C–H bond activation reactions by TpMe2Ir(III) centres. Generation of Fischer-type carbenes and development of a catalytic system for H/D exchange†
Laura L.
Santos
a,
Kurt
Mereiter
b,
Margarita
Paneque
a,
Christian
Slugovc
c and
Ernesto
Carmona
*a
aInstituto de Investigaciones Químicas, Departamento de Química Inorgánica, Universidad de Sevilla, Consejo Superior de Investigaciones Científicas, Av. Américo Vespucio s/n, Isla de la Cartuja, 41092, Sevilla, Spain. E-mail: guzman@us.es; Fax: (+34) 954460565
bDepartment of Chemistry, Vienna University of Technology, Getreidemarkt 9/164, A-1060, Vienna, Austria
cTU-Graz, Institute of Chemical Technology of Organic Materials (ICTOS), Stremayrgasse 16, A-8010, Graz, Austria
First published on 28th November 2002
Abstract
The unsaturated [TpMe2Ir(C6H5)2] fragment, readily generated from [TpMe2Ir(C6H5)2(N2)], or from [TpMe2Ir(C2H4)2] and C6H6, is able to induce the regioselective cleavage of two sp3 C–H bonds of anisole, with formation of a Fischer-type carbene, 1. The process involves additionally ortho-metallation of the anisole aromatic ring, hence three C–H bonds are sequentially broken, the last one in the course of an α-H elimination reaction. Phenetole (ethyl phenyl ether) gives an analogous product, 3, despite the possibility of competitive α- or β-H eliminations in the last step. For C6H5NMe2, two hydride-carbenes, 5a and 5b, are produced. In the latter, the aniline phenyl ring is also metallated, but the former contains a C6H5 aryl group and a C6H5N(Me)CH carbene ligand. The same Ir(III) fragment, viz.
[TpMe2Ir(C6H5)2], alternatively generated from C6H6 and [TpMe2Ir(η4-CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)C(Me)CH2)], accomplishes the efficient, catalytic H/D exchange between C6D6
(used as the deuterium source) and a variety of organic and organometallic molecules that contain C–H bonds of different nature.
C(Me)C(Me)CH2)], accomplishes the efficient, catalytic H/D exchange between C6D6
(used as the deuterium source) and a variety of organic and organometallic molecules that contain C–H bonds of different nature.
Introduction
The study of the reactivity of aliphatic C–H bonds of hydrocarbons and other organic molecules with transition metal compounds continues to be a topic of renewed interest.1 Some years ago we showed that Ir(III) species like [TpMe2Ir(H)(CHCH2)(C2H4)] or [TpMe2Ir(C6H5)2(N2)], (TpMe2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =hydridotris(3,5-dimethylpyrazol-1-yl)borate) are able to induce the selective activation of the two sp3 C–H bonds of five- and six-membered cyclic ethers, with formation of Fischer-type carbenes.2 The active species were in situ generated intermediates of the kind [TpMe2Ir(R)(R′)], where R and R′ represent formally monoanionic hydrocarbyl fragments (e.g. C6H5, C2H5, CHCH2, etc.). More recently, the use of the bulky TpPh ancillary ligand (TpPh=hydridotris(3-phenylpyrazol-1-yl)borate) in related coordination environments allowed extension of this synthetic methodology to a variety of ethers and amines.3 This reactivity constitutes an unusual, seldom employed route to heteroatom-stabilized carbenes.4
=hydridotris(3,5-dimethylpyrazol-1-yl)borate) are able to induce the selective activation of the two sp3 C–H bonds of five- and six-membered cyclic ethers, with formation of Fischer-type carbenes.2 The active species were in situ generated intermediates of the kind [TpMe2Ir(R)(R′)], where R and R′ represent formally monoanionic hydrocarbyl fragments (e.g. C6H5, C2H5, CHCH2, etc.). More recently, the use of the bulky TpPh ancillary ligand (TpPh=hydridotris(3-phenylpyrazol-1-yl)borate) in related coordination environments allowed extension of this synthetic methodology to a variety of ethers and amines.3 This reactivity constitutes an unusual, seldom employed route to heteroatom-stabilized carbenes.4
Several recent studies have focused on the application of these and related findings in C–H activation chemistry to different catalytic processes.5 An interesting transformation is H/D exchange between hydrocarbons (or other substrates) and a deuterium source (D2O or a deuterated alkane or arene). Brookhart has described catalytic H/D exchange reactions between deuterated solvents and olefins bound to RhCp* centres.6 Andersen,7 Bergman7,8 and Tilley8 have found very efficient H/D exchanges catalysed by cationic IrCp* species, using C6D67 or D2O8 as the deuterium source. Another interesting example is a CH4/CD4 exchange, catalysed by a silica supported tantalum hydride.9 These reports constitute valuable additions to earlier studies in this field.10,11
In this contribution we describe recent studies on the activation of sp3 C–H bonds, which leads to the facile generation of Fischer-type carbene complexes. The bis(ethene) Ir(I) compound, [TpMe2Ir(C2H4)2],12 can be used as the reactive precursor, in reactions with aromatic ethers and amines, C6H5OR (R=CH3, C2H5) and C6H5NMe2. Our previous knowledge of this reactivity, along with the close investigation of the mechanistic aspects of the above C–H bond activation processes, has permitted the design of a catalytic system for H/D exchange, employing C6D6 as the deuterium source and the Ir(I) complex [TpMe2Ir(η4-CH2C(Me)C(Me)CH2)]13 as the catalyst precursor.
Results and discussion
Heteroatom-stabilised Ir(III)-carbenes
To exploit the lability of the coordinated molecule of dinitrogen in the Ir(III) complex [TpMe2Ir(C6H5)2(N2)],2 an isolated sample of this material was reacted with anisole, C6H5OCH3, under nitrogen, using C6H6 as the solvent (Scheme 1a). The reaction is complex and gives a mixture of products. Following column chromatography (silica gel; eluent hexane∶ether) and crystallization from CH2Cl2∶hexane mixtures, moderate yields of the hydride-carbene compound 1 were obtained. In an alternative, cleaner and more efficient procedure, this species was generated in comparable yields using the Ir(I) derivative [TpMe2Ir(C2H4)2]12 as the starting material. Under the conditions specified in Scheme 1b, the unsaturated [TpMe2Ir(C6H5)2] fragment is readily generated.2 | ||
| Scheme 1 | ||
The molecules of 1 are chiral at iridium. Asymmetry of the metal centre results in the inequivalency of the three pyrazolyl rings, which gives rise to five 1H methyl signals in the chemical shift interval from 2.41 to 0.95 ppm (two of these resonances are accidentally degenerated at δ 2.40), along with three C–H resonances between δ 5.9 and 5.5, in the 1H NMR spectrum. Of diagnostic value regarding the molecular complexity of 1 are both the high-field 1H resonance at δ
−17.45, due to the Ir–H unit, and the low-field 1H and 13C signals associated with the Ir-carbene functionality (1H resonance at 14.32 ppm; 13C signal at 257.5 ppm, 1JCH=164 Hz). Other relevant NMR data for 1 are collected in the Experimental Section and need no further discussion.
Despite the simultaneous presence of hydride, aryl and carbene ligands in the metal coordination sphere, no indications for migratory insertion chemistry can be discerned either at room temperature or at 60–80°C. It seems that the carbene atom of 1 exhibits low electrophilicity, most likely due to the presence of the adjacent oxygen atom. Nevertheless, when 1 is heated in neat acetonitrile at 60°C, a five-membered heterometallacyclic species, 2, is produced (quantitatively by 1H NMR; see Scheme 1). The characteristic hydride and carbene resonances of 1 are replaced by those corresponding to a coordinated molecule of NCMe and to an iridium-bound CH2 group (see Experimental Section). Hence, NCMe induces migration of the hydride onto the carbene carbon to give an iridium-alkyl, as part of a metallacyclic unit. This reactivity is in accord with expectations, based on both experimental and theoretical grounds.14 Due to differences in the spatial properties of the σ-orbitals of the migratory groups (hydride or aryl), hydride migration is expected to be much faster than aryl migration. This is because the 1s orbital of the H atom, with spherical symmetry, allows easier access to the three centre transition state suggested for this migratory insertion,14b than the more directional sp2 orbital of the aryl carbon atom. Besides, the product of the latter rearrangement would contain a highly-strained cyclic unit, in contrast with the unstrained five-membered ring present in the structure of 2.

As already hinted, the formation of complex 1 under the conditions of Scheme 1 (1a and 1b) proceeds, regardless of the iridium complex precursor used, through the intermediacy of the unsaturated species [TpMe2Ir(C6H5)2]. Previous work from our laboratory has demonstrated the capacity of this intermediate to induce the double C–H bond cleavage of tetrahydrofuran, and other cyclic, aliphatic ethers, in reactions that proceed with coordination of the ether to iridium.2 A similar mechanism could be proposed for 1. However, since arene C–H bond activation by this Ir(III) fragment is much faster than THF C–H bond activation,2 it appears plausible that the first of the three C–H bonds of C6H5OCH3 that must be broken to form complex 1 is an aromatic C–H bond. Obviously, not only the ortho, but also the meta and the para C–H bonds can be cleaved, although when employing C6H6 as the reaction solvent, these single activations would remain undetected. Eventually, O-coordination of the ether moiety in an intermediate like B, which results from the activation of one of the ortho hydrogens, could lead to a metallacyclic intermediate of type C that would render 1 by means of an α-H elimination. In all, three C–H bonds, one of sp2 and the other two of sp3 character, are sequentially broken. Although some examples that involve the facile rupture of three C–H bonds have been reported recently,3a,15 this remains a very unusual observation. It seems that both the chelate effect and the stability of the five-membered metallacyclic unit of 1 are responsible for the formation of this complex, under the experimental conditions specified in Scheme 1.
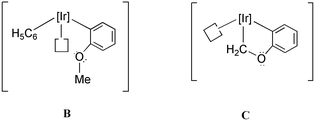
The above proposal regarding the last step of the C–H bond activation reaction of ethers to generate iridium carbenes is supported by earlier isotopic labelling studies from our group, using d8-tetrahydrofuran and different Tp′Ir(III) fragments.2,3a In the present system the intermediate that immediately precedes the product, i.e.C, has no β-hydrogens. Hence, the alternative, and ubiquitous, β-H elimination, that would produce a hydride-olefin compound, obviously cannot occur. To ascertain whether α-H elimination is preferred over β-H elimination in this kind of substrate, the analogous activation of C6H5OCH2CH3 has been investigated.
Once more, both [TpMe2Ir(C2H4)2] and [TpMe2Ir(C6H5)2(N2)] react with the ether, using benzene as the solvent, with essentially identical results. However, the reaction seems to be more complex that for C6H5OMe, since a mixture of two products, in a ca. 3∶1 ratio, is obtained. The minor component is a hydride-carbene derivative but it has not yet been fully characterized. However, the major product is a yellow solid with spectroscopic properties similar to those of 1, and can therefore be formulated as the hydride-carbene 3 (Scheme 2; see Experimental Section for relevant 1H and 13C NMR data). Exchange of the hydride ligand of 3 by chloride is facile and allows the isolation of the chloro-carbene derivative 4, which exhibits a characteristic 13C carbene resonance at δ 278.6 (271.4 ppm in the parent hydride). It is clear from these results that α-H elimination is more favourable than β-H elimination.
 | ||
| Scheme 2 | ||
Repeated attempts to grow crystals of 3 suitable for an X-ray study have finally met with success (despite our efforts crystals of the minor product have not yet been obtained). Fig. 1 shows an ORTEP view of the molecules of this compound, Table 1 contains relevant bond distances and angles. Similar to other six-coordinate Tp′ complexes, the N–Ir–N angles are close to the ideal value of 90°. This is in line with the tendency of this family of ligands to enforce octahedral coordination.16 The Ir–N bond trans to the hydride is somewhat longer than the other two (ca. 2.17 Å vs. 2.15 Å, see Table 1), as a reflection of the higher trans influence of this ligand, in comparison with those exerted by the aryl and the carbene units. The Ir–Caryl and Ir–Ccarbene bonds have normal lengths, comparable to those reported in the literature for related compounds.2,3a
 | ||
| Fig. 1 Structural view of 3 (50% ellipsoids, C- and B-bound H atoms omitted). | ||
| Bond distances | |||
| Ir–C(7) | 1.902(2) | C(1)–C(2) | 1.406(3) |
| Ir–C(1) | 2.013(2) | C(2)–C(3) | 1.400(3) |
| Ir–N(12) | 2.145(2) | C(3)–C(4) | 1.386(4) |
| Ir–N(22) | 2.153(2) | C(4)–C(5) | 1.390(4) |
| Ir–N(32) | 2.168(2) | C(5)–C(6) | 1.384(3) |
| Ir–H(1) | 1.51(3) | C(7)–C(8) | 1.492(3) |
| C(1)–C(6) | 1.390(3) | O–C(7) | 1.348(2) |
| O–C(6) | 1.402(3) | ||
| Bond angles | |||
| C(7)–Ir–C(1) | 79.5(1) | N(22)–Ir–N(32) | 87.9(1) |
| C(7)–Ir–N(12) | 174.7(1) | C(7)–Ir–H(1) | 86.0(10) |
| C(7)–Ir–N(22) | 99.3(1) | C(1)–Ir–H(1) | 85.0(10) |
| C(7)–Ir–N(32) | 99.5(1) | N(12)–Ir–H(1) | 88.8(10) |
| C(1)–Ir–N(12) | 98.6(1) | N(22)–Ir–H(1) | 92.2(10) |
| C(1)–Ir–N(22) | 177.0(1) | N(32)–Ir–H(1) | 174.4(10) |
| C(1)–Ir–N(32) | 95.1(1) | C(6)–C(1)–Ir | 112.4(2) |
| N(12)–Ir–N(22) | 82.3(1) | O–C(7)–Ir | 119.5(2) |
| N(12)–Ir–N(32) | 85.6(1) | C(7)–O–C(6) | 112.3(2) |
| C(1)–C(6)–O | 115.2(2) | ||
The generality of the double C–H bond activation reaction at the α carbon with respect to a donor atom has been demonstrated using N,N-dimethylaniline as the substrate. Analogously to the results found for TpPhIr(η4-isoprene)3a a mixture of two N-substituted Fischer carbenes, 5a and 5b is generated (Scheme 3).
 | ||
| Scheme 3 | ||
Column chromatography of the crude reaction mixture and routine crystallization procedures allow isolation of pure samples of these compounds. Microanalytical and extensive 1D and 2D NMR studies provide unequivocal evidence for their molecular complexity. 5a exists in solution, at room temperature, as a mixture of two fast equilibrating carbene rotamers. The 1H NMR spectrum shows broad resonances at ca.
−18.4 and 13.8 ppm, assignable to the hydride and the CH unit of the carbene ligand, respectively. The carbene carbon resonates at δ 222.0 and is characterized by a one-bond C–H coupling of 146 Hz. The iridium-bound phenyl group gives rise to five, well-defined resonances in the interval from 7.98 to 6.52 ppm, which appear broad at room temperature. The second phenyl group of 5a, namely that being part of the carbene ligand, originates sharp resonances in the narrower region from 7.34 to 7.16 ppm. At variance with this, only four aromatic signals are found in the 1H NMR spectrum of 5b. They are accompanied by a CH carbene resonance at δ 12.12 (13C NMR signal at δ 216.4; 1JCH=149 Hz) as well as by a hydride signal at −20.14 ppm.
The formation of the hydride-carbenes 5a and 5b during the activation of C6H5NMe2 contrasts with the observation of only compound 1 in the analogous activation of C6H5OMe. The latter compound contains a metallated aromatic ring from the ether functionality and therefore it may be considered as the analog of 5b. The ratio of 5a∶5b
(see Experimental Section) produced under the conditions of Scheme 3 does not vary with time. Furthermore, 5a remains unaltered after prolonged heating at 100°C (cyclohexane solution, 12 h). This rules out the intermediacy of 5a in the formation of the ortho-metallated compound 5b and suggests instead that the two hydride-carbenes form through different, competitive reaction pathways. Additional evidence for this stems from the observation that 5b does not convert into 5a when heated in C6H6 in the presence of N,N-dimethylaniline. A plausible proposal is that N-coordination of the amine to iridium assists the double C–H bond activation that originates 5a, whereas activation of an ortho-hydrogen atom of C6H5NMe2 is the first step of the reaction leading to 5b. Only the latter process appears to take place during the activation of C6H5OMe, perhaps as a consequence of the lower coordination ability of the ether, in comparison with the amine.
Before closing this section it is worth emphasizing the facility with which Tp′Ir(III) fragments may induce the formation of Fischer-type carbenes by means of the regioselective, double C–H bond activation of the C–H bonds in α position with respect to an ether or amine functionality. An alkyl intermediate of type C precedes the hydride-carbene product, which therefore derives from an α-H elimination. Whereas in C and the analogous intermediate of the C6H5NMe2 activation reaction the α-H elimination appears to be the only low-energy reaction pathway for further rearrangement in the case of CH3CH2OC6H5 the possibility of β-H elimination, to give a hydride-olefin compound, does exist but it is not observed. It is our view that additional studies on these and related systems are needed to gain a better understanding of the factors that govern the course of these reactions, in particular the still unusual observation of more facile α- than β-H elimination.
Catalytic H/D exchange reactions
During attempts to extend the above results regarding the activation of ethers to TpMe2Ir(I) precursors other than TpMe2Ir(C2H4)2, the diene derivative [TpMe2Ir(η4-2,3-dimethylbutadiene)], 6, was tested in the reaction with THF. Following the reaction of 6 and THF (3 equiv.; C6D6, 70°C) by 1H NMR spectroscopy, a progressive decline in the intensity of the α-CH2 protons of THF, and a concomitant increase of the C6D5H signal became apparent. This observation points to an ongoing H/D exchange reaction which has been investigated under conditions of catalysis. Although a variety of systems is known that effect thermal H/D exchange between hydrocarbon or other substrates and a deuterium source,5–11 general procedures of wide applicability are scarce.
A typical catalytic procedure follows: a mixture of ferrocene (0.23 mmol) and 6
(7.5 mol%) in C6D6
(0.5 mL) as the solvent was heated for 5 h in a sealed NMR tube at 90°C (bath temperature). The progress of the reaction was checked by 1H-NMR spectroscopy at regular periods of time, until the deuterium incorporation stopped. Although the typical reaction time was 20 h, all runs reached the maximum deuterium incorporation after approximately 3.5 h. Degradation after this time was not observed.
The extent of deuteration was obtained by integration of the 1H-NMR spectra, comparing the intensity of the signals due to the substrate and the protons from C6D5H in the initial spectra (before heating) and the final spectra (after heating). As a verification of these results, at the end of the reaction 20 μL of TMS were added and acquisition and integration of the probe rerun. The deuterium incorporation was calculated from the integrals obtained. In the case of 7-d9.5, and 8-d11.4 the products were isolated by column chromatography (neutral Al2O3; eluent n-hexane, sampling the deep-red band) and analyzed by 1H, 13C{1H}, and 2H-NMR spectroscopy.
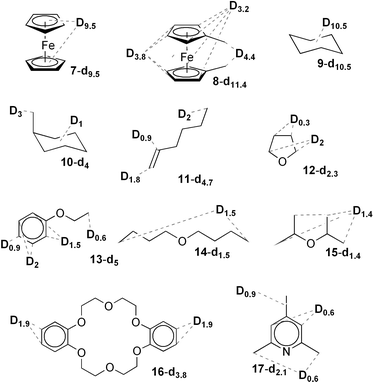
Table 2 summarizes the results of the H/D exchange catalysis, which suggest a kinetic control of the reaction, in agreement with previous literature data.5 In general, the most accessible C–H bonds take part in the reaction, whereas weaker (albeit less exposed) C–H bonds are reluctant to undergo exchange. For example, for methylcyclohexane (entry 4) or di(n-butyl) ether (entry 8) only the methyl groups experience significant exchange while the CH2 hydrogens remain unaltered. The reaction is only effective for the poorly coordinating substrates like ethers (entries 6, 7, 8, 9, and 10) or sterically hindered N-donor ligands (entry 11). 2,6-Lutidine is deuterated mostly in the meta- and para-positions, and to a lesser extent in the methyl groups. Contrarily, pyridine poisons the catalytic system by strongly binding to iridium so that no deuterium incorporation takes place. It is worth mentioning that the exclusive deuteration of dibenzo-18-crown-6 in the 3 and 3′ positions of the benzene rings illustrates the potential of this catalytic system to introduce deuterium as markers into more sophisticated molecules. Finally, in accord with expectations, the exchange of deuterium for hydrogen is also possible, as shown by the reaction of 7-d9.5, or 8-d11.4, with C6H6 (in the presence of freshly added 6 and otherwise equal reaction conditions). Virtually undeuterated 7 or 8 was isolated.
| Substrate | mmol | Catalyst/mol% | Product | TON/D | |
|---|---|---|---|---|---|
|
a Precatalyst: TpMe2Ir(2,3-dimethylbutadiene)
(6)
(0.01 g; 0.017 mmol); solvent: C6D6
(0.5 mL); reaction time: 2.5 h; bath temperature: 90°C.
|
|||||
| 1 | Ferrocene (7) | 0.228 | 7.5 | (7-d9.5) | 127 |
| 2 | 1,1′-Dimethylferrocene (8) | 0.199 | 8.5 | (8-d11.4) | 100 |
| 3 | Cyclohexane (9) | 0.463 | 3.7 | (9-d10.5) | 286 |
| 4 | Methylcyclohexane (10) | 0.509 | 3.3 | (10-d4) | 120 |
| 5 | 1-Hexene (11) | 0.594 | 2.9 | (11-d4.7) | 164 |
| 6 | THF (12) | 0.693 | 2.4 | (12-d2.3) | 94 |
| 7 | Phenyl ethyl ether (13) | 0.409 | 4.1 | (13-d5) | 120 |
| 8 | Di(n-butyl) ether (14) | 0.384 | 4.4 | (14-d1.5) | 34 |
| 9 | Di(isopropyl) ether (15) | 0.489 | 3.4 | (15-d1.4) | 40 |
| 10 | Dibenzo-18-crown-6 (16) | 0.139 | 12.2 | (16-d3.8) | 31 |
| 11 | 2,6-Lutidine (17) | 0.475 | 3.5 | (17-d2.1) | 59 |
| 12 | Pyridine (18) | 0.632 | 2.7 | (18) | — |
The fate of precatalyst 6 and the catalytic cycle for the H/D exchange, can be proposed to be as depicted in Scheme 4. Conversion of 6 into the suggested active catalyst, E, occurs by means of a well-established equilibrium in Tp′Ir(I) compounds3,17 between κ3- and κ2-TpMe2 coordination, followed by rearrangement into the Ir(III) intermediate D.18 C–D activation of two molecules of C6D613 by this Ir(III) species would then yield E, hence the original diene acts as a sacrificial deuterium acceptor being extruded as CH2DC(Me)C(Me)CH2D.3 In accord with the results presented in the previous section, E would readily activate the substrate S–H giving F, which by exchange with C6D6 would form S–D and regenerate E. In excellent agreement with this proposal, the use of [TpMe2Ir(C6H5)2(N2)]13 as the precatalyst yields results identical within experimental error with those collected in Table 2.
 | ||
| Scheme 4 | ||
Catalyst degradation may occur through either of the following pathways. Firstly, strongly coordinating substrates (e.g. pyridine, phosphines, etc.) react with 6 to provide stable Ir(III) adducts18 related to D in Scheme 4, which are catalytically ineffective. Alternatively, some substrates like THF and phenetole (ethyl phenyl ether; entries 6 and 7 in Table 2) react with 6 to form Fischer-type carbenes (e.g.G in Scheme 4) which are also unable to promote the H/D exchange catalysis, even after prolonged heating at higher temperatures (90°C, 24 h).
In conclusion, the unsaturated Ir(III) species, [TpMe2Ir(C6H5)2], which can be generated in situ from C6H6 and readily available Ir(I) precursors like [TpMe2Ir(C2H4)2] or [TpMe2Ir(η4-CH2C(Me)C(Me)CH2)], activates ether and amine substrates under mild conditions, with formation of Fischer-type carbenes. In this transformation, the regioselective cleavage of two C–H bonds in α with respect to the O- or N-donor atom is effected. Most remarkably, the second C–H bond scission, which is an α-H elimination, occurs even in the presence of β-hydrogens. [TpMe2Ir(C6H5)2] acts additionally as a powerful, efficient catalyst for H/D exchange between C6D6
(as the deuterium source) and a variety of substrates. The use of this system for the catalytic functionalization of non-activated hydrocarbons remains an exciting avenue to explore.
Experimental
General details
All syntheses and operations were done under an inert atmosphere, following previously described procedures.18Compound 1. To a solution of [TpMe2Ir(C2H4)2] in C6H6 (0.44 g, 0.82 mmol; 12 ml) CH3OC6H5 (0.44 ml, ca. 4 mmol) was added and the mixture stirred at 60
°C for 12 h. After removing the solvent under reduced pressure, the product was purified by column chromatography on silica gel, using a 10∶1 mixture of hexane∶diethyl ether as eluent (Rf=0.68). Pure complex 1 was isolated, as a yellow microcrystalline solid, in ca. 37% yield (0.18 g). It can be crystallized from hexane∶CH2Cl2 mixtures (2∶1) at −20°C. IR (Nujol): ν(Ir–H) 2192 cm−1. 1H NMR (CDCl3, 25°C): δ 14.32 (s, 1 H, IrCH), 7.39, 7.25, 6.97 (d, d, m, 1∶1∶2 H, 3JHH≈8 Hz, 4 CHar), 5.89, 5.86, 5.55 (s, 1 H each, 3 CHpz), 2.41, 2.40, 2.36, 2.25, 0.95 (s, 1∶2∶1∶1∶1, 6 Mepz), −17.45 (s, 1 H, IrH). 13C{1H} NMR (CDCl3, 25°C): δ 257.5 (IrCH, 1JCH=164 Hz), 168.2, 129.0 (Cqar), 151.9, 151.5, 150.9, 144.5, 144.2, 143.7 (Cqpz), 137.7, 124.4, 122.8, 110.9 (CHar), 107.0, 106.6, 106.5 (CHpz), 16.0, 15.7, 13.1, 13.0, 12.9, 12.0 (Mepz). Anal. Calcd. for C22H28BN6OIr: C, 44.4; H, 4.5; N, 14.1; Found: C, 44.1; H, 4.8; N, 13.6%.
Compound 2. A solution of 1 (0.042 g, 0.07 mmol) in NCMe (4 ml) was stirred at 80
°C for 12 h, after which time the solvent was distilled off under reduced pressure. At this stage, 1H NMR monitoring demonstrated quantitative formation of the acetonitrile adduct 2, which was isolated as a pure, white crystalline solid, by crystallization from hexane∶dichloromethane (3∶2) at −20°C (0.037 g, isolated yield 83%). IR (Nujol): ν(CN) 2285 cm−1. 1H NMR (CDCl3, 25°C): δ 6.97, 6.81, 6.63, 6.46 (d, t, d, t, 1 H each, 3JHH≈7 Hz, 4 CHar), 6.63, 6.42 (d, d, 1 H each, 2JHH≈7 Hz, Ir–CH2), 5.90, 5.80, 5.53 (s, 1 H each, 3 CHpz), 2.42, 2.39, 2.37, 2.33, 2.32, 1.44 (s, 3 H each, 6 Mepz), 2.37 (s, 3 H, NCMe). 13C{1H} NMR (CDCl3, 25°C): δ 170.9, 122.7 (Cqar), 152.0, 150.4, 149.5, 143.3, 143.1 (1∶1∶1∶1∶2, Cqpz), 137.0, 123.7, 116.7, 106.5 (CHar), 114.7 (NCCH3), 108.6, 107.0, 106.7 (CHpz), 54.4 (Ir–CH2, 1JCH=143 Hz), 14.7, 13.8, 13.3, 12.7, 12.5, 4.2 (1∶1∶1∶1∶2∶1, Mepz+NCCH3). Anal. Calcd. for C24H31BN7OIr·1/2CH2Cl2: C, 43.3; H, 4.7; N, 14.4; Found: C, 43.4; H, 4.9; N, 13.8%.
Compound 3. [TpMe2Ir(C2H4)2] (0.30 g, 0.55 mmol) was reacted with C6H5OCH2CH3 (0.33 ml, 2.75 mmol) in C6H6 (12 ml) at 60
°C for 12 h. After this time the colour of the solution has changed from essentially yellow to dark-green. Removal of the solvent under reduced pressure and 1H NMR monitoring revealed the formation of two compounds, 3 and a minor product which despite our efforts remains uncharacterized (the unidentified product is also a hydride-carbene; its proportion in the reaction mixture varies from one preparation to another; typically it is of the order of 20%, but in isolated instances it can be as high as 40%). Separation of the two components was achieved by column chromatography on silica gel, using a 40∶1→5∶1 mixture of hexane∶Et2O as eluent (Rf=0.53 (10∶1; hexane∶EtO2)), to yield 0.11 g (ca. 33%) of pure compound 3. Yellow crystals of 3 may be obtained from its concentrated solutions in hexane∶CH2Cl2
(ca. 2∶1 mixtures) upon cooling at −20°C. IR (Nujol): ν(Ir–H) 2148 cm−1. 1H NMR (CDCl3, 25°C): δ 7.29, 7.21, 6.98, 6.89 (d, d, t, t, 1 H each, 3JHH≈8 Hz, 4 CHar), 5.89, 5.85, 5.59 (s, 1 H each, 3 CHpz), 2.55 (s, 3 H, IrCCH3), 2.43, 2.41, 2.40, 2.28, 2.24, 1.05 (s, 3 H each, 6 Mepz), −17.74 (s, 1 H, IrH). 13C{1H} NMR (CDCl3, 25°C): δ 271.4 (IrC), 168.0, 129.6 (Cqar), 151.5, 150.1, 144.5, 144.4, 143.4 (2∶1∶1∶1∶1, Cqpz), 137.3, 123.4, 122.2, 109.8 (CHar), 106.8, 106.3, 106.2 (CHpz), 39.6 (IrCCH3), 16.5, 15.9, 13.2, 13.0, 12.9, 12.0 (Mepz). Anal. Calcd. for C23H30BN6OIr: C, 45.3; H, 4.9; N, 13.8; Found: C, 45.5; H, 5.0; N, 14.0%.
Compound 4. A solution of the above hydride-carbene 3 in CCl4 (0.04 g, 0.066 mmol; 2 ml) was heated at 80
°C for 5 days, whereupon clean, quantitave conversion into the new product 4 was ascertained by 1H NMR. The crude solid that resulted from the evaporation of the CCl4 under reduced pressure was dissolved in 7 ml of a 4∶3 Et2O∶CH2Cl2 mixture and filtered. Addition of 4 ml of hexane and cooling at −20°C gave 0.03 g (71%) of a yellow microcrystalline solid. 1H NMR (CDCl3, 25°C): δ 7.37, 7.08, 7.03, 6.93 (d, d, t, t, 1 H each, 3JHH≈8 Hz, 4 CHar), 5.90, 5.86, 5.50 (s, 1 H each, 3 CHpz), 2.92 (s, 3 H, IrCCH3), 2.57, 2.46, 2.45, 2.41, 2.39, 0.95 (s, 3 H each, 6 Mepz). 13C{1H} NMR (CDCl3, 25°C): δ 278.6 (IrC), 168.6, 125.9 (Cqar), 153.3, 151.5, 151.4, 143.7, 143.6, 143.5 (Cqpz), 136.8, 124.5, 123.5, 111.6 (CHar), 108.3, 108.0, 107.9 (CHpz), 39.2 (IrCCH3), 15.8, 15.5, 13.0, 12.7, 12.6, 11.9 (Mepz). Anal. Calcd. for C23H29BN6ClOIr·1/2 CH2Cl2: C, 41.1; H, 4.4; N, 12.2; Found: C, 41.4; H, 4.5; N, 11.9%.
Compounds 5a and 5b. 0.20 g (0.37 mmol) of [TpMe2Ir(C2H4)2] were dissolved in 10 ml of C6H6. 0.23 ml of C6H5NMe2 were then added and the resulting solution was heated at 60
°C, overnight. The yellow solution that resulted was evaporated under reduced pressure to give a crude solid consisting of a ca. 3∶1 mixture of 5a∶5b
(as shown by 1H NMR studies). Even if this proportion may change from one preparation to another, for a specific preparation it does not change with time, indicating that 5a and 5b form through different, competitive reaction pathways. Separation of the two components was accomplished by column chromatography on silica gel, using a 20∶1→1∶1 mixture of hexane∶Et2O as eluent (Rf(5a)=0.27, Rf(5b)=0.15 (15∶1; hexane∶EtO2)). 0.064 g of 5a
(ca. 26%) and 0.044 g of 5b
(20%) were obtained in this way. The two isolated compounds can be crystallized from hexane–Et2O mixtures at −20°C.
Data for 5a. IR (Nujol): ν(Ir–H) 2129 cm−1. 1H NMR (CDCl3, 25
°C): δ 13.77 (brs, 1 H, IrCH), 7.34, 7.29, 7.16 (brt, brt, brd, 2∶1∶2, 3JHH≈8 Hz, 5 CHar, N–Ph), 7.98, 6.96, 6.76, 6.64, 6.52 (brs, brs, t, brs, brs, 1 H each, 3JHH≈7 Hz, o, m, p, m, o-CHar, Ir–Ph), 5.78, 5.75, 5.67 (s, 1 H each, 3 CHpz), 2.89 (brs, 3 H, N–CH3), 2.50, 2.41, 2.38, 2.08, 1.61, 1.58 (s, 3 H each, 6 Mepz), −18.38 (brs, 1 H, IrH). 13C{1H} NMR (CDCl3, 25°C): δ 222.0 (IrCH, 1JCH=146 Hz), 152.0, 149.6, 149.3, 143.5, 143.1, 142.9 (Cqpz), 150.4 (Cqar(N–Ph)), 141.4, 140.3, 127.1, 119.7 (1∶1∶2∶1, o, o, m, p-CHar(Ir–Ph)), 138.4 (Cqar(Ir–Ph)), 129.5, 125.7, 121.4 (2∶1∶2, m, p, o-CHar(N–Ph)), 106.0, 105.6, 104.9 (CHpz), 42.5 (N–CH3), 15.3, 14.8, 14.0, 12.7 (1∶1∶1∶3, Mepz). Anal. Calcd. for C29H37BN7Ir: C, 50.7; H, 5.4; N, 14.3; Found: C, 50.6; H, 5.5; N, 14.1%.
Data for 5b. IR (Nujol): ν(Ir–H) 2135 cm−1. 1H NMR (CDCl3, 25
°C): δ 12.12 (s, 1 H, IrCH), 7.40, 7.04, 6.96, 6.89 (d, d, t, t, 1 H each, 4 CHar), 5.86, 5.85, 5.50 (s, 1 H each, 3 CHpz), 3.66 (s, 3 H, N–CH3), 2.40, 2.40, 2.39, 2.35, 2.27, 0.91 (s, 3 H each, 6 Mepz), −20.14 (s, 1 H, IrH). 13C{1H} NMR (CDCl3, 25°C): δ 216.4 (IrCH, 1JCH=149 Hz), 154.9, 138.4 (Cqar), 151.4, 150.8, 150.4, 143.6, 143.4. 142.8 (Cqpz), 137.7, 124.4, 120.1, 111.8 (CHar), 106.4, 105.9, 105.8 (CHpz), 43.1 (N–CH3), 15.9, 15.5, 12.7, 12.6, 10.8 (1∶1∶2∶1∶1, Mepz). Anal. Calcd. for C23H31BN7Ir: C, 45.4; H, 5.1; N, 16.1; Found: C, 45.6; H, 5.2; N, 15.8%.
General procedure for catalytic H/D exchange reactions
In a typical deuteration experiment, a solution of 6 (0.01 g, 0.017 mmol) and the substrate (50 μl) in C6D6 (0.5 ml) was prepared under N2 and transferred into an NMR tube, that was subsequently sealed. The tube was heated in an oil bath at 90°C (bath temperature) and the reactions were monitored by 1H NMR spectroscopy. At the end of the deuteration procedures, 13C{1H} NMR spectra, and, where appropriate, 2H NMR spectra were recorded. Below are given details for a specific example (ferrocene) as well as selected 1H NMR data.
A mixture of ferrocene (0.23 mmol) and compound 6 (7.5 mol%) in C6D6 (0.5 ml) was heated at the temperature given above. At specified periods of time the progress of the reaction was checked by 1H NMR spectroscopy. Deuterium incorporation reached its maximum after 3.5 h, although degration was not observed after this time. The extent of deuteration was measured by integration of the 1H NMR spectrum, comparing the intensity of the Fe(C5H5)2 signal and that of the deuterobenzene before and after the heating period. In this particular case the ferrocene was purified by column chromatography on alumina (neutral alumina, n-hexane eluent) and characterized by 1H and 13C{1H}, 1D- and 2D-NMR spectroscopy.
°C): δ 3.89 (m, 0.2 H), 3.86 (m, 0.8 H), 1.82 (m, 1.6 H). 13C{1H} NMR (C6D6, 25°C): δ 83.2 (m), 69.6 (s), 67.9–67.2 (m), 14.2–13.8 (m).
°C): δ 1.67–1.58 (m, 4 H), 1.23–1.14 (m, 3 H), 0.87–0.82 (m, 3 H). 13C{1H} NMR (C6D6, 25°C): δ 35.3 (s), 32.5 (m), 26.4–25.6 (m), 22.7–22.1 (m).
°C): δ 6.84 (m, 0.6 H), 3.57 (q, 2 H), 1.10 (m, 2.4 H). 13C{1H} NMR (C6D6, 25°C): δ 159.2 (s), 129.3–128.5 (m), 120.1–119.5 (m), 114.3–113.7 (m), 62.7 (m), 14.5–13.8 (m).
°C): δ 3.28 (t, 4 H), 1.53 (m, 2 H), 1.36 (m, 2 H), 0.86 (m, 4.5 H). 13C{1H} NMR (C6D6, 25°C): δ 69.8 (s), 33.1 (s), 19.5 (s), 14.1–13.7 (m).
°C): δ 6.72–6.65 (m, 4.2 H), 4.15 (m, 8 H), 3.85 (m, 8 H). 13C{1H} NMR (CDCl3, 25°C): δ 149.4 (s), 121.6 (m), 114.2 (s), 70.6 (s), 69.5 (s).
Crystallography
=609.54, yellow prism (0.40×0.18×0.12 mm) from hexane∶CH2Cl2, monoclinic, space group C2/c
(no. 15), a=18.963(2)
Å, b=10.2605(12)
Å, c=24.556(3)
Å, β=100.160(2)°, V=4712.1(10)
Å3, Z=8, Dx=1.718 Mg m−3, λ(Mo Kα)=0.71073 Å, μ=5.69 mm−1, T=150(2) K. X-Ray diffraction data were collected on a Bruker SMART diffractometer equipped with a CCD detector, graphite monochromated Mo Kα radiation, and a Kryoflex low-temperature device.19 Four sets of frames covering a complete sphere of the reciprocal space were recorded (4×606 frames, ω-scans, Δω=0.3°, time per frame 10 s). Data reduction up to θ=30° by program SAINT, corrections for absorption with program SADABS, 32347 reflections measured, 6676 independent, Rint=0.023.19 Structure solution with direct methods and program SHELXS97, structure refinement on F2 using program SHELXL97.20 All non-hydrogen atoms were refined anisotropically. C- and B-bound hydrogen atoms had isotropic temperature factors and rode on the atoms to which they were bonded (orientation refinement for CH3 groups). The Ir-bound hydrogen atom H(1), located on a difference Fourier map, was refined without restraints. Final refinement with 6676 data, no restraints, and 302 parameters gave R1=0.0242, wR2=0.0400 (all data), and R1=0.0188, wR2=0.0386 [I>2σ(I)]. CCDC reference number 195911. See http://www.rsc.org/suppdata/nj/b2/b209324c/ for crystallographic data in CIF or other electronic format.
References
- (a) G. I. Nikonov, P. Mountford, S. K. Ignatov, J. C. Green, M. A. Leech, L. G. Kuzmina, A. G. Razuvaev, N. H. Rees, A. J. Blake, J. A. K. Howard and D. A. Lemenovskii, J. Chem. Soc., Dalton Trans., 2001, 2903 RSC; (b) Y. Guari, S. Sabo-Etienne and B. Chaudret, Eur. J. Inorg. Chem., 1999, 1047 CrossRef CAS; (c) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1699 CrossRef CAS; (d) H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953 CrossRef CAS; (e) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (f) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS.
- E. Gutiérrez-Puebla, A. Monge, M. C. Nicasio, P. J. Pérez, M. L. Poveda and E. Carmona, Chem. Eur. J., 1998, 4, 2225 CrossRef CAS.
- (a) C. Slugovc, K. Mereiter, S. Trofimenko and E. Carmona, Angew. Chem., Int. Ed., 2000, 39, 2158 CrossRef CAS; (b) C. Slugovc, K. Mereiter, S. Trofimenko and E. Carmona, Helv. Chim. Acta, 2001, 84, 2868 CrossRef CAS.
- (a) D.-H. Lee, J. Chen, J. W. Faller and R. H. Crabtree, Chem. Commun., 2001, 213 RSC; (b) J. N. Coalter III, J. C. Huffmann and K. C. Caulton, Chem. Commun., 2001, 1158 RSC; (c) G. Ferrando-Miguel, J. N. Coalter III, H. Gérard, J. C. Huffman, O. Eisenstein and K. C. Caulton, New J. Chem., 2002, 26, 687 RSC.
- (a) H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS; (b) H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995 CrossRef CAS; (c) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (d) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168 CrossRef CAS.
- C. P. Lenges, P. S. White and M. Brookhart, J. Am. Chem. Soc., 1999, 121, 4385 CrossRef CAS.
- J. T. Golden, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 5837 CrossRef CAS.
- S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092 CrossRef CAS.
- L. Lefort, C. Copéret, M. Taoufik, J. Thivolle-Cazat and J.-M. Basset, Chem. Commun., 2000, 663 RSC.
- (a) J. L. Garnett and R. J. Hodges, J. Am. Chem. Soc., 1967, 89, 4546 CrossRef CAS; (b) J. L. Garnett and R. J. Hodges, Chem. Commun., 1967, 1001 Search PubMed; (c) J. L. Garnett and J. C. West, Aust. J. Chem., 1974, 27, 129 CAS.
- A. E. Shilov and A. A. Shteinman, Coord. Chem. Rev., 1977, 24, 97 CrossRef CAS.
- Y. Alvarado, O. Boutry, E. Gutiérrez, A. Monge, M. C. Nicasio, M. L. Poveda, P. J. Pérez, C. Ruíz, C. Bianchini and E. Carmona, Chem. Eur. J., 1997, 3, 860 CrossRef CAS.
- O. Boutry, M. L. Poveda and E. Carmona, J. Organomet. Chem., 1997, 528, 143 CrossRef CAS.
- (a) G. Parkin, E. Bunel, B. J. Burger, M. S. Trimmer, A. van Asselt and J. E. Bercaw, J. Mol. Catal., 1987, 41, 21 CrossRef CAS; (b) E. A. Carter and W. A. Goddard III, Organometallics, 1988, 7, 675 CrossRef CAS.
- (a) J. E. Kickham, F. Guérin, J. C. Stewart and D. W. Stephan, Angew. Chem., Int. Ed., 2000, 39, 3263 CrossRef CAS; (b) P. Barrio, R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2001, 20, 2635 CrossRef CAS.
- (a) M. D. Curtis, K. B. Shiu and W. M. Butler, J. Am. Chem. Soc., 1986, 108, 1550 CrossRef CAS; (b) M. D. Curtis, K. B. Shiu, W. M. Butler, M. F. Huff, A. L. Rheingold and B. S. Haggerty, J. Am. Chem. Soc., 1992, 114, 579 CrossRef CAS.
- M. Paneque, S. Sirol, M. Trujillo, E. Carmona, E. Gutierrez-Puebla, M. A. Monge, C. Ruiz, F. Malbosc, C. Serra-Le Berre, P. Kalck, M. Etienne and J. C. Daran, Chem. Eur. J., 2001, 7, 3868 CrossRef CAS.
- M. Paneque, M. L. Poveda, V. Salazar, E. Gutiérrez-Puebla and A. Monge, Organometallics, 2000, 19, 3120 CrossRef CAS.
- Bruker, Programs SMART, version 5.054; SAINT, version 6.2.9; SADABS version 2.03; XPREP, version 5.1; SHELXTL, version 5.1./Bruker AXS Inc., Madison, WI, USA, 2001.
- G. M. Sheldrick, Programs SHELXS97 (crystal structure solution) and SHELXL97 (crystal structure refinement), University of Göttingen, Germany, 1997.
Footnote |
| † Dedicated to Professor Domingo González-Alvarez on the occasion of his retirement. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |