Examination of cobalt, nickel, copper and zinc(II) complex geometry and binding affinity in aqueous media using simple pyridylsulfonamide ligands†
Aileen
Congreve
,
Ritu
Kataky
,
Mark
Knell
,
David
Parker
*,
Horst
Puschmann
,
Kanthi
Senanayake
and
Lisa
Wylie
Department of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE. E-mail: david.parker@dur.ac.uk
First published on 15th November 2002
Abstract
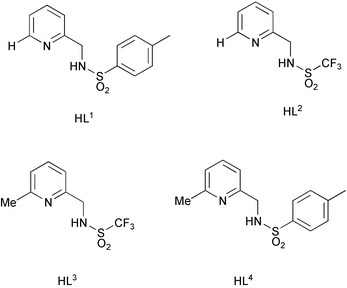
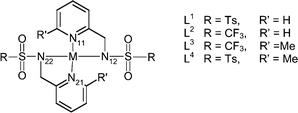
The sixteen neutral ML2 complexes of Co, Ni, Cu and Zn(II) with the p-toluenesulfonamide and trifluoromethylsulfonamide derivatives of 2-aminomethylpyridine (L1, L2) and its 6-Me homologue (L3, L4) have been characterised by low temperature X-ray crystallography (100–120 K). Complexes of Co and Zn invariantly adopted a distorted tetrahedral geometry and whilst Cu(II) complexes of L2, L3 and L4 also took up a distorted tetrahedral geometry, that with L1 was square planar. A database survey of the distortion from limiting tetrahedral/square planar geometry has been carried out, aided by a simple geometric analysis. The trifluoromethylsulfonamide ligands (L2 and L3) were less basic, e.g. log K1 7.51(3) for L2vs. 12.23(6) for L1 (80% MeOH/H2O) and afforded a weaker ligand field, exemplified by the position of the visible d–d transition in Cu(II) complexes and the ease of reduction of the Cu(II) centre: E1 /2 values (MeCN vs. Ag/AgCl) are −430, −137, +55 and −240 mV for Cu(L1)2, Cu(L2)2, Cu(L3)2 and Cu(L4)2. Ligand protonation and stepwise formation constants have been measured for L1–L3 and derived species distribution diagrams reveal that for complexes with L2 and L3, the predominant species present at pH 7.4 when zinc was in the nanomolar range was ZnL2.
The binding of the Zn ion in aqueous media has attracted attention recently in three different areas of chemistry. Firstly, stimulated by the need to probe the biological rôle of kinetically labile zinc1 in synaptic neurotransmission2 and excitotoxicity,3,4 a range of luminescent zinc probes has been devised in which a chromophore or fluorophore is integrated into the ligand structure.5–11 Such ligands need to bind the zinc ion with high affinity in the physiological pH range and examples have been reported with micromolar,11 nanomolar4,5,8 or sub-picomolar12 apparent dissociation constants. Secondly, more lipophilic zinc binding ligands have been sought that allow the selective extraction of the zinc ion from acidic aqueous media into a non-polar organic phase. Examples have included aza-carboxylates and phosphinates13,14 in which a tetrahedral binding geometry is imposed on the zinc by ligand design. Finally, as a consequence of the pivotal role of zinc in gene transcription and metalloenzyme function,1,15 a great deal of work has been directed at devising zinc-binding ligands that inhibit enzyme function and hence may allow development of therapeutic agents. In just one example, inhibition of carbonic anhydrase activity has been observed with a series of sulfonamide ligands which bind to zinc via nitrogen.16 Thus, various thiadiazole sulfonamides17,18 are active inhibitors, as is the simple example trifluoromethylsulfonamide.19 Such work has led to the parallel development of sulfonamide probes for the fluorescence anisotropy detection of zinc with a carbonic-anhydrase-based biosensor.20
We set out to compare the co-ordination chemistry of the series of simple pyridyl-sulfonamide ligands L1–L4. The introduction of the α-methyl substituent was expected to sterically inhibit formation of a square planar ML2 complex. The differing sulfonamide substituents (CF3vs. tosyl) alter the electron donating ability of the sulfonamide nitrogen. Furthermore, the lower protonation constants of trifluoromethylsulfonamides (ca. 7.5) vs. arylsulfonamides (ca. 12) was expected to inhibit protonation of the ML and ML2 complexes and enhance complex stability at ambient pH. Our aim was to identify a model ligand system from this series capable of forming a well-defined neutral complex with zinc under ambient pH conditions, when the Zn concentration is of the order of 100–0.1 nM. Such a system could then form the basis of new luminescent or MR probes.
Experimental
Solvents were dried from an appropriate drying agent where required using standard procedures. Water was purified by the “PuriteSTILL plus” system. Thin layer chromatography was carried out using fluorescent (254 nm) silica plates (Merck Art 5554). Preparative column chromatography was carried out using silica (Merck silica gel 60, 230–400 mesh). Mass spectra (ES MS) were recorded using a VG II Platform spectrometer (Fisons Instruments) with methanol as the carrier solvent. FAB spectra were recorded by the EPSRC Mass Spectrometry Service at the University of Wales at Swansea. NMR spectra were recorded on a Varian Unity 300 spectrometer at 299.91 MHz (1H) and 75.41 MHz (13C) or a Varian Mercury 200 spectrometer at 199.99 MHz (1H), 50.29 MHz (13C) or 188.18 MHz (19F) or a Bruker AC250 spectrometer at 62.90 MHz (13C). Chemical shifts are quoted with reference to the residue residual protonated solvent and are given in ppm with coupling constants in Hz. Infra-red spectra were recorded on a Perkin-Elmer FT-IR 1720X spectrometer with GRAMS Analyst operating software. Ultraviolet absorbance spectra of the complexes (1 mM) were recorded in acetonitrile on a Unicam UV2 spectrometer operating with Vision software. Melting points were determined on a Reichert-Koefler block melting point apparatus and are uncorrected. Cyclic voltammetry of the complexes (0.001 M) was carried out in a background electrolyte of tetrabutylammonium perchlorate, TBAP (0.01 M), in acetonitrile controlled with an EG&G PARC Model 273 potentiostat. Computer control and data storage were achieved using EG&G PARC Model 270 Research Electrochemistry software. The auxiliary electrode was made of platinum foil, (area 1 cm2), connected to a copper wire mounted in a glass body. The working electrode was made of glassy carbon, purchased from BAS. The reference electrode was a non-aqueous silver/silver chloride electrode, self-assembly kit, purchased from BAS. The electrodes were mounted in a circular Teflon cap and placed in a cylindrical cell (diameter 20 mm, length 65 mm), which was filled with 10 ml of the desired solution and purged with argon. The cell assembly was placed inside a Faraday cage to eliminate stray field interference.Potentiometric analyses were carried out using an apparatus described previously.21,22 The stepwise protonation and metal formation constants were evaluated by analysis of data acquired using a computer-controlled alkalimetric titration at 298 K(water-jacketed titration cell), using a calibrated Corning pH glass electrode, under an argon atmosphere. To solutions of the ligand (typically 2 mM) in tetramethylammonium nitrate solution (0.1 M, 80% aqueous methanol) was added a fixed volume of 1.0 M HCl solution (Analar) to give the hydrochloride salt. The titrant was degassed sodium hydroxide solution (0.05 M, 80% MeOH/water) and the burette function (volume increments, total volume delivered and the time interval allowed for equilibration between each reading) was computer controlled allowing smaller increments of titrant to be added towards the end-point. Titration data for protonation equilibria were collected between pH 3 and 11, typically acquiring 80 data points, used in the subsequent analysis. In measuring the metal complex formation constants, separate titrations were carried out at 1∶1 and 1∶2 metal/ligand ratios, and the pH range examined was from 3 to ca. 7.5, at which point formation of the metal hydroxide was visually discerned. Data were analysed using HYPERQUAD23 and corrections to pKw were applied to allow for solvent composition.24 Each of the metal salt hydrolysis constants used in the data analysis was taken from the IUPAC Stability Constants database Version 5.12, published by IUPAC and Academic Software, 2000. Values of protonation and metal complex formation constants, determined by iterative fitting in Hyperquad, refer to the mean of three independent titrations and gave satisfactory statistical parameters (σca. 2.5 to 3.5 and χ2 of the order of 8 to 12).
Single crystal X-ray diffraction experiments were carried out using SMART CCD area detectors and graphite-monochromated Mo-Kα radiation. The structures were solved by direct methods and refined against F2 of all data, using SHELXTL programs.25 A summary of cell and refinement data is given in Table 1.
CCDC reference numbers 186296–186312. See http://www.rsc.org/suppdata/nj/b2/b206279h/ for crystallographic data in CIF or other electronic format.
| Complex | a/Å | b/Å | c/Å | α/° | β/° | γ/° | Volume/Å3 | System | Space group | Z | μ/mm−1 | T/K | R int (%) | Rw (%) | R (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Remaining solvent electron density was refined to 0.13 molecules of water. b These crystals were badly twinned, despite appearing perfectly well formed. Satisfactory anisotropic refinement was not possible. | |||||||||||||||
| L1 | 26.831(4) | 5.958(1) | 16.576(2) | 90 | 99.967(3) | 90 | 2610.1(6) | Monoclinic | C2/c | 8 | 0.249 | 110 | 3.05 | 9.20 | 3.84 |
| Zn(L1)2 | 30.505(2) | 8.010(1) | 27.309(1) | 90 | 123.815(3) | 90 | 5543.8(9) | Monoclinic | C2/c | 8 | 1.081 | 100 | 8.30 | 10.94 | 4.83 |
| Zn(L2)2 | 8.296(1) | 9.721(1) | 13.035(1) | 93.193(4) | 95.817(4) | 107.145(4) | 995.27(17) | Triclinic |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
2 | 1.529 | 100 | 2.06 | 6.64 | 2.72 |
| Zn(L3)2 | 7.973(1) | 9.432(1) | 14.746(1) | 81.984(1) | 81.130(1) | 81.744(10) | 1076.38(5) | Triclinic |
P |
2 | 1.419 | 105 | 2.56 | 8.35 | 2.99 |
| Zn(L4)2 | 28.129(1) | 12.033(1) | 16.726(1) | 90 | 91.701(1) | 90 | 5658.54(17) | Monoclinic | I2/a | 8 | 1.057 | 120 | 2.48 | 6.65 | 2.69 |
| Cu(L1)2 | 7.293(1) | 17.089(1) | 9.912(7) | 90 | 99.176(4) | 90 | 1219.56(15) | Monoclinic | P21/c | 2 | 1.11 | 100 | 4.14 | 8.99 | 3.31 |
| Cu(L2)2b | 9.04(5) | 10.29(5) | 20.70(5) | 90 | 91.71(5) | 90 | 1923(10) | Monoclinic | Pc | 4 | 1.442 | 100 | 5.60 | 36.05 | 14.10 |
| Cu(L3)2 | 10.450(1) | 14.023(1) | 15.197(1) | 90 | 106.221(1) | 90 | 2138.19(10) | Monoclinic | P21/c | 4 | 1.301 | 105 | 5.26 | 8.17 | 3.45 |
| Cu(L4)2 | 26.673(1) | 14.539(1) | 14.980(1) | 90 | 102.051(1) | 90 | 5680.9(3) | Monoclinic | C2/c | 8 | 0.96 | 110 | 15.03 | 18.56 | 7.32 |
| Ni(L1)2 | 7.335(1) | 17.058(1) | 9.822(1) | 90 | 99.139(1) | 90 | 1213.25(8) | Monoclinic | P21/c | 2 | 1.015 | 120 | 5.46 | 20.24 | 6.99 |
| Ni(L2)2 | 10.458(1) | 16.052(2) | 29.546(4) | 90 | 90 | 90 | 4960.1(11) | Orthorhombic | Pbca | 8 | 1.038 | 100 | 4.21 | 15.38 | 5.89 |
| Ni(L3)2 | 7.936(1) | 9.450(1) | 14.716(1) | 80.519(0) | 80.609(1) | 81.548(1) | 1065.77(6) | Triclinic |
P |
2 | 1.191 | 100 | 2.42 | 9.33 | 3.36 |
| Ni(L4)2 | 32.016(1) | 11.932(4) | 16.782(8) | 90 | 118.86(2) | 90 | 5615(4) | Monoclinic | C2/c | 8 | 0.881 | 120 | 4.23 | 11.8 | 4.35 |
| Co(L1)2 | 29.967(6) | 7.977(1) | 26.183(5) | 90 | 121.349(5) | 90 | 5345.2(17) | Monoclinic | C2/c | 8 | 0.838 | 100 | 5.23 | 9.00 | 3.73 |
| Co(L2)2 | 8.270(1) | 9.801(1) | 13.050(1) | 93.015(1) | 95.599(1) | 108.418(1) | 994.84(6) | Triclinic |
P |
2 | 1.158 | 110 | 3.19 | 9.47 | 3.69 |
| Co(L3)2 | 8.000(1) | 9.372(1) | 14.736(1) | 82.625(1) | 81.527(1) | 81.726(1) | 1074.89(6) | Triclinic |
P |
2 | 1.076 | 100 | 2.90 | 9.25 | 3.59 |
| Co(L4)2 | 32.449(1) | 12.065(1) | 16.738(1) | 90 | 119.72(1) | 90 | 5694.0(3) | Monoclinic | C2/c | 8 | 0.790 | 110 | 5.26 | 11.7 | 3.82 |
Ligand synthesis
6-Methylpyridine-2-aldoxime was prepared as described in the literature.26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C was slowly added (2-aminomethyl)pyridine (2.00 g, 0.018 mmol) in pyridine (5 ml). The resulting reaction mixture was stirred at −10°C for 3 hours and was held at 5°C overnight. The reaction mixture was poured onto crushed ice resulting in the precipitation of a yellow solid, which was removed by filtration and washed with water. The precipitate was dissolved in dichloromethane, dried (NaSO4) and the solvent evaporated yielding pale yellow crystals (2.12 g, 45%), mp 76–77°C; 1H NMR (200 MHz, CD3OD): δH 2.40 (s, 3H, CH3), 4.15 (s, 2H, CH2), 7.22–7.45 (4H, m, H3, H5, H3′, H5′), 7.69 (3H, m, H4, H4′, H6′), 8.37 (1H, d, J 4.4, H6); 13C NMR (50.29 MHz, CDCl3): δc 21.15 (Me), 47.47 (CH2), 122.00 (C3 or C5), 122.32 (C3 or C5), 126.75 (C3′+C5′), 129.29 (C2′+C6′), 136.72 (C4), 142.92 (C4′), 148.59 (C2), 155.34 (C1′); m/z ES+: 546.7 (100%, 2M+Na), 284.5 (85%, M+Na); νmax
(KBr)/cm−1 3250 (ν NH), 1599 (ν py), 1574 (ν py), 1441 (ν py), 1385 (δ NH), 1329 (νa SO2), 1165 (νs SO2), 1111 (δ CH), 1089 (δ CH), 1007, 901 (ν N–S), 763 (δ py), 662 (γ NH), 543 (δ SO2); Found: C, 57.12; H, 5.58; N, 10.25. C13H14N2O2S.0.5H2O requires C, 57.54; H, 5.57; N, 10.32%.
°C was added dropwise, over 10 minutes, to a stirred solution of trifluoromethanesulfonyl chloride (1.0 g, 5.93 mmol) in pyridine (10 ml) at −40°C. The resulting bright yellow reaction mixture was stirred at −40°C for 2 hours and kept at 5°C overnight. The mixture was poured slowly onto crushed ice and stirred. The precipitate that formed was separated by filtration and washed with water. This precipitate was dissolved in dichloromethane (50 ml), washed with water (2×25 ml) and the organic phase was dried (MgSO4), filtered and evaporated under reduced pressure to give a solid which was recrystallised from ethyl acetate and hexane to yield pale brown crystals (0.72 g, 50%), mp 80–84°C; 1H NMR (300 MHz, CDCl3): δH 4.49 (2H, s, CH2), 7.24 (2H, m, H3+H5), 7.69 (1H, t of d, J 1.8, 7.8, H4), 8.46 (1H, d, J 5.1, H6); 13C NMR (50.3 MHz, CDCl3): δC 48.05 (CH2), 123.10 (C2), 123.87 (C4), 138.20 (C5), 149.30 (C6), 154.47 (q, CF3); 19F NMR (188 MHz, CD3CN): δF
−79.44 (s, CF3); m/z
(ES+): 263 (100%, M+Na+), 241 (20%, M+H+); νmax
(KBr)/cm−1 1601 (ν py), 1434 (ν py), 1379 (δ NH), 1367 (νa SO2), 1176 (νs SO2), 1143 (δ CH), 1087 (δ CH), 599 (γ NH); Found: C, 35.30; H. 3.01; N. 11.52. C7H7N2O2SF3 requires C. 35.00; H. 2.94; N. 11.66%.
°C was slowly added a solution of 2-aminomethyl-6-methylpyridine (123 mg, 1.0 mmol) in pyridine (1 ml). The resulting yellow reaction mixture was stirred at −10°C for 3 hours and was held at 5°C overnight. The reaction mixture was poured onto crushed ice, which was allowed to melt resulting in the formation of an oil. A precipitate formed upon scratching. The solid was removed by filtration, dissolved in dichloromethane (5 ml) and washed with water (2×5 ml). The organic phase was dried (MgSO4), filtered and evaporated under reduced pressure to yield a yellow oil which was crystallised from ethanol and water (1%) to form a white solid (0.53 g, 53%); mp 83–84°C; 1H NMR (200 MHz, CDCl3): δH 2.38 (3H, s, CH3), 2.46 (3H, s, CH3), 4.18 (2H, d, J 5.2, CH2), 5.92 (1H, s, NH), 6.97 (2H, t, J 8.4, H3, H5), 7.22 (2H, d, J 7.8, H3′, H5′), 7.47 (1H, t, J 7.8, H4), 7.72 (2H, d, J 6.6, H2′, H5′); 13C NMR (50.3 MHz, CDCl3): δc 21.37 (Me), 24.02 (Me), 47.35 (CH2), 118.74 (C4′), 121.94 (C1′), 127.08 (C3′+C5′), 129.45 (C2′+C6′), 136.63 (q, C6), 136.96 (C2), 143.17 (C3), 154.09 (C4), 157.68 (C2); m/z ES+: 574.7 (25%, 2M+Na+), 298.7 (100%, M+Na+); νmax
(KBr)/cm−1 1599 (ν py), 1458 (ν py), 1325 (νa SO2), 1160 (νs SO2), 1090 (δ CH), 816, 662 (γ NH), 551 (δ SO2). Found: C, 60.57; H, 5.77; N, 10.39. C13H14N2O2S requires C, 60.85; H, 5.84; N, 10.14%.
°C. The resulting yellow solution was stirred at ∼−40°C for two hours then kept at −18°C overnight before being added slowly to crushed ice. The ice was stirred and allowed to melt resulting in the formation of a green precipitate, which was filtered off under suction. This precipitate was dissolved in dichloromethane (10 ml), washed with water (2×10 ml), the combined organic phase was dried (MgSO4), filtered and the solution evaporated under reduced pressure to yield a yellow oil. The aqueous phase was extracted with dichloromethane (3×50 ml), the combined organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure to yield a yellow oil which was purified by flash column chromatography (SiO2, 1∶1 ethyl acetate∶hexane). Clear crystals formed on the evaporation of the reduced eluting solvent (0.25 g, 30%), mp 73–74°C. 1H NMR (200 MHz, CDCl3): δH 2.47 (3H, s, CH3), 4.53 (2H, s, CH2), 7.08 (2H, t, J 7, H3, H5), 7.61 (1H, t, J 7.8, H4); 13C NMR (50.3 MHz, CDCl3): δC 12.60 (CH3), 47.80 (CH2), 116.72 (q, C6), 119.49 (C5), 123.17 (C3), 137.64 (C4), 152.77 (C2), 158.34 (CF3); 19F NMR (188 MHz, CDCl3): δF
−77.70 (s, CF3); m/z
(ES+): 255 (100%, MH+); νmax
(KBr)/cm−1 1606 (ν py), 1369 (νa SO2), 1191 (νs SO2), 1145 (δ CH), 1069 (δ CH), 611 (γ NH); Found: C, 37.73; H. 3.55; N. 10.91. C8H9N2O2SF3 requires C. 37.80; H. 3.57; N. 11.02%.
°C was slowly added (2-aminomethyl)pyridine (2.00 g, 0.018 mmol) in pyridine (5 ml). The resulting reaction mixture was stirred at −10°C for 3 hours and was held at 5°C overnight. The reaction mixture was poured onto crushed ice resulting in the precipitation of a yellow solid, which was removed by filtration and washed with water. The precipitate was dissolved in dichloromethane, dried (NaSO4) and the solvent evaporated yielding pale yellow crystals (2.12 g, 45%), mp 76–77°C; 1H NMR (200 MHz, CD3OD): δH 2.40 (s, 3H, CH3), 4.15 (s, 2H, CH2), 7.22–7.45 (4H, m, H3, H5, H3′, H5′), 7.69 (3H, m, H4, H4′, H6′), 8.37 (1H, d, J 4.4, H6); 13C NMR (50.29 MHz, CDCl3): δc 21.15 (Me), 47.47 (CH2), 122.00 (C3 or C5), 122.32 (C3 or C5), 126.75 (C3′+C5′), 129.29 (C2′+C6′), 136.72 (C4), 142.92 (C4′), 148.59 (C2), 155.34 (C1′); m/z ES+: 546.7 (100%, 2M+Na), 284.5 (85%, M+Na); νmax
(KBr)/cm−1 3250 (ν NH), 1599 (ν py), 1574 (ν py), 1441 (ν py), 1385 (δ NH), 1329 (νa SO2), 1165 (νs SO2), 1111 (δ CH), 1089 (δ CH), 1007, 901 (ν N–S), 763 (δ py), 662 (γ NH), 543 (δ SO2); Found: C, 57.12; H, 5.58; N, 10.25. C13H14N2O2S.0.5H2O requires C, 57.54; H, 5.57; N, 10.32%.
°C was added dropwise, over 10 minutes, to a stirred solution of trifluoromethanesulfonyl chloride (1.0 g, 5.93 mmol) in pyridine (10 ml) at −40°C. The resulting bright yellow reaction mixture was stirred at −40°C for 2 hours and kept at 5°C overnight. The mixture was poured slowly onto crushed ice and stirred. The precipitate that formed was separated by filtration and washed with water. This precipitate was dissolved in dichloromethane (50 ml), washed with water (2×25 ml) and the organic phase was dried (MgSO4), filtered and evaporated under reduced pressure to give a solid which was recrystallised from ethyl acetate and hexane to yield pale brown crystals (0.72 g, 50%), mp 80–84°C; 1H NMR (300 MHz, CDCl3): δH 4.49 (2H, s, CH2), 7.24 (2H, m, H3+H5), 7.69 (1H, t of d, J 1.8, 7.8, H4), 8.46 (1H, d, J 5.1, H6); 13C NMR (50.3 MHz, CDCl3): δC 48.05 (CH2), 123.10 (C2), 123.87 (C4), 138.20 (C5), 149.30 (C6), 154.47 (q, CF3); 19F NMR (188 MHz, CD3CN): δF
−79.44 (s, CF3); m/z
(ES+): 263 (100%, M+Na+), 241 (20%, M+H+); νmax
(KBr)/cm−1 1601 (ν py), 1434 (ν py), 1379 (δ NH), 1367 (νa SO2), 1176 (νs SO2), 1143 (δ CH), 1087 (δ CH), 599 (γ NH); Found: C, 35.30; H. 3.01; N. 11.52. C7H7N2O2SF3 requires C. 35.00; H. 2.94; N. 11.66%.
°C was slowly added a solution of 2-aminomethyl-6-methylpyridine (123 mg, 1.0 mmol) in pyridine (1 ml). The resulting yellow reaction mixture was stirred at −10°C for 3 hours and was held at 5°C overnight. The reaction mixture was poured onto crushed ice, which was allowed to melt resulting in the formation of an oil. A precipitate formed upon scratching. The solid was removed by filtration, dissolved in dichloromethane (5 ml) and washed with water (2×5 ml). The organic phase was dried (MgSO4), filtered and evaporated under reduced pressure to yield a yellow oil which was crystallised from ethanol and water (1%) to form a white solid (0.53 g, 53%); mp 83–84°C; 1H NMR (200 MHz, CDCl3): δH 2.38 (3H, s, CH3), 2.46 (3H, s, CH3), 4.18 (2H, d, J 5.2, CH2), 5.92 (1H, s, NH), 6.97 (2H, t, J 8.4, H3, H5), 7.22 (2H, d, J 7.8, H3′, H5′), 7.47 (1H, t, J 7.8, H4), 7.72 (2H, d, J 6.6, H2′, H5′); 13C NMR (50.3 MHz, CDCl3): δc 21.37 (Me), 24.02 (Me), 47.35 (CH2), 118.74 (C4′), 121.94 (C1′), 127.08 (C3′+C5′), 129.45 (C2′+C6′), 136.63 (q, C6), 136.96 (C2), 143.17 (C3), 154.09 (C4), 157.68 (C2); m/z ES+: 574.7 (25%, 2M+Na+), 298.7 (100%, M+Na+); νmax
(KBr)/cm−1 1599 (ν py), 1458 (ν py), 1325 (νa SO2), 1160 (νs SO2), 1090 (δ CH), 816, 662 (γ NH), 551 (δ SO2). Found: C, 60.57; H, 5.77; N, 10.39. C13H14N2O2S requires C, 60.85; H, 5.84; N, 10.14%.
°C. The resulting yellow solution was stirred at ∼−40°C for two hours then kept at −18°C overnight before being added slowly to crushed ice. The ice was stirred and allowed to melt resulting in the formation of a green precipitate, which was filtered off under suction. This precipitate was dissolved in dichloromethane (10 ml), washed with water (2×10 ml), the combined organic phase was dried (MgSO4), filtered and the solution evaporated under reduced pressure to yield a yellow oil. The aqueous phase was extracted with dichloromethane (3×50 ml), the combined organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure to yield a yellow oil which was purified by flash column chromatography (SiO2, 1∶1 ethyl acetate∶hexane). Clear crystals formed on the evaporation of the reduced eluting solvent (0.25 g, 30%), mp 73–74°C. 1H NMR (200 MHz, CDCl3): δH 2.47 (3H, s, CH3), 4.53 (2H, s, CH2), 7.08 (2H, t, J 7, H3, H5), 7.61 (1H, t, J 7.8, H4); 13C NMR (50.3 MHz, CDCl3): δC 12.60 (CH3), 47.80 (CH2), 116.72 (q, C6), 119.49 (C5), 123.17 (C3), 137.64 (C4), 152.77 (C2), 158.34 (CF3); 19F NMR (188 MHz, CDCl3): δF
−77.70 (s, CF3); m/z
(ES+): 255 (100%, MH+); νmax
(KBr)/cm−1 1606 (ν py), 1369 (νa SO2), 1191 (νs SO2), 1145 (δ CH), 1069 (δ CH), 611 (γ NH); Found: C, 37.73; H. 3.55; N. 10.91. C8H9N2O2SF3 requires C. 37.80; H. 3.57; N. 11.02%.
Synthesis of metal complexes
The following are representative methods. Full details of remaining complexes are given in the ESI.†°C (decomp.); 1H NMR (200 MHz, CD3OD): δH 8.4 (2H, d, J 5, H6), 8.0 (2H, t, J 5, H4), 7.8 (4H, d, J 8, tos), 7.5 (4H, m, H5, H3), 7.2 (4H, d, J 8, tos), 4.4 (4H, s, CH2), 2.3 (6H, s, Me); m/z
(FAB): 587 (100%, ZnL2), 431 (39%, ZnL2−tos), 325 (22%, ZnL), 263 (25%, LH); νmax
(KBr)/cm−1 1611 (ν py), 1568 (ν py), 1442 (ν py), 1277 (νa SO2), 1151 (νs SO2), 1104 (δ CH), 1087 (δ CH), 970, 762 (δ py), 670 (γ NH), 560 (δ SO2); Found: C, 52.53; H, 4.44; N, 9.40. Zn(C13H13N2O2S)2·0.5MeOH requires C, 52.69; H, 4.67; N, 9.27%.
°C; m/z
(FAB): 911 (11%, CuL3), 650 (16%, Cu2L2), 608 (32%, CuL2+Na), 586 (100%, CuL2) 325 (71%, CuL), 263 (22%, L+H); νmax
(KBr)/cm−1 1609 (ν py), 1570 (ν py), 1447 (ν py), 1276 (νa SO2), 1139 (νs SO2), 1106 (δ CH), 1085 (δ CH), 844, 813, 673 (γ NH), 557 (δ SO2); λmax(MeCN) 638 nm (ε 67 dm3 mol−1 cm−1); Found: C, 53.17; H, 4.46; N, 9.49. Cu(C13H13N2O2S)2 requires C, 53.27; H, 4.47; N, 9.55%.
°C (decomp.); m/z
(FAB): 603 (14%, NiL2+Na), 581 (63%, NiL2), 425 (9%, NiL2−tos), 263 (100%, LH); νmax
(KBr)/cm−1 1611 (ν py), 1478 (ν py), 1281 (νa SO2), 1139 (νs SO2), 1084 (δ CH), 681 (γ NH), 557 (δ SO2); Found: C, 53.57; H, 4.68; N, 9.58. Ni(C13H13N2O2S)2 requires C, 53.72; H, 4.51; N, 9.64%.
°C (decomp.); m/z FAB: 604 (37%, CoL2+Na), 582 (100%, CoL2), 426 (39%, CoL2−tos), 320 (23%, CoL); νmax
(KBr)/cm−1 1609 (ν py), 1439 (ν py), 1278 (νa SO2), 1145 (νs SO2), 1085 (δ CH), 947, 760 (δ py), 669 (γ NH), 559 (δ SO2); λmax
(MeCN)/nm 513 and 579 (ε/dm3 mol−1 cm−1 302 and 291); Found: C, 53.20; H, 4.35; N, 9.51. Co(C13H13N2O2S)2·0.25MeOH requires C, 53.48; H, 4.61; N, 9.50%.
Results and discussion
Ligand and complex synthesis and X-ray structural characterisation
The ligands L1–L4 were prepared by reaction of the appropriate 2-(aminomethyl)pyridine with p-toluenesulfonyl chloride or CF3SO2Cl in dry pyridine. The precursor 6-methyl-2-(aminomethyl)pyridine was prepared by reaction of the 6-aldehyde with hydroxylamine followed by reduction of the oxime26 by catalytic hydrogenation over Pd/C. Formation of the ML2 complex was undertaken by mixing two molar equivalents of ligand with one of the M(OAc)2 salt in boiling methanol. On cooling, crystals of the neutral complex were deposited slowly—often over a period of several days. Each complex could be recrystallised from MeOH or EtOH. In the case of [Ni(L1)2] and [Co(L1)2] it was found necessary to form the potassium salt of the ligand, prior to complex formation.The structure of each of the sixteen neutral complexes has been determined by X-ray crystallography at 100–120 K (Tables 1–3; Fig. 1). The coordination geometry at Co(II) and Zn(II) was invariably a distorted tetrahedron, reflecting the minimisation of steric congestion, and for Co(II) the slightly favourable ligand field stabilisation effect. With Cu(II), the less sterically demanding ligand L1 allowed formation of a square planar complex, whereas in complexes with L2, L3 and L4, the distorted tetrahedral arrangement that characterised zinc and cobalt complex formation is favoured. Finally, for Ni(II) complexes, once again complexes with L3 and L4 adopted distorted tetrahedral arrangements, while [Ni(L1)2] was square planar and [Ni(L2)2] took up an octahedral geometry, with two ethanol molecules cis-coordinated and the sulfonamide nitrogens trans-related.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Complex | M–N11/Å | M–N12/Å | M–N21/Å | M–N22/Å | Average M–N1 | Average M–N2 | Average M–N |
|
a In CN=4, mean ionic radii for Zn, Cu, Ni and Co(II) ions are 0.60, 0.57, 0.55 and 0.58 Å.27
[Cu(L4)2]: This complex has two independent molecules in the unit cell.
|
|||||||
| [Zn(L1)2] | 2.049(3) | 1.942(3) | 2.049(3) | 1.956(3) | 2.049 | 1.949 | 1.999 |
| [Zn(L2)2] | 2.023(2) | 1.972(2) | 2.043(2) | 1.967(2) | 2.033 | 1.969 | 2.001 |
| [Zn(L3)2] | 2.048(2) | 1.965(2) | 2.057(2) | 1.966(2) | 2.052 | 1.965 | 2.009 |
| [Zn(L4)2] | 2.050(7) | 1.954(2) | 2.089(2) | 1.929(2) | 2.069 | 1.941 | 2.004 |
| [Cu(L1)2] | 1.996(1) | 1.998(1) | 1.996(1) | 1.998(2) | 1.996 | 1.998 | 1.997 |
| [Cu(L2)2] | 1.949 | 1.939 | 1.998 | 1.916 | 1.974 | 1.928 | 1.951 |
| [Cu(L3)2] | 1.979(2) | 1.973(2) | 1.962(2) | 1.969(2) | 1.971 | 1.971 | 1.971 |
| [Cu(L4)2]A | 2.067(5) | 1.920(5) | 2.019(5) | 1.944(5) | 2.043 | 1.932 | 1.988 |
| [Cu(L4)2]B | 2.067(5) | 1.914(5) | 2.058(5) | 1.918(5) | 2.063 | 1.916 | 1.989 |
| [Ni(L1)2] | 1.924(5) | 1.960(5) | 1.924(5) | 1.960(5) | 1.924 | 1.960 | 1.942 |
| [Ni(L2)2] | 2.090(3) | 2.123(3) | 2.060(3) | 2.099(3) | 2.075 | 2.111 | 2.093 |
| [Ni(L3)2] | 2.021(2) | 1.941(2) | 2.028(2) | 1.946(2) | 2.025 | 1.944 | 1.9845 |
| [Ni(L4)2] | 2.040(2) | 1.905(2) | 2.014(2) | 1.929(2) | 2.027 | 1.917 | 1.972 |
| [Co(L1)2] | 2.031(2) | 1.960(2) | 2.037(2) | 1.937(2) | 2.034 | 1.949 | 1.9915 |
| [Co(L2)2] | 2.028(2) | 1.962(2) | 2.031(2) | 1.968(2) | 2.030 | 1.965 | 1.9975 |
| [Co(L3)2] | 2.040(2) | 1.972(2) | 2.035(2) | 1.965(2) | 2.038 | 1.969 | 2.0035 |
| [Co(L4)2] | 2.061(2) | 1.934(2) | 2.052(2) | 1.960(2) | 2.056 | 1.947 | 2.001 |
|
|
||||||
|---|---|---|---|---|---|---|
| Complex | N11–M–N12 | N11–M–N21 | N11–M–N22 | N12–M–N21 | N12–M–N22 | N21–M–N22 |
| a Twinned crystal. Structure of poor quality. ESD's are estimated at ±5 in the last digit. | ||||||
| [Zn(L1)2] | 82.96(11) | 122.40(10) | 121.54(11) | 117.73(11) | 134.92(11) | 82.59(10) |
| [Zn(L2)2] | 83.12(6) | 124.98(6) | 124.75(6) | 114.93(7) | 131.99(7) | 82.44(6) |
| [Zn(L3)2] | 82.81(6) | 113.20(6) | 125.79(7) | 123.81(7) | 131.73(7) | 83.32(6) |
| [Zn(L4)2] | 82.8(2) | 113.3(2) | 129.8(3) | 117.47(8) | 134.01(8) | 82.01(7) |
| [Cu(L1)2] | 83.23(6) | 180.00(8) | 96.77(6) | 96.77(6) | 180.00(7) | 83.23(6) |
| [Cu(L2)2]a | 82.3 | 147.3 | 105.6 | 105.6 | 154.7 | 81.0 |
| [Cu(L3)2] | 84.07(8) | 150.57(9) | 112.72(8) | 103.40(8) | 131.34(9) | 83.92(8) |
| [Cu(L4)2]A | 83.0(2) | 140.66(19) | 109.3(2) | 107.2(2) | 145.0(2) | 84.2(2) |
| [Cu(L4)2]B | 82.2(2) | 128.82(19) | 115.0(2) | 110.6(2) | 145.4(2) | 82.7(2) |
| [Ni(L1)2] | 94.4(2) | 180.00(17) | 85.6(2) | 85.6(2) | 180.0(3) | 94.4(2) |
| [Ni(L2)2] | 79.48(12) | 91.01(12) | 94.27(13) | 97.31(13) | 173.27(13) | 80.18(13) |
| [Ni(L3)2] | 82.13(7) | 108.20(7) | 124.26(8) | 124.25(7) | 137.32(8) | 82.23(7) |
| [Ni(L4)2] | 81.82(8) | 110.69(8) | 116.45(8) | 133.00(8) | 134.63(8) | 81.64(7) |
| [Co(L1)2] | 81.85(9) | 122.08(9) | 119.28(10) | 119.19(10) | 137.24(10) | 82.59(9) |
| [Co(L2)2] | 82.25(8) | 125.16(8) | 122.17(8) | 116.01(9) | 135.44(8) | 81.83(8) |
| [Co(L3)2] | 82.88(8) | 115.13(8) | 124.51(8) | 127.66(9) | 129.35(9) | 82.12(8) |
| [Co(L4)2] | 81.97(8) | 114.45(8) | 119.17(8) | 128.57(8) | 134.20(8) | 81.94(7) |
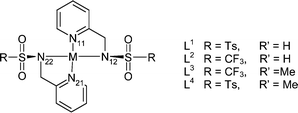
![View of the crystal structures (100 to 120 K) of the Co(ii), Ni(ii), Cu(ii) and Zn(ii) complexes of L1–L4, revealing distorted tetrahedral geometry except for [Cu(L1)2], [NiL1]
(square planar), and [Ni(L2)2(EtOH)2]
(octahedral).](/image/article/2003/NJ/b206279h/b206279h-f1.gif) | ||
| Fig. 1 View of the crystal structures (100 to 120 K) of the Co(II), Ni(II), Cu(II) and Zn(II) complexes of L1–L4, revealing distorted tetrahedral geometry except for [Cu(L1)2], [NiL1] (square planar), and [Ni(L2)2(EtOH)2] (octahedral). | ||
Geometric parameters are summarised in Tables 2 and 3. Within the series of zinc and cobalt complexes, average bond lengths to the sulfonamide N were marginally longer with the more electron poor trifluoromethyl-substituted pair of complexes. At the same time, the M–Npy bond length was slightly longer for the complexes with the 6-methyl substituent. Bond lengths to pyridyl and sulfonamide nitrogens were in line with literature values for 4- or 5-coordinate zinc complexes.28–31 For example, in the 2,2-bipyridylzinc complex of cyclohexane-1,2-diylbis(methanesulfonamide) the sulfonamide N–Zn bond length was 1.94 Å with a bpy–N–Zn length of 2.05 Å.28 With the copper(II) complexes, average bond lengths were slightly longer for the square planar example. The X-ray structure of [Cu(L1)2] has been reported independently at 293 K very recently;32,33 structural details echo those reported herein. Overall, there is a remarkable constancy in the M–N bond lengths. This can be related to the fixed chelate bite angle, associated with each ligand. Thus, the intra-ligand N–M–N′ bond angles in the series average 83° and 82° for the set of zinc and cobalt complexes respectively (Table 3). This relatively constant bite angle is also found (83±3°) in the square planar and octahedral Ni(II) complexes.
There are two limiting geometries for four-coordinate complexes: square planar and tetrahedral. In order to quantify the deviation from these limits in the complexes examined here, a search of the CSD was undertaken examining all first row four-coordinate complexes with N and O donors only. In the April 2001 version, 284 of these were for zinc complexes. The geometry around the central ion is defined by six bond angles and these were obtained from the database. For a square planar complex, the sum of these angles is 720°; around a tetrahedron the sum is 656°. However, the sum itself is not a sufficient measure of geometry, because in the limiting case of the perfect tetrahedron each of the six angles needs to be identical, whilst for a square planar arrangement two of the angles have to be 180° and the remaining four are 90°. A measure of distortion is obtained by calculating the average deviation of all six angles; for a perfect tetrahedron this is zero and for a perfect square plane it is 40°
[(2×60+4×30)/6]. A plot of the average angular deviation (y axis)
versus the sum of the six angles is given for all of the CN=4 first row transition metal complexes with N and O donors only in the CSD (Fig. 2). In this Figure, the copper complex of L1 appears in the top right hand corner, whilst the copper complex of L2 possesses an angular deviation of close to 25°. The remaining two copper structures (sum of angles 665 to 670°), each of the zinc and cobalt complexes and the three tetracoordinate nickel complexes form a cluster around 18–20°. Once again, for the complexes examined here, the degree of distortion is primarily related to the 5-ring chelate bite angle of about 82° imposed by the ligand structure.
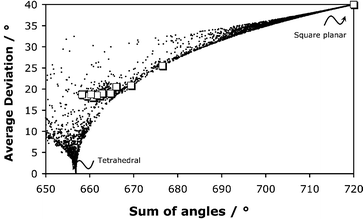 | ||
| Fig. 2 Plot of the sum of the six bond angles (x axis) versus the average deviation of these angles for all 4-coordinate first row transition metal complexes involving N and O donors (CSD 2001). The complexes in this paper are represented by squares (see Table 3). | ||
This method for analysing distortion from a regular polyhedron may be extended to related octahedral or trigonal bipyramidal structures. For the unique case of tetrahedral distortion, an alternative (1-D) analysis involves an examination of the dihedral angle between the two ML2 planes, with limits at 90° and 0° for ideal tetrahedral and square planar geometry. This method has one advantage in giving the absolute configuration for chiral systems. Such an analysis has been carried out for the 16 structures defined herein. For each of the tetrahedrally distorted cobalt, nickel and zinc complexes, the deviation from the tetrahedral (90°) limit was not more than 10°. Distortion was more evident with the copper complexes; for [Cu(L2)2] the dihedral angle was 45° whilst for [Cu(L3)2] and [Cu(L4)2], the values were 29 and 33° respectively. The 2-D plot shown in Fig. 2 intrinsically provides additional information. Tetragonal distortion of a square planar complex gives an angle sum of near to 720° and an angular distortion of >40°; no cases were found for first row elements with O and N donors, only two examples with M–M (Cu/Cu and Co/Cu) bonding fall on this limit.
Absorption spectroscopy and cyclic voltammetry
Absorption spectra were recorded for each of the coloured complexes in acetonitrile solution at a 1 mM concentration when observing the d–d bands, and in more dilute solution when charge-transfer bands were also observed. For the Co(II) complexes, the absorption spectra were similar except for [Co(L4)2], which was much more pale-coloured in the MeCN solution (Fig. 3). Molar absorption coefficients, ε, decreased in the sequence [Co(L2)2]≪[Co(L1)2]<[Co(L3)2]<[Co(L4)2]. It is generally appreciated that visible transitions in tetrahedral cobalt(II) complexes are an order of magnitude more intense than for equivalent octahedral systems. Absorption spectra tend to be dominated by the 4A2→4T1(P) transition for tetrahedral systems, and 4T1g(F)→4T1g(P) transition for octahedral examples. Fine structure is imposed by a number of transitions to doublet excited states, which gain intensity by spin–orbit coupling. Thus for [Co(L2)2], the complex must be octahedral in solution, binding to two additional solvent molecules (viz.Fig. 1 for [Ni(L2)2(EtOH)2]). Indeed, the reflectance spectra for each cobalt complex—obtained on crystalline samples—were more or less identical in intensity, supporting this idea.
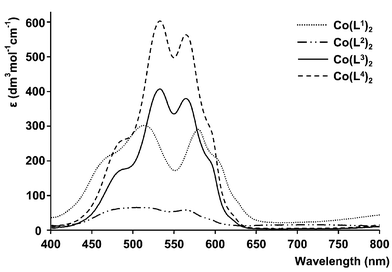 | ||
| Fig. 3 Absorption spectra of neutral cobalt(II) complexes of L1–L4 (1 mM complex, (2 mM for L2), MeCN). | ||
The tetrahedrally distorted copper complexes, [Cu(L3)2] and [Cu(L4)2], were lime-green and orange-brown in solution (Table 4 and Fig. 4), reflecting the influence of the relatively intense LMCT bands at 410 and 448 nm respectively. The portion of the d–d transition was shifted to the red in the sequence [Cu(L1)2]>[Cu(L2)2]>[Cu(L3)2]>[Cu(L4)2] associated with the increase in LFSE for square-planar complexes, with the more polarisable NTs ligand in L1 affording the greatest crystal field splitting. The CuII/CuI redox couple was examined by cyclic voltammetry (MeCN, Bu4NClO4, 295 K) for each of the four copper complexes (Fig. 5). For the complexes lacking the 6-Me substituent, the E1
/2 values (vs. Ag/AgCl) were −430 [Cu(L1)2] and −137 mV ([Cu(L2)2]), reflecting the greater stabilisation of the copper(II) state in the tosylamide complex with square planar geometry and a large ligand field stabilisation energy (Table 4). The tetrahedrally distorted complex [Cu(L3)2] is much more readily reduced, E1
/2=+55 mV, consistent with the destabilisation of the Cu(II) state. In each of these cases, quasi-reversible behaviour was exhibited (ip≈iaiia∝ν1/2), whereas for the tetrahedrally distorted complex [Cu(L4)2], more complex behaviour was noted with an apparent redox couple at ca.
−240 mV.
| Co(II) | Ni(II) | Cu(II) | |
|---|---|---|---|
| L1 | 513 (300), 579 (290) | insoluble | 639 (70) |
| L2 | 500 (45) | 367 (50), 597 (3) | 672 (230) |
| L3 | 533 (400), 564 (380) | 544 (190), 733 (50) | 410 (405), 764 (70) |
| L4 | 532 (580), 564 (540) | 544 (70), 760 (30) | 448 (2020), 784 (215) |
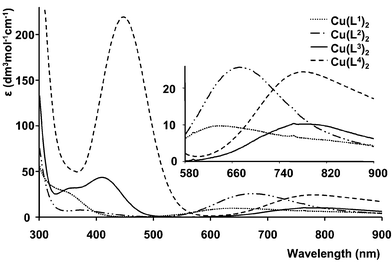 | ||
| Fig. 4 Absorption spectra of neutral copper(II) complexes of L1–L4, the inset highlights the position of the 2E2g→2T2g transition. | ||
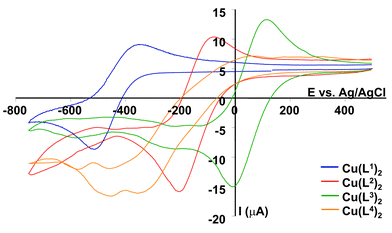 | ||
| Fig. 5 Cyclic voltammograms (295 K, 0.1 M Bu4NClO4, v=100 mV s−1) of the copper(II) complexes of L1–L4. | ||
Selected ligand protonation and metal complex formation constants: speciation at ambient pH
Equilibrium constants associated with stepwise protonation of the anionic ligands L1–L3 were measured by standard pH-metric methods in 80% MeOH/H2O, in a background of 0.1 M NMe4NO3. Data were corrected24 to allow for the variation of the water dissociation constant at this solvent composition and were analysed using the programme HYPERQUAD.23 The log K1 values (Table 5) measured for L2 and L3 were 7.51 and 7.61 respectively, similar to the literature values for simple trifluoromethylsulfonamides.19 The p-toluenesulfonyl analogue, L1, possesses a much more basic sulfonamide N, log K1=12.23, in line with data for related arylsulfonamides.34 The pyridyl nitrogen is much less basic in each case, and the introduction of the 6-methyl substituent slightly enhances proton affinity (L2vs. L3: log K2=2.69 and 3.32). Thus, around ambient pH conditions, the ligands L2 and L3 exist as almost 50% of the monoanionic species.
=0.1 M NMe4NO3, 80% MeOH–20% H2O)
| log K1a | log K2 | log KZnL | log KZnL2 | log KCuL | log KCuL2 | |
|---|---|---|---|---|---|---|
|
a Defining the successive protonation constants: K1=[LH]/[L−][H+] and K2=[LH2+]/[LH][H+].
b Values for Co complexes were: log KCoL 5.11(6); log KCoL2 5.19(5).
c Precipitation occurred under these conditions.
|
||||||
| L1 | 12.23(6) | 3.31(3) | 7.66(6) | 6.91(7) | 11.40(5) | 9.16(7) |
| L2 | 7.51(3) | 2.69(3) | 5.25(5)b | 5.12(7)b | 6.74(5) | 6.42(8) |
| L3 | 7.61(3) | 3.23(3) | c | c | 6.26(4) | 6.09(8) |
Metal complex formation constants have been measured for Cu(II) and Zn(II) complexes of L1–L3, taking account of metal ion hydrolysis.23 Separate titrations at 1∶1 and 1∶2 metal/ligand ratios were undertaken in order to measure both the ML and the ML2 formation constants. The ligand L2 formed slightly more stable complexes with copper than zinc, in accord with the Irving–Williams series,35 although a direct comparison of the tetrahedrally distorted Cu–L3/Zn–L3 systems is not possible here, owing to the insolubility of the zinc complex under the standard conditions used. The difference in stability of the copper and zinc complexes is much greater with L1, as the copper complex in that case is square planar and the zinc is tetrahedral. The relative magnitude of the stepwise formation constants (log KMLvs. log KML2) for zinc, copper and cobalt was very similar, although there was no evidence for the positive cooperativity in formation of the ML2 complex, that was apparently a feature of the behaviour of the related quinolylsulfonamides (e.g. Zinquin5). It should be noted that the analysis of the data reported in that case (“we conclude that that the 1∶1 and 2∶1 Zn/Zinquin complexes differ by at least two orders of magnitude”5) only gives a limit to the difference in values as no direct titrations were reported at 1∶1 stoichiometry and there was no direct account taken of metal ion hydrolysis in the data analysis.
The harder trifluoromethylsulfonamide N favours binding to Cu(II) less than the more basic and softer tosylamide N in L1. Hard donors—such as phosphinates—have previously been shown to disfavour binding to Zn2+ less than to Cu2+, in related 4 or 5-coordinate aza-phosphinate complexes.21 Furthermore, systems favouring tetrahedral coordination should also enhance the relative stability of Zn(II) compared to Cu(II), as the LFSE contribution is significantly diminished for Cu(II) when deviations from planarity or square pyramidal geometry occur.13,14,22
Using the data in Table 5, species distribution diagrams as a function of pH and M/L concentration may be obtained, in which the percentage of the insoluble metal hydroxide is calculated by extrapolation. Considering the Zn2+/L1 and L2 systems (Figs. 6 and 7), the pH-dependent distribution of complex species has been calculated at fixed ligand concentrations of 10 mM, 1 mM and 0.1 mM, with varying [Zn2+] values. Distributions at a fixed 2∶1 ligand to metal ratio are given in the ESI.† With the Zn2+/L2 system, a particular feature is that the [Zn(L2)2] species predominates at ambient pH and even with [L2]tot=0.1 mM and [Zn2+]=0.1 mM, more than 80% of all Zn bound species is [Zn(L2)2]. On the other hand, with the tosylamide ligand L1
(log K1=12.2) even at 10 mM [L1]tot and 1 mM [Zn2+]tot, the major species at pH 7.4 is the metal hydroxide with significant [ZnL1] formed. At lower ligand concentrations, only a small percentage fraction of the [ZnL1] species is present. Given that L2 and L3 may be readily derivatised at C-6 or alpha to the sulfonamide N, these ligands offer some scope as the basis for Zn2+ probes in neutral aqueous media.
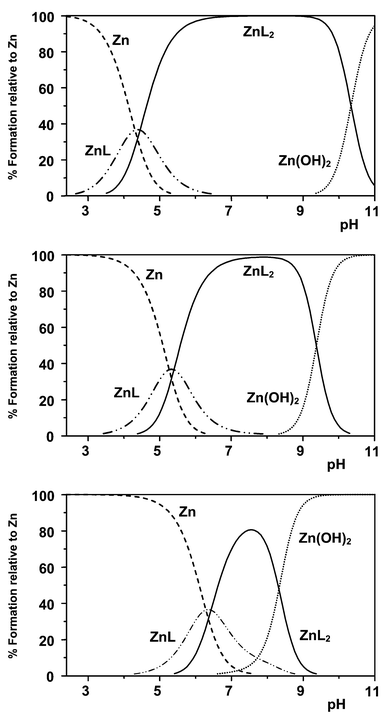 | ||
| Fig. 6 Species distribution plots for Zn2+/L2: upper, 10 mM ligand, 1 mM Zn2+; centre, 1 mM L2, 0.1 μM Zn2+; lower, 0.1 mM L2, 0.1 μM Zn2+ (80% MeOH, 20% H2O; 298 K; 0.1 M Me4NNO3). Plots at a fixed L/M ratio of 2 are given in the ESI.† | ||
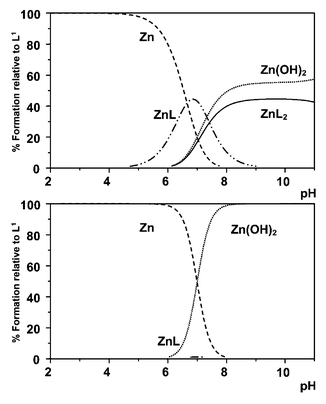 | ||
| Fig. 7 Species distribution plots for Zn2+/L1: upper, 10 mM ligand, 1 mM Zn2+; lower, 0.1 mM L1, 10 μM Zn2+. | ||
For the Cu(II)/L1 system, exhibiting, as expected, the highest stepwise formation constants for any of the systems examined here, it is possible to vary the pH over the range 4 to 8 in order to control the relative proportion of [CuL1] and [Cu(L2)2] species (Fig. 8left). Thus, for a ligand concentration of 10 mM (or 1 mM), at pH 5 the predominant species is [CuL1], whereas at pH>7, [Cu(L2)2] is the major species. With this in mind, cyclic voltammetry was used to examine the Cu(II)/Cu(I) redox couple as a function of pH (Fig. 8right). At pH 4.9 (10 mM L1; 5 mM Cu(CF3SO3)2; 298 K; 0.1 M NMe4NO3, 80% MeOH/H2O), the reversible wave observed at ca.
−10 mV may be ascribed to [CuL1], whereas at pH 8.8, the redox couple shifts to −315 mV and is associated with the square planar complex [Cu(L2)2]. At the intermediate pH value of 5.65, both species are present in nearly equal amounts, and the observation of separate redox waves for each species is consistent with the rate of electron transfer being faster than any associative ligand exchange process involving [CuL1] and [Cu(L2)2]. Given that different redox active ML species may be observed simultaneously, for example using differential pulse voltammetry, and that their relative concentration is pH dependent, such work suggests that by immobilising ligands related to L1 at an electrode surface, the selective detection and assay of a mixture of metal ions may be expedited using sensitive stripping voltammetric techniques.
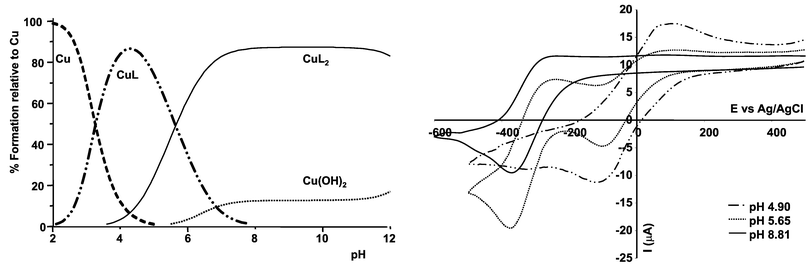 | ||
| Fig. 8
Left: Species distribution plot for Cu2+/L1
(10 mM L1, 5 mM Cu2+, 298 K, 80% MeOH, 20% H2O, 0.1 M NMe4NO3); right: cyclic voltammograms recorded under the same conditions at pH 4.9, 5.65 and 8.81 (v=100 mV s−1). | ||
In summary, ligands L2 and L3 are suitable basic ligand systems that form ML2 complexes at ambient pH when the free zinc concentration is in the micro to nanomolar range. Suitable derivatives are therefore being evaluated allowing their integration into practicable luminescent or MRI probes.
We thank EPSRC (AC, HP, MK) and the University of Durham for support.
References
- R. J. P. Williams and J. J. R. Frausto da Silva, Coord. Chem. Rev., 2000, 200–202, 247 CrossRef CAS.
- D. W. Choi and J. Y. Koh, Annu. Rev. Neurosci., 1998, 21, 347 CrossRef CAS.
- L. M. T. Canzoniero, D. M. Turetsky and D. W. Choi, J. Neurosci., 1999, 19, RC31 Search PubMed.
- J. H. Weiss, S. L. Sensi and J. Y. Koh, Trends Pharm. Sci., 2000, 21, 395 CrossRef CAS.
- C. J. Fahrni and T. V. O'Halloran, J. Am. Chem. Soc., 1999, 121, 11448 CrossRef CAS.
- K. R. Gee, Z.-L. Zhou, W.-J. Qian and R. Kennedy, J. Am. Chem. Soc., 2002, 124, 776 CrossRef CAS.
- T. Hirano, K. Kikuchi, Y. Urano, T. Higuchi and T. Nagano, Angew. Chem., Int. Ed., 2000, 39, 1052 CrossRef CAS; T. Hirano, K. Kikuchi, Y. Urano, T. Higuchi and T. Nagano, J. Am. Chem. Soc., 2000, 122, 12399 CrossRef CAS.
- G. K. Walkup, S. C. Burdette, S. J. Lippard and R. Y. Tsien, J. Am. Chem. Soc., 2000, 122, 5644 CrossRef CAS.
- P. D. Zalewski, S. H. Millard, I. J. Forbes, O. Kapaniris, A. Slavotinek, W. H. Betts, A. D. Ward, S. F. Lincoln and I. Mahadevan, J. Histochem. Cytochem., 1994, 42, 877 Search PubMed.
- W. D. Qian, C. A. Aspinwall, M. A. Battiste and R. T. Kennedy, Anal. Chem., 2000, 72, 711 CrossRef CAS.
- O. Reany, T. Gunnlaugsson and D. Parker, J. Chem. Soc., Perkin Trans. 2, 2000, 1819 RSC.
- Y. Hitomi, C. E. Outten and T. V. O'Halloran, J. Am. Chem. Soc., 2001, 123, 8614 CrossRef CAS.
- C. D. Edlin, D. Parker, J. J. B. Perry, C. Chartroux and K. Gloe, New J. Chem., 1999, 23, 819 RSC.
- G. B. Bates, D. Parker and P. A. Tasker, J. Chem. Soc., Perkin Trans. 2, 1996, 1117 RSC.
- J. M. Berg and Y. Shi, Science, 1996, 271, 1081 CAS.
- T. Koike, E. Kimura, I. Nakamura, Y. Hashimoto and M. Shiro, J. Am. Chem. Soc., 1992, 114, 7338 CrossRef CAS; U. Hartmann and H. Vahrenkamp, Inorg. Chem., 1991, 30, 4676 CrossRef CAS.
- E. E. Chufan, S. Garcia-Granda, M. R. Diaz, J. Borras and J. C. Pedregosa, J. Coord. Chem., 2001, 54, 303 Search PubMed.
- C. T. Supuran and A. Scozzafava, Eur. J. Med. Chem., 2000, 35, 867 CrossRef CAS.
- K. Hakansson and A. Liljas, FEBS Lett., 1994, 350, 319 CrossRef CAS.
- D. Elbaum, S. K. Nair, M. W. Patcham, R. B. Thompson and D. W. Christianson, J. Am. Chem. Soc., 1996, 118, 8381 CrossRef CAS.
- G. B. Bates, E. Cole, R. Kataky and D. Parker, J. Chem. Soc., Dalton Trans., 1996, 2693 RSC.
- G. B. Bates and D. Parker, J. Chem. Soc., Perkin Trans. 2, 1996, 1109 RSC.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS; L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311 CrossRef CAS; P. Gans, A. Sabatini and A. Vacca, J. Chem. Soc., Dalton Trans., 1985, 1195 RSC; P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739 CrossRef CAS.
- S. Rondini, P. R. Mussini and T. Mussini, Pure Appl. Chem., 1987, 59, 1549.
- SHELXTL version 6.12, Bruker AXS, Madison, Wisconsin, USA, 2001.
- O. Fuentes and W. W. Pandler, J. Org. Chem., 1975, 40, 1210 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. C, 1976, 32, 751 CrossRef.
- S. E. Denmark, S. P. O'Connor and S. R. Wilson, Angew. Chem., Int. Ed., 1998, 37, 1149 CrossRef CAS.
- S. Z. Haider, K. M. A. Malik, M. B. Hursthouse and S. Das, Acta Crystallogr., Sect. C, 1984, 40, 1147 CrossRef.
- U. Hartmann and H. Vahrenkamp, Z. Naturforsch. B, 1994, 49, 1725 Search PubMed.
- S. L. Sumalan, J. Casanova, G. Alzuet, J. Borras, A. Castineiras and C. T. Supuran, J. Inorg. Biochem., 1996, 62, 31 CrossRef CAS.
- L. Gutierrez, G. Alzuet, J. A. Real, J. Cano, J. Borras and A. Castineiras, Inorg. Chem., 2000, 39, 3608 CrossRef CAS.
- L. Gutierrez, G. Alzuet, J. Borras, M. Liu-Gonzalez, F. Sanz and A. Castineiras, Polyhedron, 2001, 20, 703 CrossRef CAS.
- B. Nyasse, L. Grehn, U. Ragnarsson, H. L. S. Maia, L. S. Monteiro, I. Leito, I. Koppel and J. Koppel, J. Chem. Soc., Perkin Trans. 1, 1995, 2025 RSC.
- H. M. N. H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details for [M(L2)2], [M(L3)2] and [M(L4)2] (M=Zn, Cu, Ni, Co); species distribution plots. See http://www.rsc.org/suppdata/nj/b2/b206279h/ |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |