Synthesis and optical characterisation of platinum(II) poly-yne polymers incorporating substituted 1,4-diethynylbenzene derivatives and an investigation of the intermolecular interactions in the diethynylbenzene molecular precursors†
Muhammad S. Khan*a, Muna R. A. Al-Mandharya, Mohammed K. Al-Sutia, Timothy C. Corcorana, Yaqoub Al-Mahrooqia, J. Paul Attfieldb, Neil Feederb, William I. F. Davidc, Kenneth Shanklandc, Richard H. Friendd, Anna Köhlerd, Elisabeth A. Marseglia*d, Emilio Tedescod, Chiu C. Tange, Paul R. Raithby*f, Jonathan C. Collingsg, Karl P. Roscoeg, Andrei S. Batsanovg, Lorna M. Stimsong and Todd B. Marder*g
aDepartment of Chemistry, Sultan Qaboos University, P.O. Box 36, Al Khod 123, Sultanate of Oman. E-mail: msk@squ.edu.om
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW
cISIS Facility, Rutherford Appleton Laboratory, Chilton, Didcot, Oxon, UK OX11 0QX
dCavendish Laboratory, University of Cambridge, Madingley Road, Cambridge, UK CB3 0HE
eCLRC Daresbury Laboratory, Daresbury, Warrington, UK WA4 4AD
fDepartment of Chemistry, University of Bath, Claverton Down, Bath, UK BA2 7AY. E-mail: p.r.raithby@bath.ac.uk
gDepartment of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE. E-mail: Todd.Marder@durham.ac.uk
First published on 25th November 2002
Abstract
A series of 1,4-diethynylbenzene (1) derivatives, H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R–CC–H with R
C–R–CC–H with R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =C6H3NH2
(2), C6H3F (3), C6H2F2-2,5 (4), C6F4
(5), C6H2(OCH3)2-2,5 (6) and C6H2(OnC8H17)2-2,5 (7) has been synthesised and their crystal structures determined by single crystal (2–5) or powder (6, 7) X-ray diffraction. The CCH⋯πCC hydrogen bonds dominating structure 1 are gradually replaced by CC–H⋯F ones with the increase of fluorination (3→5), or completely replaced by CCH⋯N and NH⋯πCC bonds in 2, and CCH⋯O in 6 and 7. The related platinum-based polymers, trans-[–Pt(PnBu3)2–CC–R–CC–]n
(R=as above and C6H4,) have been prepared and characterised by spectroscopic methods and thermogravimetry, which show that the amino- and methoxy-derivatives have lowest thermal stability while the fluorinated ones exhibit increasing thermal stability with increasing fluorination. Optical spectroscopic measurements reveal that substituents on the aromatic spacer group do not create strong donor–acceptor interactions along the rigid backbone of the organometallic polymers.
=C6H3NH2
(2), C6H3F (3), C6H2F2-2,5 (4), C6F4
(5), C6H2(OCH3)2-2,5 (6) and C6H2(OnC8H17)2-2,5 (7) has been synthesised and their crystal structures determined by single crystal (2–5) or powder (6, 7) X-ray diffraction. The CCH⋯πCC hydrogen bonds dominating structure 1 are gradually replaced by CC–H⋯F ones with the increase of fluorination (3→5), or completely replaced by CCH⋯N and NH⋯πCC bonds in 2, and CCH⋯O in 6 and 7. The related platinum-based polymers, trans-[–Pt(PnBu3)2–CC–R–CC–]n
(R=as above and C6H4,) have been prepared and characterised by spectroscopic methods and thermogravimetry, which show that the amino- and methoxy-derivatives have lowest thermal stability while the fluorinated ones exhibit increasing thermal stability with increasing fluorination. Optical spectroscopic measurements reveal that substituents on the aromatic spacer group do not create strong donor–acceptor interactions along the rigid backbone of the organometallic polymers.
Introduction
The last decade has witnessed a growing interest in the use of organic semiconductor materials in light-emitting diodes, lasers, photovoltaic cells and field-effect transistors.1 Conjugated polymers have shown considerable promise in this context. The need to optimise their processability and so enhance their commercial exploitation has prompted several investigations into the relationship between the chemical and electronic structure. Of particular interest are a series of organic poly-yne polymers that have been employed recently in successful liquid crystal display and photocell technology.2 A number of publications have also been dedicated to the chemical and optical characterisation of related tailored organic3 and organometallic4,5 oligomers and polymers.Recently, attention has also been directed toward oligomers, taken not only as model compounds, but also as potential materials for device applications.6 They are more crystalline than the corresponding polymers, allowing for a detailed structural analysis and, thus, an assessment of the structure–electronic property relationship.7 There is some evidence that the solid state arrangement of these molecules represents an important factor in determining their optical behaviour.5–8 While the π–π stacking interaction between adjacent oligomer molecules or polymer chains has been identified as a key ‘interchain’ interaction in solids,9 it is possible that hydrogen bonding can also play an important rôle in this context.
Hydrocarbon groups are generally poor donors of hydrogen bonds, but the ethynyl moiety with its acidic H atom is an exception.10 Furthermore, the concentration of electron density in the triple bond can act as an acceptor, albeit a rather weak one, of an H-bond.11 Thus ethynyl groups can form co-operative H-bonds,12 similarly to OH groups.13 Such a cooperative effect is evident in the crystal structure14 of 1,4-diethynylbenzene 1. In order to investigate the effect of various substituents on the crystalline architecture of these organic poly-yne precursors, particularly the stability of the CCH⋯πCC‡ H-bonding in competition15 with other intermolecular interactions, e.g. X–H⋯πCC
(X=N, C) and C–H⋯Y (Y=N, O, F16), we synthesised a series of amino-, fluoro- and alkoxy-derivatives of 1,4-diethynylbenzene (2–7, see Scheme 1) and structurally characterised them by single crystal and powder X-ray diffraction. The structures of 4 and 5 were determined at variable temperature, to investigate the supposed effects of the latter on weak hydrogen bonds.17
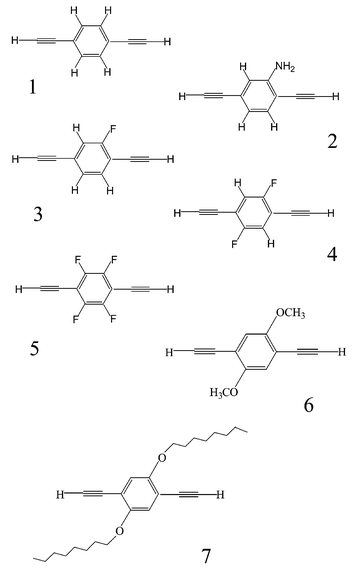 | ||
| Scheme 1 Compounds HCC–R–CCH. | ||
In addition, these substituted diethynylbenzene ligands have been incorporated into rigid-rod platinum(II)-containing polymers and their thermal and opto-electronic properties investigated systematically.
Results and discussion
Synthesis
The substituted 1,4-diethynylbenzene derivatives were synthesised by a sequence of coupling and protodesilylation reactions. The trimethylsilyl-protected bis(ethynyl) ligand precursors were synthesised by adopting improved procedures for the palladium-catalysed coupling of trimethysilylethyne with dibromo/diiodoaryls18 (Scheme 2). Conversion of the ligand precursors into their diterminal alkynes 2–7 was accomplished by smooth removal of the trimethylsilyl protecting groups with dilute aqueous KOH in MeOH/THF or with Bu4NF in THF. The products were purified by silica column chromatography and characterised by elemental analyses and by IR, NMR (1H and 13C) spectroscopy and mass spectrometry. In order to obtain the diethynylaryls, isolation of the bis(silylethynyl) derivatives is unnecessary. Thus, after the coupling reaction of the dihalide with the terminal alkyne (Scheme 2) was complete, the solvent was evaporated and the resultant residue was treated directly with dilute alkali to give the diethynyl compounds.![Reagents: (i) Pd(OAc)2, PPh3, CuI, iPr2NH/CH2Cl2; (ii) KOH, MeOH/THF, or Bu4NF, THF; (iii)
trans-[PtCl2(PnBu3)2], CuI, iPr2NH/CH2Cl2. R = C6H41, 8; C6H3NH22, 9; C6H3F 3, 10; C6H2F2-2,5 4, 11; C6F45, 12; C6H2(OMe)2-2,5 6, 13; C6H2(OnC8H17)2-2,5 7, 14.](/image/article/2003/NJ/b206946f/b206946f-s2.gif) | ||
| Scheme 2 Reagents: (i) Pd(OAc)2, PPh3, CuI, iPr2NH/CH2Cl2; (ii) KOH, MeOH/THF, or Bu4NF, THF; (iii)
trans-[PtCl2(PnBu3)2], CuI, iPr2NH/CH2Cl2. R=C6H41, 8; C6H3NH22, 9; C6H3F 3, 10; C6H2F2-2,5 4, 11; C6F45, 12; C6H2(OMe)2-2,5 6, 13; C6H2(OnC8H17)2-2,5 7, 14. | ||
Compared to the parent 1, the amino, fluoro and the alkoxy derivatives are relatively stable at low temperature in the absence of light and air; if exposed to either, the initially colourless solids slowly turn off-white, yellow, light brown or light purple. Long storage times at ambient temperature and under aerobic conditions led to the formation of some insoluble material (<5%), which was presumed to be a polymerisation product.
The platinum-containing poly-yne polymers were obtained by the organometallic polycondensation reaction of the diethynylbenzene derivatives with one equivalent of trans-[Pt(PnBu3)2Cl2] in CH2Cl2/iPr2NH solution in the presence of catalytic quantities of CuI (Scheme 2). The resultant polymers were purified by passing through a short alumina column, and isolated as off-white or yellow solids. The materials were characterised by spectroscopic techniques. The IR spectra showed a single ν(CC) stretching frequency at 2095 cm−1 consistent with the trans-arrangement of the ethynyl groups. The 31P NMR spectrum of each polymer displayed the expected singlet signal at approximately δ
−138 ppm with JPt-P coupling confirming the trans-configuration of the phosphine ligands. The 1H NMR spectra of the polymers show the expected resonances corresponding to the substituted 1,4-diethynylbenzene derivatives. The weight average molecular weights (Mw) of the polymers indicate a high degree of polymerisation. The number average molecular weight (Mn) values are in the range of 74000 to 95000 g mol−1. These values correspond to between 95 and 128 repeat units in the polymer chain. The molecular weights should be viewed with caution in view of the difficulties associated with utilizing GPC for rigid-rod polymers. GPC is not a direct measure of molecular weight but a measure of the hydrodynamic volume. Rod-like polymers in solution possess very different hydrodynamic properties from flexible polymers. Thus, by GPC using randomly coiled polystyrene standards, the observed average molecular weights of rigid rod poly-ynes are likely to be inflated to some extent relative to the actual molecular weights. However, the lack of discernable resonances that could be attributed to end groups in the NMR spectra provides support for the view that there is a high degree of polymerisation in these poly-ynes.
Crystal structures of 2–7
Structures of compounds 2–5 were determined by single-crystal X-ray diffraction, and those of the alkoxy derivatives 6 and 7 by powder X-ray diffraction. The molecular structures of 2–7 (see ESI) are unexceptional; the geometry of the 1,4-diethynylbenzene moiety is little affected by the substituents, but the crystal packing and H-bond patterns are significantly different. Comparison with the structure of 1 is instructive. In it the potential donor (ethynyl H) and acceptor (CC bonds) H-bonding sites are not only congruous (moderately polar) but also equal in numbers. This favours the formation of a two-dimensional net of co-operative H-bonds, linking molecules into a puckered layer (Fig. 1, Table 1). In both recent studies this layer was described as parallel to the (1 0 0), i.e. bc, plane.14b,c In fact, as can be seen from Fig. 2, it is parallel to the (1 0 −2) plane. It is noteworthy that the same mistake was repeated in the description of three crystal structures of p-XC6H4CCH (X=Cl, Br or I), pseudo-isomorphous with 1.19 There are infinite π–π stacks between molecules of adjacent layers (related by the a translation), at normal interplanar separations (d in Table 1).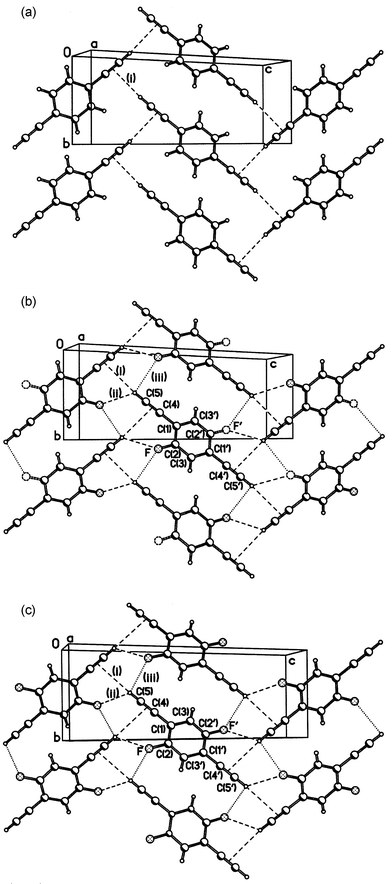 | ||
| Fig. 1 Crystal structures of 1 (a), 3 (b) and 4 (c); projections on the (1 0 −2) plane. | ||
| Compound | 114b | 114c | 3 | 4(A) | 4(B) | 5(A) | 5(B) | 5(C) |
|---|---|---|---|---|---|---|---|---|
| a See notation in Figs. 1 and 4.b Mean interplanar separation in the π–π stack.c Probably erratum in the original, cannot be verified as atomic coordinates are unavailable. | ||||||||
| T/K | 295 | 125 | 180 | 180 | 120 | 273 | 180 | 150 |
| (i)a H⋯π/Å | 2.68 | 2.60 | 2.75 | 2.87 | 2.85 | 2.94 | 2.93 | 2.89 |
| (i) C–H⋯π/° | 176 | 175 | 168 | 161 | 160 | 129 | 127 | 127 |
| (ii) H⋯F/Å | 2.66 | 2.60 | 2.58 | |||||
| (ii) C–H⋯F/° | 121 | 122 | 122 | |||||
| (iii) H⋯F/Å | 2.86 | 2.94 | 2.88 | 2.49 | 2.41 | 2.41 | ||
| (iii) C–H⋯F/° | 90 | 116 | 117 | 132 | 136 | 134 | ||
| db/Å | 3.71c | 3.53 | 3.47 | 3.43 | 3.41 | 3.63 | 3.60 | 3.59 |
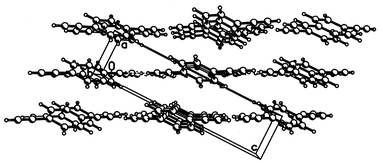 | ||
| Fig. 2 Crystal structure of 1, viewed down the y axis. | ||
Introduction of a polar (amino) group in 2 contributes two additional “active” H atoms, but only one potential acceptor site (the lone pair of N). The prominent feature of this structure (Fig. 3) is a ring system of co-operative H-bonds, involving four molecules. The ring (having crystallographic Ci symmetry) includes two relatively strong N–H⋯πCC bonds (H⋯π=2.51 Å, N–H⋯π=145°)§ and two CCH⋯N bonds (H⋯N=2.44 Å, C–H⋯N=159°), directed towards the lone pair of the substantially pyramidalised N atom (the valent bond angles at N average 116°). As each molecule participates in two such tetrameric systems, this generates a molecular layer, parallel to the crystallographic (1 0 −2) plane. The other amino-H atom is involved in a bifurcated interaction with two (crystallographically non-equivalent) CC bonds of two different molecules (H⋯π=2.89 and 2.87 Å, N–H⋯π=128 and 140°, respectively), belonging to the next layer. The ethynyl group not involved in the co-operative H-bonding points almost perpendicularly towards the benzene ring of one of these molecules (C–H vector/ring plane angle of 81°), with the shortest contacts H⋯C(4) 3.04 and H⋯C(5) 2.98 Å typical for van der Waals interactions.20 The outcome is a peculiar orientation of molecular planes: normal to each other but all normal to the layer plane. Molecules belonging to different layers (as defined by cooperative H-bonds) are nevertheless engaged in π–π stacking with each other (interplanar separations of 3.56 Å), albeit with considerable offset. Unlike all other compounds reported herein, molecule 2 has no crystallographic symmetry and is slightly ‘bent’, the directions of the two bonds forming an angle of 170.8(2)°.
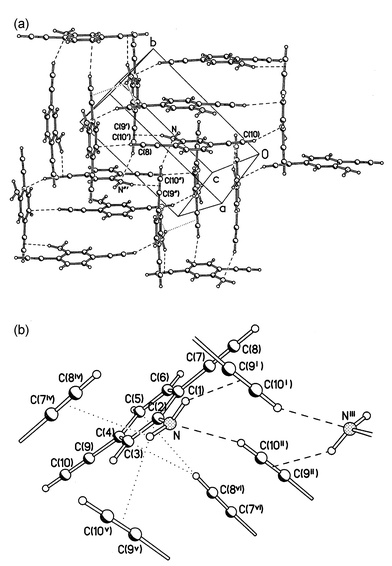 | ||
| Fig. 3 (a) Crystal structure of 2, projected on the (1 0 −2) plane; (b) H-bonds and intermolecular contacts. Symmetry transformations: (i) 1−x, ½+y, ½−z; (ii) 2−x, 1−y, 1−z; (iii)
x+1, ½−y, ½+z; (iv)
x−1, y, z; (v)
x, ½−y, ½+z; (vi) 2−x, y−½, ½−z. | ||
Crystal structures of 3 and 4
(Fig. 1) are isomorphous with that of 1. In each case, the molecule lies at a crystallographic inversion centre, which means that in 3 the fluorine atom is disordered, being distributed equally between two positions, related by this centre. Hydrogen bonding in 1, 3 and 4 is compared in Table 1. The ‘organic’
(i.e. bonded to carbon) fluorine atom, notwithstanding its high formal electronegativity, is known to be very poor H-bond acceptor, and is able to participate in them only in the absence of competition from stronger ones, e.g. oxygen.21 However, structures 3, 4 and 5 satisfy the conditions for the formation of C–H⋯F hydrogen bonds, as pointed out by Desiraju et al. in a study on fluorobenzenes:22 they contain only C, H and F atoms, and the H atoms are bonded to sp2 or sp-hybridised C atoms. Indeed, in 3 and 4 the CCH⋯πCC H-bond of 1 is weakened, or rather replaced by a bifurcated H-bond to both the CC group and the fluorine atom of the same molecule, the CCH⋯F interaction being the stronger of the two. Thus, the H⋯π distance increases by ca. 0.2 Å from that in 1 to that in 4. There is also a much longer, but probably electrostatically attractive, contact (iii) with another F atom. Comparison of the structure of 4 at different temperatures (180 and 120 K) shows similar (ca. 0.02 Å) thermal expansions of the H-bonds (i) and (ii) and the interplanar separations (d) in the π–π stack, but a larger extension of contact (iii). The layers in all three structures are similarly puckered: the dihedral angles between H-bonded molecules are 48°
(1), 52°
(3) and 54°
(4).
The increased degree of fluorination in compound 5 gives rise to completely different crystal packing (Fig. 4). The molecule also possesses crystallographic Ci symmetry, but the principal synthon in structure 5 is the pair of inversion-related, parallel and nearly coplanar molecules, linked by two CCH⋯F hydrogen bonds (iii), shorter than in 3 and 4
(see Table 1). Thus molecules are linked into an infinite ‘ribbon’, parallel to the (1 0 −1) direction. There are no continuous π–π stacks of molecules, although each molecule has one neighbour contacting it in a π–π fashion. These molecules, related by an inversion centre, have a longitudinal (parallel to the CC bond) shift of ca. 4.3 Å, so that a CC bond of one molecule overlaps with the benzene ring of another. The interplanar separation contracts from 3.63 Å at 273 K to 3.59 Å at 150 K.
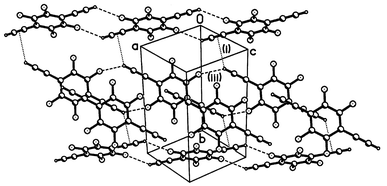 | ||
| Fig. 4 Crystal structure of 5. | ||
This double ribbon is surrounded on all sides by four other ribbons, propagating in the same direction but with perpendicular molecular planes. Each ethynyl H atom participates in a rather long inter-ribbon CCH⋯πCC contact (i)
(Table 1); the interaction thus can be interpreted as a very asymmetric bifurcated H-bond. Structure determinations at 273, 180 and 150 K did not reveal any substantial change, although thermal expansion in the direction of the strongest H-bond (iii) is somewhat greater than in other directions.
The crystal packing of di-alkoxy derivatives 6 and 7 is shown in Figs. 5 and 6. Both molecules have crystallographic Ci symmetry and adopt planar (6) or nearly planar (7) conformations. Structure 6 comprises puckered layers, parallel to the (1 0 −2) plane and broadly similar to those observed in structures 3 and 4, in which the molecules are linked by CCH⋯O hydrogen bonds (H⋯O=2.34 Å, C–H⋯O=146°), while the CCH⋯πCC interaction between the same molecules is much weaker than in structures 3 and 4
(H⋯π=3.07 Å, C–H⋯π=137°). Unlike 1–4, structure 6 contains no π–π stacks, molecules of adjacent layers contacting in a herring-bone fashion.
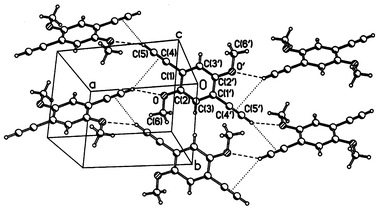 | ||
| Fig. 5 Crystal structure of 6. | ||
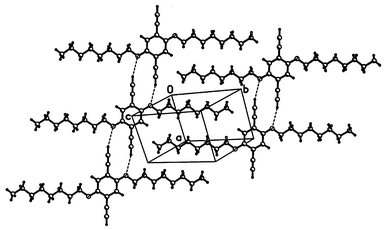 | ||
| Fig. 6 Crystal structure of 7. | ||
Structure 7 contains nearly-flat layers, in which a synthon, broadly similar to that of structure 5, can be identified: a centrosymmetric pair of molecules, linked by two CCH⋯O hydrogen bonds. These, however, are much weaker than in 6
(H⋯O=2.80 Å, C–H⋯O=167°). Probably, the crystal structure of 7 is dominated by the close packing of (all-trans) n-alkyl chains, to which the H-bonding pattern has to adjust. This structure is in good agreement with the structural model proposed recently for the corresponding polymer.3b
Thermal analysis
All the polymer samples exhibited an exotherm coincident with mass loss due to decomposition. Decomposition onset was defined as a mass loss of 2%. The peak decomposition temperature was defined as the first inflection point in the thermogravimetic curve, corresponding to a peak in the derivative of the TG data. This peak usually preceded the peak of the exotherm in the DTA data. The decomposition exotherms were broad with multiple peaks, and the thermogravimetic curve suggests a stepwise process. The results are shown in Table 2 and Fig. 7. Polymers 9 and 13 exhibited the lowest onset decomposition temperatures, while the fluorinated polymers 10–12 exhibited increasing decomposition temperatures with increasing fluorination. Polymer 14 also exhibited one of the highest decomposition temperatures, since the octyloxy groups likely encourage stronger interchain van der Waals bonding.°C) from thermal analysis
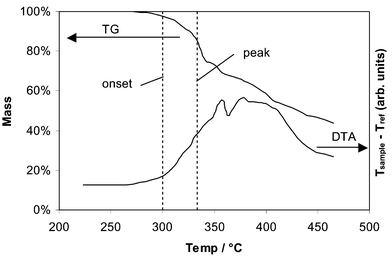 | ||
| Fig. 7 Simultaneous thermogravimetric (TG) curve (above) and differential thermal analysis (DTA) curve (below) for 10. The onset and peak decomposition temperatures, as defined in the text, are marked. | ||
Optical spectroscopy
The effect of the substitution of the aromatic spacer group on the electronic structure of the platinum(II) poly-yne polymers has been investigated. The absorption and emission spectra taken at room temperature from thin films of polymers 8, 12 and 13 are shown in Fig. 8. The first absorption band of polymer 8 peaking at 3.26 eV (380 nm) has its origin mostly in the π–π* transitions of the conjugated polymer backbone and is associated with the first singlet excited state denoted S1.23 Substitution by fluorine or by methoxy groups leads to a weak red shift of 0.05 eV (6 nm) and 0.15 eV (20 nm), respectively, indicating only slightly more conjugated backbones. Substitution with less than four fluorines, as in 10 and 11, leads to a correspondingly smaller red shift (not shown here).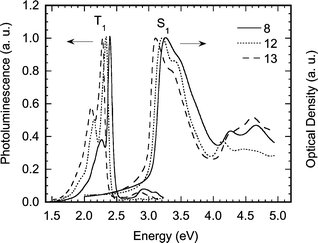 | ||
| Fig. 8 Absorption and luminescence spectra of the platinum-containing poly-yne polymers 8, 12 and 13 taken from thin films at room temperature with excitation at 3.4 eV. | ||
The emission spectra consist of two bands. The band just below the onset of absorption at 3 eV is emission from the S1 state (fluorescence). The band below 2.5 eV has been well characterised for the polymer 8 and other aromatic/hetero-aromatic spacers by time-resolved luminescence measurements and is assigned to emission from the triplet excited state T1 (phosphorescence).8,24 Substitution of the aromatic ring shifts the energy of the T1 emission by about the same amount as the S1 emission, so that the S1–T1 energy difference remains constant. This can be used to accurately fine-tune the energy of the T1 state for energy harvesting purposes25 particularly in analogous organic polymers, where T1 emission (phosphorescence) is spin-forbidden and cannot be observed. While such a constant S1–T1 energy gap has been observed for these organometallic and analogous organic polymers with a variety of spacers its cause is not yet fully understood.23,25
The emission spectra normalised to the fluorescence peak for the polymers with one, two and four fluorine substituents (polymers 10, 11 and 12, respectively) are shown in Fig. 9. The relative intensity of phosphorescence increases strongly with the fluorine content.
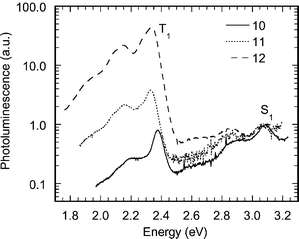 | ||
| Fig. 9 Room temperature thin-film luminescence spectra of platinum-containing poly-yne polymers 10, 11 and 12 normalised to the peak of the S1 emission and shown on a logarithmic scale. | ||
Conclusions
A number of substituted derivatives of 1,4-diethynylbenzene with electron donating and accepting substituents have been studied, and their structural characterisation performed by both single-crystal and powder X-ray diffraction. An interesting result of this work has been the ability to obtain comparable structural information from conventional techniques (single crystal) and from powder X-ray diffraction.The packing analysis of the structures of the polymer precursors has also thrown light on the co-presence of weak hydrogen bonding in model compounds of polymers that are of technological interest. In summary the ethynylenic moiety provides a good hydrogen bond donor and acceptor, which is able to compete in terms of intermolecular interactions and as structure defining synthons, with more electronegative atoms, such as nitrogen. CCH⋯πCC hydrogen bonds dominate the crystal structure of 1, and are gradually replaced by CC–H⋯F ones with the increase of fluorination (3→5), as has been noted previously,19 or completely replaced by CCH⋯N and NH⋯πCC bonds in 2, and CCH⋯O in 6 and 7. However, it is hard to judge, based solely on geometrical measurements, the relative strength of the C–H⋯F interactions in these derivatives. A C–H⋯πCC bond in 1 has been estimated being of ca. 4 kJ mol−1.26 When alkoxy substituents are present, C–H⋯O interactions occur, but the presence of long alkyl chains in the structure can impede the formation of these short contacts.
The substituted 1,4-diethynyl benzene ligands have also been incorporated into rigid-rod Pt(II) polymers and their thermal and opto-electronic properties investigated. The substituents on the aromatic spacer have a major effect on the thermal stability of the polymeric complexes but their electronic properties are affected only to a minor extent.
Experimental
General
All reactions were performed under a dry nitrogen atmosphere using standard Schlenk or glove box techniques. Solvents were pre-dried and distilled before use by standard procedures.27 All chemicals, except where stated otherwise, were obtained from Sigma Aldrich and checked for purity by GC/MS prior to use. 1,4-Diiodo-2,5-bis(octyloxy)benzene was prepared by adaptation of the literature method.28NMR spectra were recorded on Bruker WM-250 or AM-400 spectrometers in CDCl3 and the 1H and 13C{1H} were referenced to solvent resonances. Infrared spectra were recorded as CH2Cl2 solutions, in a NaCl cell, on a Perkin-Elmer 1710 FT-IR spectrometer, mass spectra on a Kratos MS 890 spectrometer by the electron impact (EI) technique. Microanalyses were performed in the Department of Chemistry, University of Cambridge. Column chromatography was performed in Kieselgel 60 (240–400 mesh) silica gel. GC/MS analyses were performed on a Hewlett-Packard 5890 Series II/5971A MSD instrument equipped with an HP 7673A autosampler and a fused silica column (30 m×0.25 mm×0.25 μm, cross-linked 5% phenylmethyl silicone). The following operating conditions were used: injector, 260°C; detector, 280°C; oven temperature was ramped from 70° to 260°C at the rate of 20°C min−1; helium (UHP grade) was used as the carrier gas; toluene (BDH, 99.7%) was used as an internal integration standard.
Synthesis of 1,4-diethynylbenzene derivatives
Coupling. To an ice-cooled solution of 1,4-dibromo-2-aminobenzene (3.01 g, 12 mmol) in diisopropylamine/THF (75 cm3, 1∶4 v/v) under nitrogen were added CuI (29 mg, 0.15 mmol), Pd(OAc)2 (27 mg, 0.12 mmol) and PPh3 (157 mg, 0.60 mmol). The solution was stirred for 0.5 h, trimethylsilylethyne (2.95 g, 30 mmol) was then added over 10 min to the vigorously stirred solution; during the addition a white precipitate formed. The suspension was stirred for 30 min in an ice-bath before being warmed to room temperature. After reacting for 30 min at room temperature the mixture was heated to 80
°C for 20 h when TLC, GC analysis and IR spectroscopy indicated that all the starting material had been consumed and the coupling reaction was completed. After being cooled to room temperature, the mixture was filtered to eliminate the ammonium salt and the solvent mixture was removed in vacuo.Protodesilylation. The solid residue was dissolved in THF (50 cm3) with stirring. Methanol (15 cm3) and aqueous KOH (1.50 g, 27 mmol in 5 cm3 water) were added at room temperature to the stirred solution. The reaction mixture was stirred for 2 h. The completion of the desilylation reaction was verified by TLC, GC analysis and IR spectroscopy. The mixture was filtered and the solvent was removed under reduced pressure to leave a yellow residue. This residue was dissolved in the minimum amount of dichloromethane and subjected to column chromatography on silica using hexane–dichloromethane (2∶1 v/v) as eluent to afford a pale yellow solid. Recrystallization from hexane with activated charcoal yielded pale yellow micro-crystals (0.62 g, 76% yield). IR (CH2Cl2): ν/cm−1 2108 (–C
C–), 3300 (CCH), 3428 (–NH–). 1H NMR (250 MHz, CDCl3): δ 3.12 (s, 1H, CC–H), 3.50 (s, 1H, CC–H), 4.70 (br s, 2H, NH2), 7.21 (d, 2H, ar), 7.73 (dd, 1H, ar). 13C NMR (100.6 MHz, CDCl3): δ 78.2, 80.6, 83.1, 84.0 (CC), 107.2 (C1), 117.5 (C3), 121.4 (C4), 123.0 (C6), 132.2 (C5), 148.0 (C2). EI mass spectrum: m/z 141 (M+). Found: C, 85.03; H, 4.94; N, 9.90; Calc. for C10H7N: C, 85.08; H. 5.00; N, 9.92%.Coupling. To a solution of 1,4-dibromo-2-fluorobenzene (2.5 g, 10 mmol) in diisopropylamine/THF (100 cm3, 1∶4 v/v) at reflux under nitrogen was added a catalyst mixture of CuI (24 mg, 0.13 mmol), Pd(OAc)2 (22 mg, 0.10 mmol) and PPh3 (131 mg, 0.50 mmol). Trimethylsilylethyne (2.45 g, 25 mmol) was then added over the course of 10 min to the vigorously stirred solution; during the addition a white precipitate formed. The reaction mixture was stirred at reflux for 8 h and the completion of the reaction was verified by TLC, GC analysis and IR spectroscopy. The reaction mixture was allowed to cool to room temperature and the ammonium iodide precipitate was filtered off. The yellow filtrate was evaporated to dryness.
Protodesilylation. Removal of the trimethylsilyl protecting groups was accomplished as for the synthesis of 2 described above. Compound 3 was obtained as an off-white solid in 82% yield. Good quality crystals were obtained by sublimation (15 mmHg, ice-water cooled). IR (CH2Cl2): ν/cm−1 2107 (–C
C–) and 3300 (–CC–H). 1H NMR (250 MHz, CDCl3): δ 3.21 (s, 1H, CC–H), 3.38 (s, 1H, CC–H), 7.22 (d, 2H, ar) and 7.46 (d, 1H, ar). 13C NMR (100.6 MHz, CDCl3): δ 77.1, 80.2, 81.8, 84.1 (CC), 111.51 (C1), 119.02 (C3), 124.36 (C4), 128.10 (C6), 134.20 (C5) and 162.65 (C2). EI mass spectrum: m/z 144 (M+). Found: C, 83.29; H, 3.58. Calc. for C10H5F: C, 83.32; H, 3.50%.C–) and 3299 (–CC–H). 1H NMR (250 MHz, CDCl3): δ 3.41 (s, 2H, CCH), 7.24 (d, 2H, ar), 13C NMR (100.6 MHz, CDCl3): δ 75.7, 85.5 (CC), 112.8 (C1,4), 120.2 (C3,6), 158.9, (C2,5). EI mass spectrum: m/z 162 (M+). Found C, 74.11; H, 2.45; Calc. for C10H4F2: C, 74.08; H. 2.49%.Coupling17e,28. 1,4-Diiodo-2,3,5,6-tetrafluorobenzene (4.02 g, 10 mmol) and trimethylsilylethyne (2.45 g, 25 mmol) were reacted in diisopropylamine/THF (100 cm3, 1∶4 v/v) in the presence of catalytic amounts of CuI (24 mg), Pd (OAc)2 (22 mg) and PPh3 (131 mg). The coupling reaction was completed after 1 h of reflux. The crude product was worked up, as before, to yield a pale-yellow residue.
Protodesilylation. A similar procedure to that for 4 was adopted to obtain an off-white solid in 82% yield. Good quality crystals were obtained by sublimation (15 mmHg, ice-water cooled). IR (CH2Cl2) ν/cm−1: 2106 (–C
C–) and 3297 (–CC–H). 1H NMR (250 MHz, CDCl3): δ 3.72 (s, 2H, CCH). 13C NMR (100.6 MHz, CDCl3): δ 68.4, 91.5 (CC), 104.5 (C1,4), 147.5 (C2,3,5,6). EI mass spectrum: m/z 198 (M+). Anal. Found: C, 60.71; H, 1.04; Calc. for C10H2F4: C, 60.63; H, 1.02%.Coupling29. Similar coupling procedures as in 5 were adopted using 1,4-diiodo-2,5-bis(methoxy)benzene (3.90 g, 10 mmol) to produce the crude trimethylsilylethynyl compound.
Protodesilylation. The solid residue was dissolved in THF (50 cm3) and a solution of tetrabutylammonium fluoride (1.1 M, 2 equiv.) was added. The reaction mixture was stirred for 5 min at ambient temperature when TLC, GC analysis and IR spectroscopy indicated that all the starting material had been consumed and the desilylation reaction was completed. The reaction mixture was chromatographed on a small silica gel column using THF as eluent. The solvent was removed in vacuo and the crude product was recrystallized in methanol to yield an off-white solid in 86% yield. IR (CH2Cl2): ν/cm−1 2107 (–C
C–) and 3299 (–CC–H). 1H NMR (250 MHz CDCl3): δ 3.33 (s, 2H, CC–H), 3.92 (s, 6H, OCH3), 7.19 (s, 2H, ar). 13C NMR (100.6 MHz, CDCl3): δ 69.89 (OCH3), 79.96, 82.57 (CC), 113.41 (C1,4), 117.89 (C3,6), 154.14 (C2,5). EI mass spectrum: m/z 186 (M+). Found: C, 77.46; H, 5.43; Calcd for C12H10O2: C, 77.40; H, 5.41%.Coupling. Similar coupling procedures as in 5 were adopted using 1,4-diiodo-2,5-bis(octyloxy)benzene (5.87 g, 10 mmol) to produce the crude trimethylsilylethynyl compound.
Protodesilylation. Similar desilylation procedures as in 6 were adopted to obtain a pale yellow solid in 79% yield. IR (CH2Cl2): ν/cm−1 2107 (–C
C–) and 3299 (–CC–H). 1H NMR (250 MHz CDCl3): δ 0.86 (t, 6H, CH3), 1.26, 1.40 (both m, 20H, CH2), 1.78 (m, 4H, CH2), 3.32 (s, 2H, CC–H), 3.95 (t, 4H, OCH2), 6.93 (s, 2H, ar). 13C NMR (100.6 MHz, CDCl3): δ 14.29 (CH3), 22.85, 26.09, 29.31, 29.42, 29.48, 31.99 (all CH2), 69.84 (OCH2), 79.98, 82.60 (CC), 113.45 (C1,4), 117.92 (C3,6), 154.18 (C2,5). EI mass spectrum: m/z 382 (M+). Found: C, 81.52; H, 10.04; Calc. for C26H38O2: C, 81.63; H, 10.01%.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C–C6H3(NH2)–CC)–]n9. CuI (10 mg) was added to a mixture of trans-[Pt(PnBu3)2Cl2]
(0.670 g, 1.0 mmol) and 1,4-diethynyl-2-amimobenzene (0.141 g, 1.0 mmol) in CH2Cl2/iPr2NH (50 cm3, 1∶1 v/v). The yellow solution was stirred at room temperature over a period of 15 h, after which all volatile components were removed under reduced pressure. The residue was dissolved in dichloromethane and filtered through a short alumina column. After removal of solvent by rotary evaporator, an off-white solid of polymer 9 was obtained in 85% yield (0.750 g). Further purification can be accomplished by precipitating the polymer from dichloromethane solution in methanol. IR (CH2Cl2): ν/cm−1 2094 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.87 (t, 18H, CH3), 1.40 ((sex, 12H, CH2), 1.65 (brs, 12H, CH2), 2.25 (m, 12H, PCH2), 4.18 (s, 2H, NH2), 6.58 (d, 2H, ar), 7.12 (d, IH, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.08, JPt-P=2330 Hz. Found: C, 55.17; H, 8.14; Calc. for (C34H59NP2Pt)n: C, 55.26; H. 8.05%. GPC (THF): Mn=94700 g mol−1
(n=128), Mw=151500 g mol−1, polydispersity=1.6.C–C6H3F–CC)–]n10. Off-white solid (82% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC). 1H NMR (250 MHz, CDCl3): δ 0.82 (t, 18H, CH3), 1.38 (sex, 12H, CH2), 1.58 (brs, 12H, CH2), 2.29 (m, 12H, PCH2), 7.38 (d, 2H, ar), 7.28 (d, IH, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.50, JPt-P=2330 Hz. Found: C, 55.15; H, 7.74; Calc. for (C34H57P2FPt)n: C, 55.05; H. 7.76%. GPC (THF): Mn=94650 g mol−1
(n=128), Mw=160900 g mol−1, polydispersity=1.7.C–C6H2F2–CC)–]n11. Off-white solid (78% yield). IR (CH2Cl2): ν/cm−1 2094 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.77 (t, 18H, CH3), 1.34 (sex, 12H, CH2), 1.52 (brs, 12H, CH2), 2.20 (m, 12H, PCH2), 7.28 (d, 2H, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.50, JPt-P=2330 Hz. Found: C, 53.65; H, 7.44; Calc. for (C34H56P2F2Pt)n: C, 53.74; H. 7.43%. GPC (THF): Mn=88230 g mol−1
(n=116), Mw=158810 g mol−1, polydispersity=1.8.C–C6F4–CC–]n12. Off-white solid (72% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.75 (t, 18H, CH3), 1.38 (sex, 12H, CH2), 1.58 (brs, 12H, CH2), 2.20 (m, 12H, PCH2). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.50, JPt-P=2330 Hz. Found: C, 51.36; H, 6.79; Calc. for (C34H54P2F4Pt)n: C, 51.31; H. 6.84%. GPC (THF): Mn=82500 g mol−1
(n=103). Mw=156770 g mol−1, polydispersity=1.9.C–C6H4(OCH3)2–CC–]n13. Light yellow solid (85% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.86 (t, 18H, CH3), 1.38 (sex, 12H, CH2), 1.58 (brs, 12H, CH2), 2.20 (m, 12H, PCH2), 3.70 (s, 6H, OCH3), 7.10 (s, 2H, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.10, JPt-P=2330 Hz. Found: C, 55.22; H, 8.03; Calc. for (C36H62P2O2Pt)n: C, 55.16; H. 7.97%. GPC (THF): Mn=74100 g mol−1
(n=95), Mw=111200 g mol−1, polydispersity=1.5.C–C6H4(OC8H17)2–CC–]n14. Light yellow solid (90% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.84 (t, 24H, CH3), 1.22–1.38 (m, 32H, CH2), 1.62 (m, 16H, CH2), 2.20 (m, 12H, PCH2), 3.75 (t, 4H, OCH2), 6.82 (s, 2H, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.10, JPt-P=2330 Hz. Found: C, 61.36; H, 9.29; Calc. for (C50H90P2O2Pt)n: C, 61.26; H. 9.25%. GPC (THF): Mn=94850 (n=97) g mol−1, Mw=151750 g mol−1, polydispersity=1.6.°C running in THF at 1 cm3 min−1 with a Roth Mocel 200 high precision pump. A DAWN DSP (Wyatt Technology) Multi-Angle Laser Light Scattering (MALLS) apparatus with 18 detectors and auxiliary Viscotek model 200 differential refractometer/viscometer detectors was used to calculate the molecular weights (referred to GPC LS).
C–C6H3(NH2)–CC)–]n9. CuI (10 mg) was added to a mixture of trans-[Pt(PnBu3)2Cl2]
(0.670 g, 1.0 mmol) and 1,4-diethynyl-2-amimobenzene (0.141 g, 1.0 mmol) in CH2Cl2/iPr2NH (50 cm3, 1∶1 v/v). The yellow solution was stirred at room temperature over a period of 15 h, after which all volatile components were removed under reduced pressure. The residue was dissolved in dichloromethane and filtered through a short alumina column. After removal of solvent by rotary evaporator, an off-white solid of polymer 9 was obtained in 85% yield (0.750 g). Further purification can be accomplished by precipitating the polymer from dichloromethane solution in methanol. IR (CH2Cl2): ν/cm−1 2094 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.87 (t, 18H, CH3), 1.40 ((sex, 12H, CH2), 1.65 (brs, 12H, CH2), 2.25 (m, 12H, PCH2), 4.18 (s, 2H, NH2), 6.58 (d, 2H, ar), 7.12 (d, IH, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.08, JPt-P=2330 Hz. Found: C, 55.17; H, 8.14; Calc. for (C34H59NP2Pt)n: C, 55.26; H. 8.05%. GPC (THF): Mn=94700 g mol−1
(n=128), Mw=151500 g mol−1, polydispersity=1.6.C–C6H3F–CC)–]n10. Off-white solid (82% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC). 1H NMR (250 MHz, CDCl3): δ 0.82 (t, 18H, CH3), 1.38 (sex, 12H, CH2), 1.58 (brs, 12H, CH2), 2.29 (m, 12H, PCH2), 7.38 (d, 2H, ar), 7.28 (d, IH, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.50, JPt-P=2330 Hz. Found: C, 55.15; H, 7.74; Calc. for (C34H57P2FPt)n: C, 55.05; H. 7.76%. GPC (THF): Mn=94650 g mol−1
(n=128), Mw=160900 g mol−1, polydispersity=1.7.C–C6H2F2–CC)–]n11. Off-white solid (78% yield). IR (CH2Cl2): ν/cm−1 2094 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.77 (t, 18H, CH3), 1.34 (sex, 12H, CH2), 1.52 (brs, 12H, CH2), 2.20 (m, 12H, PCH2), 7.28 (d, 2H, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.50, JPt-P=2330 Hz. Found: C, 53.65; H, 7.44; Calc. for (C34H56P2F2Pt)n: C, 53.74; H. 7.43%. GPC (THF): Mn=88230 g mol−1
(n=116), Mw=158810 g mol−1, polydispersity=1.8.C–C6F4–CC–]n12. Off-white solid (72% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.75 (t, 18H, CH3), 1.38 (sex, 12H, CH2), 1.58 (brs, 12H, CH2), 2.20 (m, 12H, PCH2). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.50, JPt-P=2330 Hz. Found: C, 51.36; H, 6.79; Calc. for (C34H54P2F4Pt)n: C, 51.31; H. 6.84%. GPC (THF): Mn=82500 g mol−1
(n=103). Mw=156770 g mol−1, polydispersity=1.9.C–C6H4(OCH3)2–CC–]n13. Light yellow solid (85% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.86 (t, 18H, CH3), 1.38 (sex, 12H, CH2), 1.58 (brs, 12H, CH2), 2.20 (m, 12H, PCH2), 3.70 (s, 6H, OCH3), 7.10 (s, 2H, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.10, JPt-P=2330 Hz. Found: C, 55.22; H, 8.03; Calc. for (C36H62P2O2Pt)n: C, 55.16; H. 7.97%. GPC (THF): Mn=74100 g mol−1
(n=95), Mw=111200 g mol−1, polydispersity=1.5.C–C6H4(OC8H17)2–CC–]n14. Light yellow solid (90% yield). IR (CH2Cl2): ν/cm−1 2095 (–CC–). 1H NMR (250 MHz, CDCl3): δ 0.84 (t, 24H, CH3), 1.22–1.38 (m, 32H, CH2), 1.62 (m, 16H, CH2), 2.20 (m, 12H, PCH2), 3.75 (t, 4H, OCH2), 6.82 (s, 2H, ar). 31P{1H} NMR (101.3 MHz, CDCl3): δ
−138.10, JPt-P=2330 Hz. Found: C, 61.36; H, 9.29; Calc. for (C50H90P2O2Pt)n: C, 61.26; H. 9.25%. GPC (THF): Mn=94850 (n=97) g mol−1, Mw=151750 g mol−1, polydispersity=1.6.°C running in THF at 1 cm3 min−1 with a Roth Mocel 200 high precision pump. A DAWN DSP (Wyatt Technology) Multi-Angle Laser Light Scattering (MALLS) apparatus with 18 detectors and auxiliary Viscotek model 200 differential refractometer/viscometer detectors was used to calculate the molecular weights (referred to GPC LS).X-Ray crystallography
![[small lambda, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0cc.gif) =1.54178 Å); for 4(B) and 5(C) on a SMART 3-circle diffractometer with a 1K CCD area detector, using graphite-monochromated Mo-Kα radiation (=0.71073 Å). In all cases, crystals were cooled with Cryostream (Oxford Cryosystems) open-flow N2 gas cryostat. Absorption corrections were performed for 3 and 4
(A and B) by a semi-empirical method, based on ψ-scans. The structures were solved by direct methods using the SHELXS-86 program31
(or SIR9232 for 5) and refined by full-matrix least squares against F2, using SHELXL-97 software.33
=1.54178 Å); for 4(B) and 5(C) on a SMART 3-circle diffractometer with a 1K CCD area detector, using graphite-monochromated Mo-Kα radiation (=0.71073 Å). In all cases, crystals were cooled with Cryostream (Oxford Cryosystems) open-flow N2 gas cryostat. Absorption corrections were performed for 3 and 4
(A and B) by a semi-empirical method, based on ψ-scans. The structures were solved by direct methods using the SHELXS-86 program31
(or SIR9232 for 5) and refined by full-matrix least squares against F2, using SHELXL-97 software.33CCDC reference numbers 196099–196102. See http://www.rsc.org/suppdata/nj/b2/b206946f/ for crystallographic data in CIF or other electronic format.
| 2 | 3 | 4(A) | 4(B) | 5(A) | 5(B) | 5(C) | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|---|
| a Total number of inequivalent reflections in the powder pattern;b Rwp for the Rietveld fits;c Rp for the Rietveld fits. | |||||||||
| Formula | C10H7N | C10H5F | C10H4F2 | C10H2F4 | C12H10O2 | C26H38O2 | |||
| Mw/g mol−1 | 141.17 | 144.14 | 162.13 | 198.12 | 186.20 | 380.55 | |||
| T/K | 180 | 180 | 180 | 120 | 273 | 180 | 150 | 290 | 290 |
| λ/Å | 1.54178 | 1.54178 | 1.54178 | 0.71073 | 1.54178 | 1.54178 | 0.71073 | 1.2996 | 1.54178 |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic | monoclinic | triclinic | |||
| Space group | P21/c | P21/c | P21/c | P21/c | P21/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | |||
| a/Å | 5.919(2) | 3.878(1) | 3.8535(7) | 3.828(1) | 6.1510(6) | 6.112(1) | 6.109(2) | 9.1718(1) | 7.6769(8) |
| b/Å | 12.030(4) | 5.947(1) | 5.906(1) | 5.887(1) | 11.524(1) | 11.463(2) | 11.456(4) | 6.04823(7) | 11.682(2) |
| c/Å | 11.026(3) | 15.800(5) | 16.221(5) | 16.204(4) | 5.8204(7) | 5.764(1) | 5.751(2) | 9.54469(1) | 6.926(2) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 104.29(1) |
| β/° | 104.17(2) | 93.46(3) | 95.23(3) | 95.21(1) | 98.71(1) | 99.27(2) | 99.35(2) | 107.428(1) | 96.637(8) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 99.671(9) |
| V/Å3 | 761.2(4) | 363.7(2) | 367.6(2) | 363.7(1) | 397.1(2) | 398.6(1) | 397.1(2) | 505.17(1) | 586.0(2) |
| Z | 4 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 1 |
| μ/mm−1 | 0.57 | 0.77 | 1.03 | 0.12 | 1.42 | 1.45 | 0.16 | 0.67 | 0.51 |
| Refls. total | 1043 | 920 | 510 | 4764 | 753 | 510 | 4179 | ||
| Refls. unique | 959 | 460 | 466 | 966 | 691 | 506 | 907 | 243a | 70a |
| Rint | 0.031 | 0.026 | 0.026 | 0.020 | 0.040 | 0.059 | 0.023 | — | — |
| Refls., I>2σ(I) | 777 | 369 | 431 | 902 | 601 | 333 | 807 | ||
| wR(F2) | 0.136 | 0.122 | 0.073 | 0.106 | 0.129 | 0.072 | 0.085 | 0.070b | 0.031b |
| R[I>2σ(I)] | 0.050 | 0.045 | 0.028 | 0.034 | 0.043 | 0.035 | 0.031 | 0.045c | 0.024c |
=1.2996 Å) using a flat plate geometry, that for 7 on a Stoe powder X-ray diffractometer equipped with a linear PSD, using Cu-Kα radiation (=1.5406 Å) from a sealed-tube source. Both diffraction patterns were indexed using the DICVOL91 program34 and each structure solved using the DASH suite of programs.35 The structure solution details are summarised in Table 4.
| 6 | 7 | |
|---|---|---|
| Data range used/degrees | 5–40 | 7–34 |
| Number of intensities extracted | 74 | 66 |
| χ2 for Pawley profile fit | 7.67 | 2.94 |
| Number of atoms in model, excluding dummy | 12 | 33 |
| Internal degrees of freedom | 1 | 8 |
| Initial simulation temperature (χ2 units) | 300 | 100 |
| Final simulation temperature (χ2 units) | 280 | 4.5 |
| Initial χ2 for intensities | 3740 | 1825.6 |
| Final χ2 for intensities | 63.1 | 24.4 |
| χ2 for Rietveld profile fit of SA model (refine scale+ITF only) | 18.17 | 5.61 |
It was clear from the space groups and lattice parameters that the molecules in both structures lie at crystallographic inversion centres and have no translational degree of freedom. The trial structures were subjected to an optimisation in which torsion angles were the only internal degrees of freedom, and limits on the external degrees of freedom were derived from the Euclidean normalisers of the relevant space groups,36 while the molecule was anchored on a dummy atom lying at the origin of the unit cell. Structure solutions were deemed to have been obtained when the χ2 figure of merit fell below a predetermined value.
Finally, the fractional coordinates obtained at the end of the simulated annealing runs were verified by Rietveld refinements using the GSAS program.37 In both cases, the scale factor, cell constants, and parameters describing a linear interpolated background function and the diffraction peak shape were varied. The highly-resolved synchrotron powder pattern of 6 contained sufficient data for the atomic coordinates and isotropic U-factors of all non-hydrogen atoms to be refined independently while the H atom positions were restrained to idealised positions, giving an excellent fit with the final χ2=5.32. The powder diffraction pattern of 7 contained an amorphous component at 2θ>34°, therefore only the data below this limit were used in the solution and subsequent Rietveld refinement. The refinement of atomic coordinates being unstable, these were fixed and an overall isotropic U-factor was refined, giving a good fit with the final χ2=2.24.
SHELXTL software38 was used for analysing the geometry and for graphical presentation of all structures (1–7).
°C min−1 to 465°C. The thermocouple readings were calibrated using a series of DTA standard materials: KNO3, In, Sn, Ag2SO4, and K2SO4 as well as Pb and Al as secondary standards, using the same heating rates as the samples.Acknowledgements
We thank Mr. Jon P. Wright (Department of Chemistry, University of Cambridge) for helpful discussions during indexing and Rietveld refinement of the data on compounds 6 and 7. M. S. K. gratefully acknowledges Sultan Qaboos University Research Grant No. IG/SCI/CHEM/02/02, EPSRC (U.K.) for a Visiting Fellowship and Sultan Qaboos University for a research leave. A. K. thanks Peterhouse, Cambridge for a Research Fellowship and the Royal Society for a University Research Fellowship. We also acknowledge the EPSRC grant GR/L52581 (J. P. A.) and GR/L92761 (P. R. R.) and for a studentship (to J. C. C.).References
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS; N. Tessler, G. J. Denton and R. H. Friend, Nature, 1996, 382, 695 CrossRef CAS; J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS; G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS; F. Garnier, R. Hajlaoui, A. Yassar and P. Srivastava, Science, 1994, 265, 1684.
- A. Montali, C. Bastiaansen, P. Smith and C. Weder, Nature, 1998, 392, 261 CrossRef CAS; C. Weder, C. Sarwa, A. Montali, C. Bastiaansen and P. Smith, Science, 1998, 279, 835 CrossRef CAS.
- (a) C. Weder and M. S. Wrighton, Macromolecules, 1996, 29, 5157 CrossRef CAS; (b) T. Yamamoto, K. Honda, N. Ooba and S. Tomaru, Macromolecules, 1998, 31, 7 CrossRef CAS; (c) H. Li, D. R. Powell, R. K. Hayashi and R. West, Macromolecules, 1998, 31, 52 CrossRef CAS; (d) A. Beeby, K. Findlay, P. J. Low and T. B. Marder, J. Am. Chem. Soc. Search PubMed , in the press; (e) C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W. Clegg and C. Viney, Chem. Commun., 1999, 2493 RSC; (f) M. Biswas, P. Nguyen, T. B. Marder and L. R. Khundkar, J. Phys. Chem. A, 1997, 101, 1689 CrossRef CAS; (g) P. Nguyen, G. Lesley, T. B. Marder, I. Ledoux and J. Zyss, Chem. Mater., 1997, 9, 406 CrossRef CAS.
- D. Beljonne, H. F. Wittman, A. Kohler, S. Graham, M. Younus, J. Lewis, P. R. Raithby, M. S. Khan, R. H. Friend and J. L. Bredas, J. Chem. Phys., 1996, 105, 3868 CrossRef CAS; J. Lewis, N. J. Long, P. R. Raithby, G. P. Shields, W.-Y. Wong and M. Younus, J. Chem. Soc., Dalton Trans., 1997, 4283 RSC; M. Younus, A. Köhler, S. Cron, N. Chawdhury, M. R. A. Al-Mandhary, J. Lewis, N. J. Long, R. H. Friend and P. R. Raithby, Angew. Chem., Int. Ed., 1998, 37, 3036 CrossRef CAS; N. Chawdhury, A. Köhler, R. H. Friend, M. Younus, N. J. Long, P. R. Raithby and J. Lewis, Macromolecules, 1998, 31, 722 CrossRef CAS; M. C. B. Colbert, J. Lewis, N. J. Long, P. R. Raithby, M. Younus, A. J. P. White, D. J. Williams, N. N. Payne, L. Yellowlees, D. Beljonne, N. Chawdhury and R. H. Friend, Organometallics, 1998, 17, 3034 CrossRef CAS; J. Lewis, P. R. Raithby and W.-Y. Wong, J. Organomet. Chem., 1998, 556, 219 CrossRef CAS; M. S. Khan, M. R. A. Al-Mandhary, M. K. Al-Suti, N. Feeder, S. Nahar, A. Köhler, R. H. Friend, P. J. Wilson and P. R. Raithby, J. Chem. Soc., Dalton Trans., 2002, 2441 RSC; P. Li, B. Ahrens, K.-H. Choi, M. S. Khan, P. R. Raithby, P. J. Wilson and W.-Y. Wong, CrystEngComm, 2002, 4, 405 Search PubMed.
- N. Chawdhury, A. Köhler, R. H. Friend, W.-Y. Wong, J. Lewis, M. Younus, P. R. Raithby, T. C. Corcoran, M. R. A. Al-Mandhary and M. S. Khan, J. Chem. Phys., 1999, 110, 4963 CrossRef CAS.
- K. Mullen and G. Wegner, Electronic Materials: The Oligomer Approach, Wiley-VCH, New York, 1998 Search PubMed.
- U. H. F. Bunz, V. Enkelmann, L. Kloppenburg, D. Jones, K. D. Shimizu, J. B. Claridge, H.-C. zur Loye and G. Lieser, Chem. Mater., 1999, 11, 1416 CrossRef CAS.
- F. Wittmann, R. H. Friend, M. S. Khan and J. Lewis, J. Chem. Phys., 1994, 101, 2693 CrossRef CAS.
- J. Cornil, D. A. dos Santos, X. Crispin, R. Silbey and J. L. Bredas, J. Am. Chem. Soc., 1998, 120, 1289 CrossRef CAS.
- G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1990, 454 RSC; P. J. Langley, J. Hulliger, R. Thaimattam and G. R. Desiraju, New J. Chem., 1998, 22, 1307 RSC; J. M. A. Robinson, D. Philp, B. M. Kariuki and K. D. M. Harris, Chem. Commun., 1999, 329 RSC.
- T. Steiner, J. Chem. Soc., Chem. Commun., 1995, 95 RSC; D. Philp and J. M. A. Robinson, J. Chem. Soc., Perkin Trans. 2, 1998, 1643 RSC.
- S. Kumar, K. Subramanian, R. Srinivasan, K. Rajagopalan, A. M. M. Schreurs, J. Kroon, G. Koellner and T. Steiner, J. Mol. Struct., 1998, 448, 51 CrossRef CAS.
- T. Steiner, B. Lutz, J. van der Maas, N. Veldman, A. M. M. Schreurs, J. Kroon and J. A. Kanters, Chem. Commun., 1997, 191 RSC.
- (a) N. A. Ahmed, A. I. Kitaigorodsky and M. Sirota, Acta Crystallogr., Sect. B, 1972, 28, 2875 CrossRef CAS; (b) J. M. A. Robinson, B. M. Kariuki, R. J. Gough, K. D. M. Harris and D. Philp, J. Solid State Commun., 1997, 134, 203 Search PubMed; (c) H.-C. Weiss, D. Bläser, R. Boese, B. M. Doughan and M. M. Haley, Chem. Commun., 1997, 1703 RSC.
- T. Steiner, E. B. Starikov and M. Tamm, J. Chem. Soc., Perkin Trans. 2, 1996, 67 RSC.
- V. R. Vangala, A. Nangia and V. M. Lynch, Chem. Commun., 2002, 1304 RSC.
- T. Steiner, J. Chem. Soc., Chem. Commun., 1994, 101 RSC.
- (a) S. Thorand and N. Krause, J. Org. Chem., 1998, 63, 8551 CrossRef CAS; (b) M. S. Khan, M. R. A. Al-Mandhary, M. K. Al-Suti, A. K. Hisahm, P. R. Raithby, B. Ahrens, M. F. Mahon, L. Male, E. A. Marseglia, E. Tedesco, R. H. Friend, A. Köhler, N. Feeder and S. J. Teat, J. Chem. Soc., Dalton Trans, 2002, 1358 RSC; (c) R. Ziessel, J. Suffert and M.-T. Youinou, J. Org. Chem., 1996, 61, 6535 CrossRef CAS; (d) P. Nguyen, Z. Yuan, L. Agocs, G. Lesley and T. B. Marder, Inorg. Chim. Acta, 1994, 220, 289 CrossRef CAS; (e) M. S. Khan, A. K. Kakkar, N. J. Long, J. Lewis, P. R. Raithby, P. Nguyen, T. B. Marder, F. Wittman and R. H. Friend, J. Mater. Chem., 1994, 4, 1227 RSC; (f) M. Moroni, J. Le Moigne, T. A. Pham and J.-Y. Bigot, Macromolecules, 1997, 30, 1964 CrossRef CAS.
- H.-C. Weiss, R. Boese, H. L. Smith and M. M. Haley, Chem. Commun., 1997, 2403 RSC.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384 CrossRef CAS.
- J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613 CAS; J. D. Dunitz and R. Taylor, Chem. Eur. J., 1997, 3, 89 CrossRef CAS.
- V. R. Thalladi, H.-C. Weiss, D. Blaser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702 CrossRef CAS.
- J. S. Wilson, A. Köhler, R. H. Friend, M. K. Al-Suti, M. R. A. Al-Mandhary, M. S. Khan and P. R. Raithby, J. Chem. Phys., 2000, 113, 7627 CrossRef CAS.
- (a) V. Cleave, G. Yahioglu, P. Lebarny, R. H. Friend and N. Tessler, Adv. Mater., 1999, 11, 285 CrossRef CAS; (b) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- A. Köhler, J. S. Wilson, R. H. Friend, M. K. Al-Suti, M. S. Khan, A. Gerhard and H. Bässler, J. Chem. Phys. Search PubMed , submitted.
- D. Philp and J. M. A. Robinson, J. Chem. Soc., Perkin Trans. 2, 1998, 1643 RSC.
- Purification of Laboratory Chemicals, eds. W. L. F. Armarego and D. D. Perrin, Butterworth-Heinemann, London, 1996 Search PubMed.
- T. Sargent, A. T. Shuglin and C. A. Mathia, J. Med. Chem., 1984, 27, 1071 CrossRef CAS.
- T. X. Neenan and G. M. Whitesides, J. Org. Chem., 1988, 53, 2489 CrossRef CAS.
- S. J. Davies, B. F. G. Johnson, M. S. Khan and J. Lewis, J. Chem. Soc., Chem. Commun., 1991, 187 RSC; N. Hagihara, K. Sonogashira and S. Takahashi, Adv. Polym. Sci., 1980, 41, 149 Search PubMed; S. Takahashi, H. Morimoto, E. Murata, S. Kataoka, K. Sonogashira and N. Hagihara, J. Polym. Sci. Polym. Chem. Edu., 1982, 20, 565 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 CrossRef.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. C. Camalli, J. Appl. Crystallogr., 1994, 27, 435 CrossRef.
- G. M. Sheldrick, SHELXL97, Program for crystal structure refinement, Göttingen, 1997.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 1991, 24, 987 CrossRef CAS.
- K. Shankland, W. I. F. David and T. Csoka, Z. Kristallogr., 1997, 212, 550 Search PubMed; W. I. F. David, K. Shankland and N. Shankland, Chem. Commun., 1998, 931 RSC; R. E. Dinnebier, P. Sieger, H. Nar, K. Shankland and W. I. F. David, J. Pharm. Sci., 2000, 89, 1465 CrossRef CAS.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory, Report LAUR 86-748, 1994.
- SHELXTL, An integrated system for solving, refining and displaying crystal structures from diffraction data, Ver. 5.10. Bruker AXS, Madison, Wisconsin, USA, 1997.
Footnotes |
| † Electronic supplementary information (ESI) available: atomic cooordinates for 6 and 7. See http://www.rsc.org/suppdata/nj/b2/b206946f/ |
| ‡ πCC is the midpoint of the CC bond. |
| § All hydrogen bond parameters are calculated for idealized bond lengths C–H 1.08 Å and N–H 1.01 Å. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |