol). The thin films were obtained by spin-coating the solutions followed by drying under ambient conditions and a subsequent pyrolysis step in a horizontal furnace up to 500°C (heating rate, 10°C min−1). Typically, three deposition cycles (60 seconds) with a spinning speed of 5000 rpm were performed to obtain films with a thickness of few hundred nanometers. Finally, the samples were annealed at 500°C for 2 h. The heat-treated films were brown and green for the precursors 1 and 2, respectively. The films were weakly adherent on the substrates, especially on silicon, and could be easily scratched to obtain solid materials, which were then used for different analyses. In the following discussion, the thin film samples obtained from [Cp2MoFe2S2(CO)6]
1 and [Cp2Mo2Fe2S2(CO)8]
2 have been designated S1 and S2, respectively. The results obtained on silicon and glass substrates showed no major differences in terms of chemical composition and crystalline components, however the adhesion was weaker on silicon substrates.
Infrared spectroscopy.
The infrared spectra of the films obtained from precursor 2 are shown in Fig. 2. In the IR spectrum of the as-deposited film before heat treatment (curve a), the ν(CO) and ν(CH) absorptions due to the carbonyl and cyclopentadienyl ligands are observed at 1972–2048 and 3100–3300 cm−1, respectively, which suggests that the structure of the precursor is probably retained in the coating solution. As expected, the absorptions due to the organic ligands are absent in the thermally-treated film (curve b). However, peaks corresponding to iron–oxygen and molybdenum–oxygen stretching frequencies are observed in the typical range.
![Infrared spectra of the (a) as-deposited and (b) heat-treated films obtained from MOD of [Cp2Mo2Fe2S2(CO)8]
(2).](/image/article/2003/NJ/b206923g/b206923g-f2.gif) |
| | Fig. 2 Infrared spectra of the (a) as-deposited and (b) heat-treated films obtained from MOD of [Cp2Mo2Fe2S2(CO)8]
(2). | |
Scanning electron microscopy and energy dispersive X-ray analysis.
The metal ratios in the as-deposited and calcined films on silicon substrates were investigated by EDX analyses, which revealed the Fe/Mo ratio in sample S1 to be close to the metal stoichiometry (Fe/Mo=2∶1) of the precursor molecule. However, sample S2 showed extreme inhomogeneity with respect to the distribution and ratio of the metals, presumably due to the dissociation of the heterometal framework and/or formation of other cluster aggregates with different Fe/Mo ratios. The above observations were also corroborated by ESCA analysis (vide supra). The SEM image (Fig. 3a) of an oven-dried (150°C) film obtained from [Cp2MoFe2(μ3-S)2(CO)6]
1, exhibits an incomplete but homogeneous coverage with a regular morphology made up of spherical particles. However, the film delaminated upon calcination at 500°C in the case of S1, revealing islands of facetted crystals (Fig. 3b). A rough surface is observed in S2 upon heat-treatment where large grains are formed by agglomeration of several tiny crystallites (Fig. 3c). The films obtained on borosilicate glass substrates exhibited better coverage and higher homogeneity in terms of microstructure as observed in the SEM images of films obtained using 1 as precursor (Fig. 4a). The high-resolution image (Fig. 4b) shows that film growth occurs by island formation. The observed morphology originates from a thermal mismatch between the deposit and the substrate, which is unfavorable for the formation of a continuous film.
![SEM images of the films obtained from (a) the MOD of [Cp2MoFe2S2(CO)6]
(1) and heat-treated S1
(b) and S2
(c) samples (Magnification: ×800 and ×1500, respectively).](/image/article/2003/NJ/b206923g/b206923g-f3.gif) |
| | Fig. 3 SEM images of the films obtained from (a) the MOD of [Cp2MoFe2S2(CO)6]
(1) and heat-treated S1
(b) and S2
(c) samples (Magnification: ×800 and ×1500, respectively). | |
 |
| | Fig. 4 SEM images of the films obtained on a borosilicate glass substrate from a heat-treated S1 sample: (a) magnification 3400× and (b) magnification 52000×. | |
Powder diffractometry.
The phase characterization was performed by powder X-ray diffraction studies of the films S1 and S2 obtained after heat-treatment (500°C)
(Fig. 5). In both cases, the major crystalline component was identified to be the solid solution Fe2MoxOz
(Fe2O3∶xMoO3, where 3<x<4, z is a variable parameter depending on the exact Fe/Mo ratio (Powder Diffraction File [15-371])20). Together with a significant amorphous background, small amounts of crystalline iron and molybdenum oxides (Fe2O3, PDF [33-664])20
(MoO3, PDF [35-609])20 were observed in S1 and S2, respectively. The formation, at rather low temperature (500°C), of a crystalline mixed-metal oxide as the major phase, underlines the advantages of heterometallic clusters as precursors to complex oxides. The detection of monometallic oxides indicates partial dissociation of the mixed-metal clusters 1 and 2 either in the coating solution or on the substrate surface during the drying step. This is also evident from the colours of the films (see later). Despite the presence of metal–sulfur and metal–carbon bonds in the precursors, the resulting solid is oxidic in nature because the heat treatment was carried out in air; the ligands probably decompose to SO2 and CO2 gases, respectively.
![Powder X-ray diffractograms of the films obtained from [Cp2MoFe2S2(CO)6]
(S1) and [Cp2Mo2Fe2S2(CO)8]
(S2).](/image/article/2003/NJ/b206923g/b206923g-f5.gif) |
| | Fig. 5 Powder X-ray diffractograms of the films obtained from [Cp2MoFe2S2(CO)6]
(S1) and [Cp2Mo2Fe2S2(CO)8]
(S2). | |
X-Ray photoelectron spectroscopy.
The elemental composition of the films and the oxidation states of the constituent metals (Fe and Mo) were determined by XPS (Figs. 6 and 7). The spectra of the samples S1 and S2
(Fig. 6) show the characteristic peaks of Mo and Fe species. The experimental values for the elemental distribution in the two samples were found to be Fe, 8.53%; Mo, 5.10%; O, 59.22%; C, 10.11%; Si, 17.05% for S1 and Fe, 2.32%; Mo, 10.10%; O, 59.81%; C, 15.84%; Si, 11.93% for S2, respectively. In sample S1, the Fe/Mo element ratio (1.67∶1) is close to the expected value (2∶1), but in the case of S2, the Fe/Mo ratio (0.25∶1) is significantly different from the elemental stoichiometry present in the precursor (Fe/Mo=1∶1). Whereas the sulfur content in the films is negligible (<0.5 at.%), the amount of residual carbon was substantial (10–15 at.%) in the film thermally treated for 1 h, presumably owing to the incomplete removal of the organic residues. The carbon contamination could be reduced (<5 at.%) upon annealing the films at 500°C for 6 h. The appearance of the substrate (Si) signal in the XPS spectra (Fig. 6) indicates incomplete coverage by the film, which is also supported by the SEM images. For comparison, the Mo 3d5/2 peaks for MoO2 and MoO3 are shown in Fig. 7
(the data have been taken from the ESCA handbook).21 It is evident from Fig. 7 that both samples contain MoO3 and the characteristic MoO2 feature at 229 eV is not observed. The small peak corresponding to In 3d signal in the spectrum (Fig. 6) of sample S1 is probably due to external contamination. A comparison of Fe 2p (Fig. 7b) in S1 and S2 indicates that S2 contains besides Fe3+ small amount of Fe2+ species, which could result from partial reduction of Fe3+ due to the removal of oxygen during the sputtering process.
 |
| | Fig. 7 Mo 3d (a) and Fe 2p (b) XPS spectra of S1 and S2. | |
Absorption spectra.
The heat-treated films obtained on silicon substrates were reddish brown (S1) or yellowish green (S2). It appears that the colours of the deposits (Fe2MoxOz, where 3<x<4, is gray-green) are dominated by the presence of iron (brown) and molybdenum (yellow) oxides, which were observed in the X-ray diffractograms (Fig. 5) and also indicated by the determined metal ratios (ESCA evidence). These two metal oxides are common inorganic pigments, well known for their intense colours. Consequently, films containing Fe or Mo oxide reflect the colour of the corresponding oxides, namely, brown and yellowish green, the colour of the Fe–Mo mixed-metal oxide being masked. The absorption spectra (400–700 nm) of the two samples are shown in Fig. 8. As expected, S1 shows a non-zero absorption coefficient in the recorded wavelength range (400–600 nm) in contrast to Fe3+ oxide22 in S2.
![Absorption spectra of the films obtained from the MOD of [Cp2MoFe2S2(CO)6]
(S1) and [Cp2Mo2Fe2S2(CO)8]
(S2).](/image/article/2003/NJ/b206923g/b206923g-f8.gif) |
| | Fig. 8 Absorption spectra of the films obtained from the MOD of [Cp2MoFe2S2(CO)6]
(S1) and [Cp2Mo2Fe2S2(CO)8]
(S2). | |
Conclusion
The new cluster [Cp2MoFe2(μ3-S)2(CO)6]
(1) was obtained by the reaction of [Fe2(μ2-η2-S2)(CO)6] with [Cp2MoH2] and characterized notably by single crystal X-ray diffraction analysis. This cluster and the known tetranuclear [Mo2Fe2] cluster 2 have been used in Metal Organic Deposition (MOD) processes. Despite their inability to sublime, even at high temperature, which hampered CVD investigations, thin film deposits were obtained from MOD of both precursors on silicon and borosilicate substrates. XRD analyses of the deposits on silicon revealed in both cases the presence as the major crystalline phase of the mixed-metal oxide phase Fe2MoxOz
(3<x<4) and, in the case of 1, the thin film obtained is homogeneous (SEM) and the metal ratio of the initial precursor is nearly retained (EDX, XPS).
Experimental
All reactions were performed in standard Schlenk-type flasks under a dry nitrogen atmosphere. Solvents were dried by conventional methods and freshly distilled under nitrogen. Deuterated solvents for NMR spectroscopy were degassed, dried and stored over molecular sieves (4 Å Merck). The NMR spectra were recorded on a FT Bruker AC 300 spectrometer (1H, 300 MHz). Chemical shifts are given in ppm; they were measured at ambient temperature and are relative to TMS for 1H. The IR spectra were recorded in the region 4000–400 cm−1 on a Perkin-Elmer 1600 FT-IR instrument. The complexes [Fe2(μ2-η2-S2)(CO)6],23
[Cp2MoH2]18a and [Mo2Fe2(μ3-S)2Cp2(CO)8]32 were prepared as reported in the literature.
Room temperature powder X-ray diffraction patterns were obtained on a STOE diffractometer operating with a Cu Kα radiation. The scanning electron imaging (SEM) and energy dispersive X-ray (EDX) analysis were performed on an EDX coupled scanning electron microscope CAM SCAN S4. The XPS analyses were performed on a Surface Science Instrument (SSI M-Probe) using Al-Kα radiation. The total energy resolution of the M-Probe was about 0.8 eV. The peak positions were corrected using residual carbon as binding energy standard located at +284.6 eV. The absorption spectra were recorded on a BENTHAM optical spectrometer. A self-constructed spin coater working with NECKAR motors was used for depositing the films.
Synthesis of [Cp2MoFe2S2(CO)6]
(1).
To a stirred solution of [Cp2MoH2]
(0.35 g, 1.54 mmol) in THF (20 ml) was added slowly at −76°C a solution of [Fe2(μ2-η2-S2)(CO)6]
(0.53 g, 1.54 mmol) in THF (20 ml) under nitrogen. The mixture turned immediately dark green-brown. Under constant agitation, the temperature was slowly raised to room temperature in 2 h. The resulting dark green-brown mixture was evaporated to dryness under vacuum and the residue was chromatographed on a silica gel column. Elution with hexane gave a red-orange product (0.01 g), which was not identified further. IR (hexane): ν(CO) 2045, 2008 cm−1. Elution with a mixture of CH2Cl2/pentane (1∶1) gave a dark green product identified as 1, which was recrystallized from CH2Cl2/pentane. Yield: 0.180 g, 20%. IR (CH2Cl2): ν(CO) 2056 (m), 2018 (s), 1981 (m), 1974 (sh) cm−1. 1H NMR (C6D6): δ 4.33 (s, Cp).
Acknowledgements
We are grateful to Prof. R. Welter (ULP-Strasbourg) for the single crystal structure determination of 1, to the CNRS for support, the Ministère de la Recherche for a PhD grant to N.A. (Réseau doctoral franco-allemand) and the “Université franco-allemande” for mobility support to N.A. The Deutsche Forschungsgemeinschaft is also gratefully acknowledged for support within the Graduiertenkolleg 532 “Physical Methods for the Structural Investigations of New Materials”.
References
-
P. Braunstein and J. Rosé, Heterometallic Clusters in Catalysis, in Metal Clusters in Chemistry, eds. P. Braunstein, L. A. Oro and P. R. Raithby, Wiley-VCH, Weinheim (Germany), 1999, vol. 2, p. 616 and references cited therein Search PubMed.
- S. M. Malinak and D. Coucouvanis, Prog. Inorg. Chem., 2001, 49, 599 Search PubMed and references cited therein.
- P. Braunstein, J.-M. Jud, A. Tiripicchio, M. Tiripicchio Camellini and E. Sappa, Angew. Chem., Int. Ed. Engl., 1982, 21, 307 CrossRef.
- P. D. Williams, M. D. Curtis, D. N. Duffy and W. M. Butler, Organometallics, 1983, 2, 165 CrossRef CAS.
- T. P. Fehlner, C. E. Housecroft and K. Wade, Organometallics, 1983, 2, 1426 CrossRef CAS.
- M. D. Curtis, J. E. Penner-Hahn, J. Schwank, O. Baralt, D. J. McCabe, L. T. Thompson and G. Waldo, Polyhedron, 1988, 7, 2411 CrossRef CAS.
-
(a)
M. D. Curtis, J. Schwank, L. T. Thompson and P. D. Williams, US Patent 4,605,751, 1986; Chem. Abstr., 1986, 105, 214560t Search PubMed;
(b)
M. D. Curtis, in Report of the International Seminar on Organic Chemistry on Metal Clusters and Surfaces, S. Vittoria d'Alba, July 26–31, 1992, p. 50 Search PubMed.
- M. D. Curtis, J. Cluster Sci., 1996, 7, 247 CAS.
-
(a) W. Kuang, Y. Fan and Y. Chen, Langmuir, 2000, 16, 5205 CrossRef CAS;
(b) W. Kuang, Y. Fan and Y. Chen, J. Colloid Interface Sci., 1999, 215, 364 CrossRef CAS.
-
(a) S. Mathur, M. Veith, H. Shen and S. Hüfner, Mater. Sci. Forum, 2002, 386, 341 Search PubMed;
(b) S. Mathur, M. Veith, H. Shen, S. Hüfner and M. Jilavi, Chem. Mater., 2002, 14, 568 CrossRef CAS;
(c) M. Veith, S. Mathur, H. Shen, N. Lecerf, S. Hüfner and M. Jilavi, Chem. Mater., 2001, 13, 4041 CrossRef CAS;
(d) H.-W. T. Sun, H.-F. Wang and K.-M. Chi, J. Mater. Chem., 2000, 10, 1231 RSC;
(e) M. Veith, N. Lecerf, S. Mathur, H. Shen and S. Hüfner, Chem. Mater., 1999, 11, 3103 CrossRef CAS;
(f) R. A. Fischer and J. Weiss, Angew. Chem., Int. Ed., 1999, 38, 2830 CrossRef;
(g) J.-C. Hierso, R. Feurer and P. Kalck, Coord. Chem. Rev., 1998, 178–180, 1811 CrossRef CAS.
- E. P. Boyd, D. R. Ketchum, H. Deng and S. G. Shore, Chem. Mater., 1997, 9, 1154 CrossRef CAS.
- S.-G. Shyu, J.-S. Wu, C.-C. Wu, S.-H. Chuang and K.-M. Chi, Inorg. Chim. Acta, 2002, 334, 276 CrossRef CAS.
-
(a) P. T. Bishop, J. R. Dilworth and S. Morton, J. Organomet. Chem., 1988, 341, 373 CrossRef CAS;
(b) D. E. Barber, R. F. Bryan, M. Sabat, K. S. Bose and B. A. Averill, Inorg. Chem., 1996, 35, 4635 CrossRef CAS.
- W. H. Watson, A. Nagl, M.-J. Don and M. G. Richmond, J. Chem. Crystallogr., 1999, 29, 871 CrossRef CAS.
-
(a) M. Cowie, R. L. Dekock, T. R. Wagenmaker, D. Seyferth, R. S. Henderson and M. K. Gallagher, Organometallics, 1989, 8, 119 CrossRef CAS;
(b) D. Seyferth, R. S. Henderson and L.-C. Song, Organometallics, 1982, 1, 125 CrossRef CAS.
- A. A. Pasynskii, B. I. Kolobkov, S. E. Nefedov, I. L. Eremenko, E. S. Koltrun, A. I. Yanovshy and Y. T. Struchkov, J. Organomet. Chem., 1993, 454, 229 CrossRef CAS and references cited therein.
- Z.-G. Fang, P. M. N. Low, S.-C. Ng and T. S. A. Hor, J. Organomet. Chem., 1994, 483, 17 CrossRef CAS.
-
(a) N. D. Silavwe, M. P. Castellani and D. R. Tyler, Inorg. Synth., 1992, 29, 204 CAS;
(b)
M. J. Morris, in Comprehensive Organometallic Chemistry II, eds. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, 1995, vol. 5, p. 393 Search PubMed;
(c) H. Brunner, M. Muschiol, T. Neuhierl and B. Nuber, Chem. Eur. J., 1998, 4, 168 CrossRef CAS.
-
(a) R. L. DeKock, E. J. Baerends and A. Oskam, Inorg. Chem., 1983, 22, 4158 CrossRef CAS;
(b) R. L. DeKock, E. J. Baerends and R. Hengelmolen, Organometallics, 1984, 3, 289 CrossRef.
- JCPDS Powder Diffraction File, File Card Nos. PDF [15-371], [33-664] and [35-609], Joint Committee for Powder Diffraction Standards, 1990.
-
ESCA Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Eden Prairie, MN, 1992 Search PubMed.
- S. Mathur, M. Veith, V. Sivakov, H. Shen, V. Hoch, U. Hartmann and H.-B. Gao, Adv. Mater. Search PubMed , in press.
- P. F. Brandt, D. A. Lesch, P. R. Stafford and T. B. Rauchfuss, Inorg. Synth., 1995, 31, 112.
-
(a)
G. M. Sheldrick, SHELXS-97: program for crystal structure solution, Universität Göttingen, Germany, 1997;
(b)
G. M. Sheldrick, SHELXL-97: program for refinement of crystal structures, Universität Göttingen, Germany, 1997.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |
Click here to see how this site uses Cookies. View our privacy policy here. 
