Thermomorphic fluorous imine and thioether palladacycles as precursors for highly active Heck and Suzuki catalysts; evidence for palladium nanoparticle pathways
Christian
Rocaboy
and
J. A.
Gladysz
*
Institut für Organische Chemie, Friedrich-Alexander-Universität Erlangen-Nürnberg, Henkestrasse 42, 91054, Erlangen, Germany. E-mail: gladysz@organik.uni-erlangen.de
First published on 28th November 2002
Abstract
p-Iodobenzaldehyde is elaborated to the fluorous alcohol p-Rf8(CH2)3C6H4CH(OH)(CH2)2Rf8 (three steps/80%; Rf8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =n-C8F17), which is converted to imine p-Rf8(CH2)3C6H4C(
=n-C8F17), which is converted to imine p-Rf8(CH2)3C6H4C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(CH2)3Rf8)(CH2)2Rf8 (6, two steps/93%) and thioether p-Rf8(CH2)3C6H4CH(S(CH2)3Rf8)(CH2)2Rf8 (12, 64%). Reactions with Pd(OAc)2 (AcOH, 95°C) give palladacycles with [RC
N(CH2)3Rf8)(CH2)2Rf8 (6, two steps/93%) and thioether p-Rf8(CH2)3C6H4CH(S(CH2)3Rf8)(CH2)2Rf8 (12, 64%). Reactions with Pd(OAc)2 (AcOH, 95°C) give palladacycles with [RC![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) 6H3CR′N(R)Pd
6H3CR′N(R)Pd![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) (μ-OAc)]2 (7, 87%) and [RC6H3CHR′S(R)Pd(μ-OAc)]2 (13, 84%) cores. The former reacts with LiCl and LiI to give the corresponding bridging halide complexes (8, 9); LiCl/PPh3 affords monomeric RC6H3CR′N(R)Pd(Cl)(PPh3) (10). Palladacycles 7–9 and 13 are poorly soluble or insoluble in many solvents at 20–24°C, but much more soluble at higher temperatures. The CF3C6F11/toluene partition coefficients of 6, 7, 12, and 13 are >91∶<9 (24°C). Both 7 and 13 are excellent catalyst precursors for Heck reactions of aryl halides. Turnover numbers exceed 106 with phenyl iodide under homogeneous conditions in DMF at 140°C. The palladacycles precipitate as bridging halides upon cooling, and can in theory be recovered by liquid/solid phase separations. However, since the quantities are small, the solvent C8F17Br is added for recycling. Induction periods in both the first and second cycles, and progressively lower activities, are noted. Transmission electron microscopy indicates the formation of soluble palladium nanoparticles. Together with other data, it is proposed that the nanoparticles are the active catalysts, for which the recyclable palladacycles constitute a steady state source, until exhausted. Complex 7 similarly catalyzes the Suzuki reaction (K3PO4, toluene, 130°C).
(μ-OAc)]2 (7, 87%) and [RC6H3CHR′S(R)Pd(μ-OAc)]2 (13, 84%) cores. The former reacts with LiCl and LiI to give the corresponding bridging halide complexes (8, 9); LiCl/PPh3 affords monomeric RC6H3CR′N(R)Pd(Cl)(PPh3) (10). Palladacycles 7–9 and 13 are poorly soluble or insoluble in many solvents at 20–24°C, but much more soluble at higher temperatures. The CF3C6F11/toluene partition coefficients of 6, 7, 12, and 13 are >91∶<9 (24°C). Both 7 and 13 are excellent catalyst precursors for Heck reactions of aryl halides. Turnover numbers exceed 106 with phenyl iodide under homogeneous conditions in DMF at 140°C. The palladacycles precipitate as bridging halides upon cooling, and can in theory be recovered by liquid/solid phase separations. However, since the quantities are small, the solvent C8F17Br is added for recycling. Induction periods in both the first and second cycles, and progressively lower activities, are noted. Transmission electron microscopy indicates the formation of soluble palladium nanoparticles. Together with other data, it is proposed that the nanoparticles are the active catalysts, for which the recyclable palladacycles constitute a steady state source, until exhausted. Complex 7 similarly catalyzes the Suzuki reaction (K3PO4, toluene, 130°C).
Introduction
Fluorous biphase catalysis is now a well established technique for catalyst/product separation and recycling that exploits the temperature-dependent miscibility of organic and fluorous phases.1–3Fig. 1A shows the most common protocol. Catalysts are derivatized with “pony tails” or (CH2)m(CF2)n−1CF3 ((CH2)mRfn) segments that provide high fluorous solvent affinities. Reactions can be conducted under homogeneous conditions at the one-phase, high temperature limit. Products normally have much greater affinities for the non-fluorous solvent, and are easily separated at the two-liquid-phase, low temperature limit. A newer-generation protocol is shown in Fig. 1B.4,5 This dispenses with the fluorous solvent, and instead exploits the temperature-dependent solubility of the fluorous catalyst in the organic solvent. Such “thermomorphic”6 behavior allows homogeneous reaction conditions at higher temperatures, with catalyst recovery via liquid/solid phase separation at lower temperatures.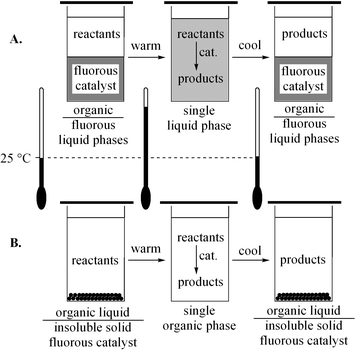 | ||
| Fig. 1 Traditional (A) and fluorous-solvent-free (B) fluorous biphase catalysis. | ||
Many fluorous transition-metal catalysts are now known. They have given impressive results in metal-catalyzed hydroformylations,7 hydrogenations,8 hydroborations,9 hydrosilylations,10 oxidations,11 carbon–carbon bond forming reactions of aryl halides,12–14 additions of Et2Zn to carbonyl compounds,15 and other processes.16 Surprisingly, there have been no attempts to develop fluorous versions of palladacycle catalysts,17 some examples of which are given in Fig. 2.18–21 These are particularly effective for sp2–sp2 carbon coupling processes such as the Heck22 and Suzuki23 reactions. One contributing factor may be that most palladacycles contain multiple aromatic rings, which typically require three Rf8 “pony tails” for effective immobilization in fluorous solvents.2,8b,24 However, since Heck reactions normally require elevated temperatures, we thought it might be feasible to employ the newer protocol in Fig. 1B, which reduces the dependence upon a high fluorous/organic solvent partition coefficient.
 | ||
| Fig. 2 Representative palladacycle catalyst precursors for the Heck reaction. | ||
Our attention was drawn to two types of cyclopalladated complexes. The first was an imine-based system reported by Milstein (B, Fig. 2), with just one arene per palladium.19 The second was a similar thioether-derived family described by Dupont (C, Fig. 2).20 Both have been applied to the Heck reaction, and the latter to the Suzuki coupling. We independently became interested in palladium complexes of simple fluorous thioethers (Rf8(CH2)n)2S (n=2, 3), which are also catalyst precursors for Suzuki coupling reactions.13d Hence, we set out to synthesize fluorous analogs of B and C, evaluate their efficacies as catalyst precursors, and attempt their recovery and reuse. A portion of this work has previously been communicated.25
Results
1. Synthesis and characterization of catalyst precursors
We first sought a fluorous imine containing at least three pony tails. As shown in Scheme 1, a synthesis was developed starting from commercial p-iodobenzaldehyde (1). We have previously shown that aromatic aldehydes undergo high-yield Wittig reactions with the ylide derived from the readily available phosphonium salt Rf8CH2CH2PPh3+I−.24,26 An analogous procedure with 1 gave the alkene 2 in 96% yield after workup (90∶10 Z/E mixture). All new compounds gave satisfactory microanalyses, and were characterized by 1H and 13C NMR, and often additional means, as summarized in the experimental section.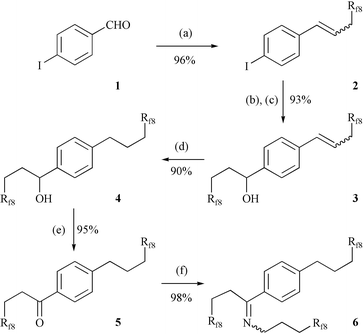 | ||
| Scheme 1 Synthesis of fluorous imine 6. Conditions: (a) Rf8CH2CH2PPh3+I−, K2CO3, p-dioxane/H2O, 95°C; (b) i-PrMgCl, THF; (c) Rf8CH2CH2CHO; (d) (Ph3P)3RhCl, H2 (75 psi), EtOH/CF3C6H5, 40°C; (e) Dess–Martin periodinane, CF3C6H5; (f) NH2CH2CH2CH2Rf8, SnCl2(H2O)2, toluene, reflux, Dean Stark. | ||
To our initial surprise, we were unable to hydrogenate the alkene moiety in 2 or closely related compounds such as the analogous bromide without simultaneous partial hydrogenolysis of the aryl halide (Pd/C, (Ph3P)3RhCl catalysts). The resulting arene was also very difficult to separate from the target aryl iodide. Accordingly, i-PrMgCl was used to effect iodine/magnesium exchange,27 and subsequent addition of the readily available fluorous aldehyde Rf8CH2CH2CHO28 gave the benzylic alcohol 3 in 93% yield after workup. The alkene moiety of this two-pony-tail system could be hydrogenated (7 mol% (Ph3P)3RhCl) without competing carbon–oxygen bond hydrogenolysis, affording 4 in 90% yield.
Oxidation of 4 with the Dess–Martin periodinane29 gave the aryl ketone 5 in 95% yield. Subsequent condensation with the readily available fluorous amine H2NCH2CH2CH2Rf828 in the presence of SnCl2(H2O)2 (20 mol%) simultaneously introduced the third pony tail and generated the imine 6, which was isolated in 98% yield (35∶65 Z/E mixture). Although the synthesis of 6 required five steps, product purifications—often the most important consideration in fluorous syntheses—were easy and the overall yield was good. The CF3C6F11/toluene partition coefficients of 5 and 6 were measured by GC as described in the experimental section. As summarized in Table 1, the fluorous phase affinity of 6 was very high (98.7∶1.3), but that of 5 was—as expected from the number of pony tails—lower.
°C)
As shown in Scheme 2, 6 underwent cyclopalladation under standard conditions (Pd(OAc)2, AcOH, 95°C). Workup gave the yellow dimeric N-donor palladacycle 7 in 87% yield. A crystallized sample melted at 78–80°C and was thermally stable to 225°C, as assayed by DSC and TGA measurements.30 When DMF or CF3C6H5 solutions of 7 were kept at 140°C (2 h) or 100°C (16 h), no decomposition was detected visually or by NMR. Reactions of 7 with LiCl or LiI gave the corresponding dimeric palladacycle halides 8 and 9 in 85–86% yields. We thought that monomeric species might be better suited for X-ray crystallography. Accordingly, reactions of (1) the palladacycle chloride 8 with PPh3, and (2) the palladacycle acetate 7 with LiCl/PPh3, gave the phosphine complex 10 in 79–81% yields. However, all crystals diffracted poorly. The thermal stabilities of 8–10 were similar to that of 7.
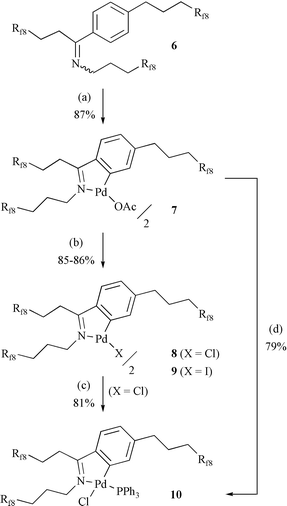 | ||
| Scheme 2 Synthesis of fluorous N-donor palladacycles. Conditions: (a) Pd(OAc)2, AcOH, 95°C; (b) LiX (X=Cl, I), CF3C6H5/MeOH; (c) PPh3, CH2Cl2; (d) LiCl, PPh3, THF. | ||
The N-donor palladacycles 7–10 showed IR νCN bands at lower frequencies than the imine 6 (1583–1587 vs. 1657 cm−1). Complex 7 gave IR νCO values (1571, 1424 cm−1) close to those of other palladacycle bridging acetates.20c,31 The 1H and 13C NMR spectra clearly indicated a trisubstituted arene ring. Palladacycle bridging acetates exhibit structures that are sharply folded about each O2C–CH3 axis.18b,20c This renders the protons of each methylene group in 7 diastereotopic, and exchange is slow on the 1H NMR time scale. Interestingly, the chemical shifts of the NCH21H NMR signals of the palladacycle halides 8 and 9 vary significantly from those of 7 (7, 2.98–3.05 (m, 1H), 3.22–3.29 (m, 1H); 8, 3.84–3.91 (m, 2H); 9, 4.15–4.18 (m, 2H).
At room temperature, 7 was soluble in fluorinated solvents such as CF3C6F11, C8F17Br, CF3C6F5, and CF3C6H5, poorly soluble in common organic solvents such as CH2Cl2, CHCl3, acetone, and THF, and insoluble in DMF. Complexes 8 and 9 were completely insoluble in organic solvents, and poorly soluble in the preceding fluorinated solvents. However, solubilities in CF3C6F5 were much higher above 50°C, allowing NMR spectra to be recorded. Complex 10 was much more soluble than 7 in CH2Cl2, CHCl3, acetone, and THF, and was very soluble in CF3C6F11, CF3C6F5, and CF3C6H5 as well. The CF3C6F11/toluene partition coefficient of 7 could be determined by HPLC, but 8 and 9 were too insoluble. As shown in Table 1, the value was slightly lower than that of imine 6, and was little changed in C8F17Br/DMF, a biphase system employed below.
We next turned our attention to fluorous thioethers that could serve as precursors to palladacycles similar to C (Fig. 2). Thiols are known to condense with benzylic alcohols in the presence of Lewis acids.32 Accordingly, the known fluorous thiol Rf8CH2CH2CH2SH33 was synthesized by a “new” route (previously applied to other fluorous thiols)34 involving thiourea and the fluorous iodide Rf8CH2CH2CH2I.11a As shown in Scheme 3, reaction with the fluorous benzylic alcohol 4 gave the triply pony-tailed fluorous thioether 12 in 64% yield. Cyclopalladation under conditions analogous to those in Scheme 2 afforded the dimeric S-donor palladacycle 13 in 84% yield.
 | ||
| Scheme 3 Synthesis of a fluorous S-donor palladacycle. Conditions: (a) Rf8CH2CH2CH2SH (11), ZnI2, CF3C6H5, 60°C; (b) Pd(OAc)2, AcOH, 95°C. | ||
Unlike the imine 6, thioether 12 is chiral. Accordingly, the dimeric palladacycle 13 contains two carbon stereocenters—as well as two sulfur stereocenters not present in the free ligand. Mixtures of diastereomers are therefore possible, and NMR spectra were much more complex than those of 7. Analogs of C with n-alkyl sulfur substituents also exist as mixtures of diastereomers.20c The palladacycle 13 exhibited solubilities similar to those of 7. However, the partition coefficient was slightly lower (Table 1), and only a single HPLC peak was observed.
2. Catalysis
We first sought to demonstrate that the fluorous N-donor and S-donor palladacycle acetates 7 and 13 were viable catalyst precursors for Heck couplings of aryl halides. Note that halide ions are generated under the reaction conditions—for example in the form of the ammonium salts Et3NH+X− when Et3N is used as the base. Thus, given the rapid reactions with LiCl and LiI in Scheme 2, both 7 and 13 should be converted to bridging halide complexes after the first turnover (even, foreshadowing a point below, if catalysis is effected by an entirely different species). In this context, it is often overlooked that the properties of the catalyst rest state, not the catalyst precursor, are of greatest relevance to recycling.35Hence, a solution of an aryl halide in freshly distilled DMF was sequentially treated with an alkene, Et3N, and a standard solution of 7 or 13 in CF3C6H5, as summarized in Table 2. In order to maximize turnover numbers (TON), the catalyst loadings were kept low (0.66–1.83×10−4 mol%). The apparently homogeneous samples were reacted at 140°C, and then cooled. GC analyses showed the expected coupling products in 49–100% yields, corresponding to TON values of 266000 to 1510000. In accord with most other studies involving dimeric palladacycles, these are based upon the molecular structure (two palladium atoms). The values place 7 and 13 among the best high-turnover Heck catalyst precursors.21 For the less reactive aryl bromide (entries 5 and 6), GC monitoring showed that the lower conversions are due to catalyst deactivation.

| Entry | X | R1 | R2 | Catalyst, mmol | t/h | Conv.c (%) | Yieldc (%) | TON |
|---|---|---|---|---|---|---|---|---|
| a Conditions for 7: ArX (ca. 5.000 mmol), alkene (ca. 1.25 equiv), Et3N (ca. 2 equiv), DMF (6.00 mL). b Conditions for 13: ArX (ca. 5.000–6.279 mmol), alkene (ca. 2 equiv), NEt3 (ca. 2 equiv), DMF (8.00 mL). c Determined by GC. d trans only. e trans∶cis 87∶13. | ||||||||
| 1 | I | H | CO2CH3 | 7, 0.00000344 | 14 | 100 | 100d | 1460000 |
| 2 | 13, 0.0000415 | 18 | 100 | 100d | 1510000 |
|||
| 3 | I | H | C6H5 | 7, 0.00000344 | 24 | 94 | 88e | 1270000 |
| 4 | 13, 0.0000415 | 48 | 100 | 100e | 1510000 |
|||
| 5 | Br | CH3CO | CO2CH3 | 7, 0.00000917 | 48 | 77 | 49d | 266000 |
| 6 | 13, 0.0000831 | 48 | 66 | 50d | 301000 |
|||
Similar reactions were conducted in which 7 was introduced as a solid, and at much higher loadings (0.5 mol%). The initially insoluble 7 dissolved as the DMF was warmed. Upon cooling, palladium complexes precipitated. In reactions of phenyl iodide, NMR analyses showed palladacycle iodide 9 to be the dominant species (>95%). The reaction products and all other materials remained soluble. These observations raised the possibility of catalyst recovery by a simple liquid/solid phase separation of the type in Fig. 1B. However, the catalyst quantities involved were impracticably small—a problem that would be ameliorated on industrial process-chemistry scales.
Accordingly, the reactions shown in Table 3 were conducted with 0.02 mol% of 7, and with periodical monitoring by GC. Since the catalyst loading was higher than in Table 2, a lower temperature (100°C) could be used. After cooling, C8F17Br was added as a “catalyst carrier”, giving a liquid/liquid biphasic sample. This fluorous solvent best dissolves the palladacycle iodide 9. Pictures of a reaction sequence are given in Fig. 3, using 0.5 mol% of 7 for better visualization. The upper DMF phase was separated, and the C8F17Br phase washed with DMF. The combined DMF phases were analyzed to give the data in Table 3. The C8F17Br was removed under vacuum, and the residue charged with fresh educts and DMF for a second reaction cycle.

| Data for 7 | Data for 13 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| R | Cycle | t/h | Conv.b (%) | Yieldb (%) | TONc | t/h | Conv.b (%) | Yieldb (%) | TONc |
| a Conditions: 7 or 13 (0.0014 g or 0.0015 g, 0.00044 mmol), phenyl iodide (0.250 mL, 2.24 mmol), alkene (ca. 1.25 equiv), Et3N (0.625 mL, 4.48 mmol), DMF (4.00 mL). b Determined by GC. c Cumulative. d trans only. e trans∶cis 87∶13. | |||||||||
| CO2Me | 1 | 2 | 100 | 100d | 5100 | 1 | 100 | 100d | 5100 |
| 2 | 2 | 100 | 100d | 10200 |
1.5 | 100 | 100d | 10200 |
|
| 3 | 2 | 77 | 55d | 13300 |
6.5 | 100 | 100d | 15300 |
|
| 4 | 10 | 100 | 100d | 18100 |
16 | 100 | 100d | 20400 |
|
| C6H5 | 1 | 5 | 92 | 85e | 4340 |
5 | 87 | 85e | 4340 |
| 2 | 15 | 88 | 80e | 8420 |
24 | 79 | 72e | 8010 |
|
| 3 | 24 | 86 | 74e | 12190 |
55 | 81 | 81e | 12140 |
|
| 4 | 24 | 74 | 52e | 14840 |
132 | 0 | 0e | 12140 |
|
 | ||
| Fig. 3 Photographs of a recycling sequence analogous to the first entry in Table 3 (7, methyl acrylate) but with 0.5 mol% 7. A, before heating, with undissolved 7; B, after 2 h at 100°C; C, after cooling to room temperature, with precipitated palladium complexes; D, after addition of C8F17Br. Reprinted from ref. 25 with permission from the American Chemical Society. | ||
The results of four cycles are given in Table 3. The data show a gradual loss of activity or turnover frequency. With 7 and methyl acrylate, conversion and yield drop in the third cycle. Only by extending the duration of the fourth cycle from 2 to 10 h are quantitative yields restored. With styrene, activity loss is evident in the second cycle (15 vs. 5 h), and then drops further. The same protocol was applied to the S-donor palladacycle 13. With methyl acrylate, reaction times must again be continually lengthened to maintain quantitative yields. With styrene, no catalyst remains after three cycles. Thus, 13 is consumed at a faster rate than 7. However, additional GC data showed that 13 gave higher TOF values than 7 in the first two cycles with methyl acetate.
These observations are consistent with several scenarios. One would be that the active catalyst is efficiently recycled, but is of limited stability, resulting in progressively slower rates. Another would be that catalyst recycling is not as efficient as anticipated—due, for example, to retention in the DMF phase. Another would be that the active catalysts are non-recyclable decomposition products of 7 or 13. The remaining palladacycle would then be recycled (as the bridging halide), with the diminishing activity representing progressively smaller quantities available for catalyst generation.
In the latter context, many metallic palladium catalysts for the Heck reaction are known.22,36–41 These include organic-solvent-soluble colloidal palladium nanoparticles,36,42 and two varieties of fluorous-solvent-soluble nanoparticles—one type imbedded in a fluorous dendrimer, and the other stabilized by absorbed fluorous molecules.37,38 Also, close relatives of the catalyst B (Fig. 2) upon which 7 is based decompose to active metallic palladium.39 Furthermore, colloidal palladium nanoparticles often impart a reddish-orange tint to DMF,36a and a similar hue is apparent in Fig. 3. We therefore considered the possibility that 7, 13, and the analogous bridging halides serve mainly as recyclable sources of soluble non-fluorous colloidal nanoparticle catalysts. To probe this model, the rate of reaction of phenyl iodide and methyl acrylate was monitored under the conditions summarized in Fig. 4.
 | ||
| Fig. 4 Conversion as a function of time for Heck reactions of phenyl iodide and methyl acrylate. | ||
Importantly, both the first and second cycles with 7 in Table 3 (Fig. 4, top; red and blue traces) appeared to show an induction period, and the second cycle was slightly slower. An induction period for the first cycle could be rationalized under any circumstances, but an induction period for the second suggests that the active catalyst is not being recycled. In order to improve the time resolution, reactions were repeated at 80°C (Fig. 4, bottom; green and orange traces). The induction periods and rate differences were markedly enhanced. The palladacycle iodide 9 showed identical activity (purple trace). In all cases, reddish-orange tints became apparent near the end of the induction period.
The first cycle with 7 in Table 3 was repeated (100°C), and the DMF separated from the DMF/C8F17Br mixture (see panel D, Fig. 3). The C8F17Br layer was washed with DMF (50% of original DMF volume). The DMF phases were combined, charged with additional educts, and heated to 100°C. As shown in Fig. 4 (top, black trace), reaction now occurred without an induction period, indicating a leached catalyst. Although the activity is somewhat less than for the first or second cycles (red and blue traces), it should be noted that all concentrations are ca. 67% lower.
Next, aliquots from the first cycle in Table 3, at the stage of panel B in Fig. 3, were examined by transmission electron microscopy. This is one of the best methods for detecting palladium nanoparticles.36,38 As shown in Fig. 5, a distribution of sizes was found, with an average diameter of ca. 10 nm. Given the above data, and as further elaborated in the discussion section, we assign the bulk of the activity of palladacycle catalyst precursors 7, 9, and 13 to DMF-soluble, non-fluorous palladium nanoparticles.
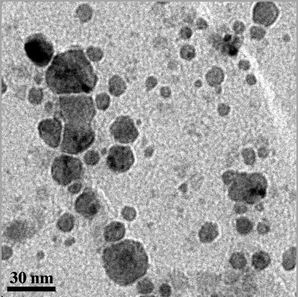 | ||
| Fig. 5 Transmission electron microscopy (TEM) image taken from panel B in Fig. 3. | ||
During the course of the above studies, but before the nature of the active catalyst was appreciated, we also conducted a variety of Suzuki reactions, some of which are summarized in Table 4. We note in passing that the insoluble nature of the base K3PO4, which is customarily used in excess, renders these reactions less suitable for catalyst recovery by liquid/solid phase separation (Fig. 1B). Although palladacycle 7 gave high TON values in the single-cycle experiments in Table 4, attempted recycling with C8F17Br always gave dramatically lower yields and activities. Indeed, many types of metallic palladium are known to catalyze the Suzuki reaction, as discussed further below.36a,38a,41

| X | R | ArX/mmol | 7/mmol | t/h | Conv.b (%) | Yieldb (%) | TON |
|---|---|---|---|---|---|---|---|
| a Conditions: ca. 1.0/1.5/2.0 ArX/PhB(OH)2/K3PO4, toluene (8.00–13.00 mL). b Determined by GC. | |||||||
| I | H | 5.023 | 0.00000458 | 38 | 65 | 40 | 440000 |
| Br | CH3CO | 5.000 | 0.00000458 | 24 | 67 | 31 | 337000 |
| Br | H | 2.374 | 0.0000115 | 24 | 98 | 82 | 170000 |
| Br | CH3 | 2.374 | 0.0000115 | 24 | 96 | 76 | 157000 |
| Br | CH3O | 2.391 | 0.0000115 | 24 | 90 | 83 | 174000 |
Discussion
Despite the careful design of the highly fluorophilic, thermomorphic palladacycle catalyst precursors 7 and 13, the preceding data clearly indicate that a non-molecular catalyst is responsible for the Heck and Suzuki reactions in Tables 2–4. All evidence is consistent with the generation, during the induction periods illustrated in Fig. 4, of highly active soluble colloidal palladium nanoparticles—which are physically detected in Fig. 5. Since the palladacycles are otherwise stable in DMF and CF3C6H5 at 100–140°C, this process must be triggered by one of the reactants (e.g., phenyl iodide, alkene, or Et3N in Table 3). As 7 and 13 or the corresponding bridging halide complexes are progressively consumed in each cycle, catalysis slows and ultimately ceases.
Interestingly, the nanoparticles preferentially partition into non-fluorous solvents, as shown by the retention of activity in DMF after extraction with C8F17Br. This suggests, in accord with much precedent,36a,42 that they are stabilized by the by-product Et3NH+X−. However, as noted above, palladium nanoparticles can also be stabilized by certain fluorous molecules, such as E–G in Fig. 6.38 Such nanoparticles exhibit high fluorous phase affinities, and those stabilized by E are highly active and easily recycled catalysts for the Heck and Suzuki reactions.38a Unfortunately, the types of fluorous molecules that are able to stabilize palladium nanoparticles can, at present, only be determined empirically. Certainly the fluorous ligands of 7 and 13, which must be extruded en route to nanoparticles, could have provided the requisite stabilization. It therefore remains possible that ligand modifications might reverse the phase affinity, giving recyclable fluorous catalysts.
 | ||
| Fig. 6 Representative fluorous compounds that stabilize palladium nanoparticles.38 | ||
What do our and other data suggest regarding the active Heck catalyst derived from the Milstein palladacycle B (Fig. 2)? Detailed kinetic studies of the closely related Blackmond/Pfaltz catalyst precursor D clearly establish homogeneous pathways, with rates dependent upon concentrations of dissolved palladium complexes.43 However, Nowotny has found that the similar polystyrene-supported palladacycle H (Fig. 7) exhibits characteristics comparable to 7 and 13.39 These include an induction period for the first cycle, little or no activity in subsequent cycles, and complete retention of activity in the supernatant after removal of the insoluble polystyrene support. Bedford has reported similar behavior for the silica-bound palladacycles I/I′ in Suzuki reactions.41a Hence, the nature of the catalytically active species appears to be an extremely sensitive function of structure.
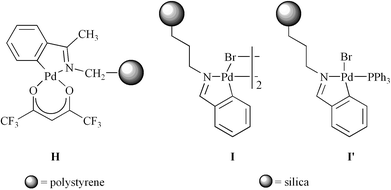 | ||
| Fig. 7 Other immobilized palladacycles that have been evaluated as catalyst precursors for Heck and Suzuki reactions.39,41a | ||
In this regard, it is worth emphasizing that no nanoparticles are detected during Heck reactions with the Herrmann/Beller P-donor palladacycle A.36a For this reason, we had thought that sulfur, another second-row donor atom, might give more robust fluorous palladacycles. However, the S-donor palladacycle 13 appears to be slightly more labile than 7. There are no data at present suggesting that Dupont's similar catalyst precursor C also functions as a palladium nanoparticle source. However, several other palladium adducts of sulfur donor ligands have been evaluated as catalyst precursors for Heck and Suzuki reactions. In many cases, such as we have described for the fluorous complexes [(Rf8(CH2)n)2S]2PdCl2 (n=2,3),13b there is good evidence that the active catalyst is a form of metallic palladium.
In view of the above data, it is in our opinion worth critically examining whether any of the fluorous palladium catalysts so far applied in carbon–carbon bond forming reactions of aryl halides12–14 are truly molecular. As pointed out in a recent commentary,35 activity losses of the type in Table 3 can be masked in several ways. One is to use a high catalyst loading. Another is to select a reaction time for the first few cycles that is much longer than required (e.g., 10 h for the 7/methyl acrylate runs in Table 3). This can allow high yields to be maintained, even when (for example) 90% of the catalyst has been lost. Rate experiments of the type in Fig. 4 unambiguously indicate the degree of retained activity and catalyst, but for some reason are not as frequently conducted. Despite these question marks, we continue to believe that highly active and easily recycled molecular fluorous catalysts for the Heck and Suzuki reactions are realistic objectives. Other types of chelate ligands appear to offer enhanced stabilities, and are currently under active investigation in our laboratory.44
This work, together with related studies mentioned above,39,41a illustrates the valuable insight that recoverable and/or immobilized compounds can offer in the elucidation of mechanism.45,46 In order to rationally design a recoverable catalyst, a model for the active species is necessary. The success or failure of the recoverable catalyst in turn bears upon the accuracy of the model. Whenever (1) induction periods are observed, and (2) activity remains in a phase different from that of the recovered “catalyst”, and (3) the recovered material continues to exhibit induction periods, together with reduced activity, it is highly probable that the immobilized species bears little relationship to the active catalyst. Naturally, there are a variety of related scenarios, such as simultaneous catalysis by an immobilized species and a leached species. Regardless, such “phase tests”45,46 deserve more frequent use in the study of catalyst mechanisms.
Conclusion
This study has established new catalyst systems for the Heck and Suzuki reactions (Tables 2–4) based upon the novel fluorous palladacycle catalyst precursors 7 and 13. From the standpoint of TON values, the Heck reactions rank with the best in the literature. Complexes 7 and 13 represent new examples of thermomorphic fluorous compounds, with little or no solubility in organic solvents at room temperature but significant solubility at elevated temperatures, thus enabling homogeneous reactions in the absence of fluorous solvents.4,5 However, in apparent contrast to closely related non-fluorous palladacycles, they act mainly as steady-state sources of extremely reactive, soluble colloidal palladium nanoparticles. Such species are of great current interest,36–42 but those obtained from 7 and 13 are not amenable to fluorous recycling protocols. Fluorous analogs of existing Heck catalysts that are expected to show greater stabilities with respect to nanoparticles, and function as molecular catalysts, will be described in future reports.44Experimental
General
All reactions were conducted under N2. Chemicals were treated as follows: THF, ether, toluene, hexanes, distilled from Na/benzophenone; Et3N, distilled from Na; CF3C6F11 (Oakwood or ABCR), distilled from P2O5; CF3C6H5 (ABCR), distilled from CaH2; i-PrMgCl (2.0 M in ether, Aldrich), standardized;47 C8F17Br (Fluorochem), (Ph3P)3RhCl, Pd(OAc)2 (2×Strem), PPh3, phenyl iodide (2×Fluka), bromobenzene, p-bromotoluene, p-bromoacetophenone, p-bromoanisole, methyl acrylate, PhB(OH)2, p-iodobenzaldehyde, aliquat 336 (8×Aldrich), styrene (Acros), CDCl3 (Cambridge Isotope or Aldrich) and other solvents, used as received. NMR spectra were recorded on 400 MHz spectrometers at ambient probe temperatures unless noted and referenced to residual internal CHCl3 (1H, δ 7.27) or CDCl3 (13C, δ 77.2). IR spectra were measured on an ASI React-IR spectrometer. GC data were acquired using a ThermoQuest Trace GC 2000 instrument fitted with a capillary column (OPTIMA-5-0.25 μm; 25 m×0.32 mm). HPLC was conducted on a Thermoquest instrument package (pump/autosampler/detector P4000/AS3000/UV6000LP). DSC and TGA data were recorded with a Mettler-Toledo DSC821 instrument and treated by standard methods.30 TEM data were obtained with a Philips CM 300 UT instrument. Elemental analyses were conducted with a Carlo Erba EA1110 instrument (in-house).
p-Rf8CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CHC6H4I (2)
CHC6H4I (2)
A flask was sequentially charged with p-iodobenzaldehyde (1; 0.724 g, 3.12 mmol), Rf8CH2CH2PPh3+I− (3.91 g, 4.68 mmol),24a K2CO3 (0.646 g, 4.68 mmol), p-dioxane (15 mL), and water (0.5 mL), fitted with a condenser, and placed in a 95°C oil bath. The mixture was stirred and monitored by TLC (1∶9 v/v EtOAc/hexanes). After 24 h, reaction was complete. The solvent was removed by rotary evaporation. Water (25 mL) was added to the yellow oil, and the mixture extracted with CH2Cl2 (2×50 mL). The extracts were dried (MgSO4) and the solvent removed by rotary evaporation. The oily residue was chromatographed on a silica gel column (eluent hexane) to give 2 as a colorless oil that solidified upon standing (1.994 g, 3.011 mmol, 96%, 90∶10 Z/E).48 Calcd for C17H8F17I: C, 30.83; H, 1.21. Found: C, 31.08; H, 1.08%.
NMR (CDCl3): 1H 2.97–3.21 (2 overlapping dt, 3JHF=18 Hz, 3JHH=7 Hz, CF2CH2, Z/E), 5.78 (dt, 3JHH=11 Hz, 3JHH=7 Hz, CH2CH, Z), 6.16 (dt, 3JHH=16 Hz, 3JHH=7 Hz, CH2CH, E), 6.55 (d, 3JHH=16 Hz, CHC6H4, E), 6.72 (d, 3JHH=11 Hz, CHC6H4, Z), 6.97 and 7.77 (2d, 3JHH=8 Hz, C6H4, Z), 7.13 and 7.68 (2d, 3JHH=8 Hz, C6H4, E); 13C{1H} (partial, Z) 30.7 (t, 2JCF=22 Hz, CF2CH2), 119.0 (t, 3JCF=5 Hz, CH2CH), 128.4, 130.4, 134.7, 135.5, l38.0 (5s, CHC6H4).
p-Rf8CH2CHCHC6H4CH(OH)(CH2)2Rf8 (3)
A Schlenk flask was charged with 2 (0.795 g, 1.200 mmol) and THF (10 mL). Then i-PrMgCl (2.4 M in ether, 0.60 mL, 1.44 mmol) was added with stirring over the course of 5 min (room temperature). The mixture warmed slightly and turned yellow. After 30 min, a solution of Rf8CH2CH2CHO (0.686 g, 1.44 mmol)28 in THF (10 mL) was added over 10 min. After an additional 2 h, aqueous HCl (0.1 M, 25 mL) was added. The mixture was extracted with ether (2×50 mL). The extracts were dried (MgSO4) and the solvent removed by rotary evaporation. The yellow oil was chromatographed on a silica gel column (1∶9 v/v ether/hexanes) to give 3 as a colorless oil that solidified upon standing (1.131 g, 1.117 mmol, 93%, 90∶10 Z/E).48 Calcd for C28H14F34O: C, 33.22; H, 1.39. Found: C, 33.08; H, 1.28%.
NMR (CDCl3): 1H 1.92 (br s, OH), 1.99–2.38 (m, CH2CH2CF2, E/Z), 3.14–2.98 (2 overlapping dt, 3JHF=18 Hz, 3JHH=7 Hz, CF2CH2CH, Z/E), 4.81 (t, 3JHH=6 Hz, CHOH), 5.78 (dt, 3JHH=11 Hz, 3JHH=7 Hz, CH2CH, Z), 6.16 (dt, 3JHH=16 Hz, 3JHH=7 Hz, CH2CH, E), 6.63 (d, 3JHH=16 Hz, CHC6H4, E), 6.83 (d, 3JHH=11 Hz, CHC6H4, Z), 7.25 and 7.37 (2d, 3JHH=8 Hz, C6H4, Z), 7.33 and 7.41 (2d, 3JHH=8 Hz, C6H4, E); 13C{1H} (partial, Z) 27.5 (t, 2JCF=22 Hz, CH2CH2CF2), 29.5 (br s, CH2CH2CF2), 30.7 (t, 2JCF=22 Hz, CF2CH2CH), 73.1 (s, CHOH), 118.6 (t, 3JCF=5 Hz, CHCH2), 126.1, 129.0, 135.2, 136.0, l43.0 (5s, CHC6H4).
p-Rf8(CH2)3C6H4CH(OH)(CH2)2Rf8 (4)
A Fisher–Porter bottle was charged with 3 (0.770 g, 0.761 mmol), (Ph3P)3RhCl (0.053 g, 0.057 mmol), and CF3C6H5/EtOH (20 mL, 1∶1 v/v). The system was purged with H2 and pressurized to 75 psig. The bottle was placed in a 40°C oil bath and the mixture stirred. After 14 h, the bath was removed, the system vented, and the solvents removed by rotary evaporation. The residue was chromatographed on a silica gel column (1∶9 v/v ether/hexanes) to give 4 as a white solid (0.695 g, 0.685 mmol, 90%), mp 89°C (capillary), 89.8°C (DSC). Calcd for C28H16F34O: C, 33.15; H, 1.59. Found: C, 32.76; H, 1.31%.
NMR (4∶1 v/v CDCl3/CF3C6F5): 1H 1.88 (br s, OH), 1.90–2.39 (m, 2CH2CH2CF2), 2.76 (t, 3JHH=7 Hz, CH2C6H4), 4.77 (t, 3JHH=6 Hz, CHOH), 7.22 and 7.33 (2d, 3JHH=8 Hz, C6H4); 13C{1H} (partial) 22.0 (br s, CF2CH2CH2CH2), 27.6 (t, 2JCF=22 Hz, CH(OH)CH2CH2CF2), 29.6 (br s, CH(OH)CH2CH2CF2), 30.7 (t, 2JCF=22 Hz, CF2CH2CH2CH2), 34.9 (s, CH2C6H4), 73.3 (s, CHOH), 126.3, 129.0, 141.0, l42.0 (4s, C6H4).
p-Rf8(CH2)3C6H4C(O)(CH2)2Rf8 (5)
A Schlenk flask was charged with 4 (0.642 g, 0.632 mmol), the Dess–Martin periodinane (0.322 g, 0.759 mmol),29 and CF3C6H5 (20 mL). The solution was stirred overnight. Ether (50 mL) was added, and the white suspension poured into a solution of Na2SO3 (1.31 g, 5.31 mmol) in saturated aqueous KHCO3 (50 mL). The biphasic mixture was vigorously stirred until the organic phase appeared clear. The organic phase was washed with saturated aqueous KHCO3 (50 mL) and dried (MgSO4). The solvent was removed by rotary evaporation. The residue was chromatographed on a silica gel column (3∶17 v/v CHCl3/hexanes) to give 5 as a white solid (0.608 g, 0.600 mmol, 95%), mp 112–113°C (capillary), 110.9°C (DSC). Calcd for C28H14F34O: C, 33.22; H, 1.39. Found: C, 33.42; H, 1.38%. IR (cm−1, thin film) νCO 1683.
NMR (4∶1 v/v CDCl3/CF3C6F5): 1H 1.96–2.15 (m, CF2CH2CH2CH2), 2.54–2.68 (m, C(O)CH2CH2), 2.80 (t, 3JHH=7 Hz, CH2C6H4), 3.30 (t, 3JHH=7 Hz, C(O)CH2), 7.32 and 7.95 (2d, 3JHH=8 Hz, C6H4); 13C{1H} (partial) 21.7 (br s, CF2CH2CH2CH2), 25.8 (t, 2JCF=22 Hz, C(O)CH2CH2CF2), 29.6 (s, C(O)CH2CH2CF2), 30.5 (t, 2JCF=22 Hz, CF2CH2CH2CH2), 35.2 (s, CF2CH2CH2CH2), 128.8, 129.1, 134.9, l47.3 (4s, C6H4), 196.3 (s, CO).
p-Rf8(CH2)3C6H4C(N(CH2)3Rf8)(CH2)2Rf8 (6)
A flask was charged with 5 (0.522 g, 0.516 mmol), H2NCH2CH2CH2Rf8 (0.492 g, 1.031 mmol),28 SnCl2(H2O)2 (0.017 g, 0.10 mmol), and toluene (50 mL), fitted with a Dean–Stark trap, and placed in a 140°C oil bath. After 14 h, the mixture was cooled and the solvent was removed by oil pump vacuum. The residue was flash chromatographed (silica gel dried for a minimum of 5 days at 120°C) using dry hexanes/THF (3∶1 v/v). The solvent was removed by rotary evaporation to give 6 as a colorless oil that solidified upon standing (0.744 g, 0.506 mmol, 98%, 35∶65 Z/E).49 Calcd for C39H20F51N: C, 31.83; H, 1.37. Found: C, 32.06; H, 1.47%. IR (cm−1, thin film) νCN 1657.
NMR (CDCl3):501H 1.81–2.30 (m, 2CH2CH2CH2CF2, Z/E, NCCH2CH2CF2, Z), 2.49–2.60 (m, NCCH2CH2CF2, E), 2.74 (t, 3JHH=8 Hz, CH2C6H4, E), 2.75 (t, 3JHH=8 Hz, CH2C6H4, Z), 2.82 (t, 3JHH=8 Hz, NCCH2, E), 3.01 (t, 3JHH=8 Hz, NCCH2, Z), 3.30 (t, 3JHH=8 Hz, NCH2, E), 3.61 (t, 3JHH=8 Hz, NCH2, Z), 7.07 and 7.25 (2d, 3JHH=8 Hz, C6H4, E), 7.24 and 7.70 (2d, 3JHH=8 Hz, C6H4, Z); 13C{1H} (partial) 19.3 (s, CH2CN, Z), 21.7 (s, NCH2CH2, Z), 21.8 (s, NCH2CH2, E), 22.1 (br s, CH2CH2C6H4, Z/E), 27.0 (t, 2JCF=22 Hz, NCCH2CH2, E), 28.8 (t, 2JCF=22 Hz, CH2CH2CH2C6H4, Z/E), 29.0 (t, 2JCF=22 Hz, NCCH2CH2, Z), 30.3 (t, 2JCF=22 Hz, NCH2CH2CH2, Z), 30.4 (t, 2JCF=22 Hz, NCH2CH2CH2, E), 31.7 (s, CH2CN, E), 34.8 (s, CH2C6H4, Z), 34.9 (s, CH2C6H4, E), 50.0 (s, CNCH2, Z), 51.4 (s, CNCH2, E), 126.7 (s, C6H4, E), 127.2 (s, C6H4, Z), 129.0 (s, C6H4, Z/E), 136.3 (s, C6H4, E), 136.8 (s, C6H4, Z), 141.7 (s, C6H4, E), 143.3 (s, C6H4, Z), 166.0 (s, CN, Z), 168.8 (s, CN, E).
Imine palladacycle acetate 7
A Schlenk flask was charged with 6 (1.019 g, 0.692 mmol), Pd(OAc)2 (0.155 g, 0.692 mmol), and AcOH (12 mL), and placed in a 95°C oil bath. The mixture was stirred (1.5 h) and cooled to room temperature. The solvent was removed by rotary evaporation, and CF3C6H5 (10 mL) was added. This was poured on top of a short column of silica gel in CF3C6H5 (10 cm×2.5 cm Ø). The column was washed with CF3C6H5 (ca. two times the column volume) and then eluted with CF3C6H5/EtOH (9∶1 v/v). The solvent was removed from the last fraction by rotary evaporation. The residue was dried by oil pump vacuum to give 7 as a dark yellow gum (0.986 g, 0.301 mmol, 87%). A solution of 7 (0.070 g), in CF3C6H5 (2 mL) was layered with toluene (10 mL) and kept at 4°C. This gave yellow needles of 7, mp 78–80°C (capillary), 78.5°C (DSC). Calcd for C82H44F102N2O4Pd2: C, 30.10; H, 1.35. Found: C, 30.15; H, 1.13%. IR (cm−1, thin film) νCN 1587, νCO20c,31 1571, 1424.
NMR (4∶1 v/v CDCl3/CF3C6F5):501H 1.35–1.45 (m, NCCH2CHH′), 1.70–1.81 (m, NCH2CHH′, NCCH2CHH′), 1.87–1.94 (m, CH2CH2C6H3), 2.08–2.25 (m, NCH2CH2CH2, CH2CH2CH2C6H3), 2.17 (s, CH3), 2.40–2.45 (m, NCH2CHH′), 2.50–2.55 (m, NCCHH′), 2.61–2.73 (m, NCCHH′, CH2C6H3), 2.98–3.05 (m, NCHH′), 3.22–3.29 (m, NCHH′), 6.88, 6.92 (2d, 3JHH=8 Hz, C6H3), 6.98 (s, C6H3); 13C{1H} (partial) 18.7 (s, NCCH2), 21.5 (s, NCH2CH2), 22.3 (s, CH2CH2C6H3), 24.0 (s, CH3), 28.8, 31.0 (2t, 2JCF=22 Hz, CH2CH2CH2C6H3, NCH2CH2CH2), 29.4 (t, 2JCF=22 Hz, NCCH2CH2), 35.8 (s, CH2C6H3), 52.2 (s, NCH2), 124.3, 126.2, 128.7, 129.1 132.9, 157.151 (6s, C6H3), 179.9 (s, CN), 181.9 (s, CO).
Imine palladacycle chloride 8
A Schlenk flask was charged with a solution of 7 (0.358 g, 0.109 mmol) in CF3C6H5 (8 mL). Then a solution of LiCl (0.070 g, 2.19 mmol) in CH3OH (5 mL) was added with stirring. After 1 h, solvent was removed from the cloudy mixture by oil pump vacuum. The yellow residue was triturated with CH3OH (10 mL), collected by filtration, washed with CH3OH (3×10 mL) and ether (2×5 mL), and dried by oil pump vacuum. This gave 8 as a beige powder (0.300 g, 0.0093 mmol, 85%), mp 176°C (capillary), 179.0°C (DSC). Calcd for C78H38F102Cl2N2Pd2: C, 29.05; H, 1.19. Found: C, 29.18; H, 1.69%. IR (cm−1, thin film) νCN 1583.
NMR (1∶1 v/v CDCl3/CF3C6F5, 57°C): 1H 1.99–2.57 (m, 2CH2CH2CH2CF2, NCCH2CH2CF2), 2.78, 2.84 (2t, 3JHH=7 Hz, CH2C6H3), 3.12–3.16 (m, NCCH2), 3.84–3.91 (m, NCH2), 7.07–7.23 (m, C6H3); 13C{1H} (partial) 19.6, 22.0, 22.3 (3s, 3CH2CH2CF2), 29.2, 30.3, 31.3 (3t, 2JCF=22 Hz, 3CH2CF2), 36.0 (s, CH2C6H3), 53.0 (s, CH2N), 125.5, 127.3, 134.1, 134.3, 145.1, 156.051 (6s, C6H3), 182.7 (s, CN).
Imine palladacycle iodide 9
Complex 7 (0.240 g, 0.073 mmol), CF3C6H5 (8 mL), LiI (0.200 g, 1.49 mmol), and CH3OH (5 mL) were combined in a procedure analogous to that given for 8. An identical workup gave 9 as a yellow powder (0.215 g, 0.063 mmol, 86%), mp 182°C (capillary), 188.5°C (DSC). Calcd for C78H38F102I2N2Pd2: C, 27.49; H, 1.12. Found: C, 27.54; H, 1.20%. IR (cm−1, thin film) νCN 1583.
NMR (1∶1 v/v CDCl3/CF3C6F5, 57°C): 1H 2.00–2.75 (m, 2CH2CH2CH2CF2, NCCH2CH2CF2), 2.80–2.85 (m, CH2C6H3), 3.15–3.19 (m, NCCH2), 4.15–4.18 (m, NCH2), 7.10–7.16, 7.22–7.24, and 7.71 (m/m/s, C6H3); 13C{1H} (partial) 20.0, 22.0, 22.6 (3s, 3CH2CH2CF2), 28.7, 30.4, 31.3 (3t, 2JCF=22 Hz, 3CH2CF2), 36.0 (s, CH2C6H3), 54.6 (s, CH2N), 125.2, 128.0, 128.2, 139.4, 146.5, 159.751 (6s, C6H3), 183.1 (s, CN).
Imine palladacycle phosphine 10
A. A Schlenk flask was charged with 7 (0.450 g, 0.137 mmol), LiCl (0.058 g, 1.38 mmol), and PPh3 (0.079 g, 0.302 mmol). THF (10 mL) was added with stirring, and 7 dissolved over ca. 0.5 h. After 1 h, the solvent was removed by oil pump vacuum. The yellow powder was extracted with CH2Cl2 (2×10 mL). The extract was filtered under N2 into a new Schlenk flask and taken to dryness by oil pump vacuum. The residue was triturated with hexanes (5 mL, 15–20 min), collected by filtration, washed with hexanes (2×10 mL) and dried by oil pump vacuum. This gave 10 as a beige powder (0.408 g, 0.217 mmol, 79%). B. A Schlenk flask was charged with 8 (0.269 g, 0.083 mmol), PPh3 (0.048 g, 0.18 mmol), and CH2Cl2 (10 mL). The suspension was vigorously stirred. After 1 h, the solvent was removed from the light yellow solution by oil pump vacuum. Hexanes (5 mL) was added to the solid, which was isolated by filtration, washed with hexanes (2×10 mL), and dried by oil pump vacuum to give 10 as a beige powder (0.254 g, 0.135 mmol, 81%), mp 135°C (capillary), 138.4°C (DSC). Ti, 228.4°C (TGA). Calcd for C57H34ClF51NPPd: C, 36.52; H, 1.82. Found: C, 36.61; H, 1.89%. IR (cm−1, thin film) νCN 1586.
NMR (CDCl3):501H 1.17–1.25 (m, CH2CH2C6H3), 1.63–1.74 (m, CH2CH2CH2C6H3), 2.03 (t, 3JHH=8 Hz, CH2C6H3), 2.10–2.18 (m, NCH2CH2), 2.23–2.36 (m, NCH2CH2CH2), 2.40–2.49 (m, NCCH2CH2), 3.06–3.10 (m, NCCH2), 4.20 (br s, NCH2), 6.35 (d, 3JPH=5 Hz, 1H of C6H3), 6.77 and 7.14 (2d, 3JHH=8 Hz, 2H of C6H3), 7.36–7.47 (m, 9H of P(C6H5)3), 7.72–7.77 (m, 6H of P(C6H5)3); 13C{1H} (partial) 19.4 (s, NCCH2), 21.3 (s, CH2CH2C6H3), 22.1 (s, NCH2CH2), 28.3 (t, 2JCF=22 Hz, NCH2CH2CH2), 29.4 (t, 2JCF=22 Hz, NCCH2CH2), 30.3 (t, 2JCF=22 Hz, CH2CH2CH2C6H3), 35.1 (s, CH2C6H3), 51.0 (s, NCH2), 124.4, 127.0 (2s, 2C of C6H3), 128.4 (d, 2JPC=11 Hz, m-PPh), 131.1 (s, p-PPh), 131.2 (d, 1JPC=50 Hz, i-PPh), 135.6 (d, 3JPC=13 Hz, o-PPh), 139.5 (d, 3JPC=11 Hz, 1C of C6H3), 143.8, 146.5, 159.951 (3s, 3C of C6H3), 181.6 (s, CN); 31P{1H} 42.9 (s, PPh3).
Rf8CH2CH2CH2SH (11)33
A round bottomed flask was charged with Rf8CH2CH2CH2I (5.02 g, 8.53 mmol),11a thiourea (0.973 g, 12.8 mmol), phase transfer catalyst aliquat 336 (0.221 g, 0.546 mmol), and water (15 mL), fitted with a condenser, and placed in a 100°C bath. The mixture was stirred and a precipitate slowly formed. After 20 h, aqueous KOH (50 mL, 0.1 M) was added. After 0.5 h, the mixture was allowed to cool to room temperature. Concentrated HCl was added until the sample was acidic. The mixture was extracted with CH2Cl2 (2×50 mL). The extract was dried (MgSO4), and the solvent was removed by rotary evaporation. The light brown oil was chromatographed (silica gel column, hexanes) to give 11 as a clear oil (3.30 g, 6.68 mmol, 78%). Calcd for C11H7F17S: C, 26.73; H, 1.42. Found: C, 26.83; H, 1.42%.
NMR (CDCl3): 1H 1.36 (t, 3JHH=8 Hz, SH), 1.87–1.95 (m, CF2CH2CH2CH2), 2.14–2.27 (m, CF2CH2CH2CH2), 2.61 (dt,3JHH=8 Hz, 3JHH=7 Hz, CF2CH2CH2CH2); 13C{1H} (partial) 23.8 (s, CF2CH2CH2CH2), 24.6 (s, CF2CH2CH2CH2), 29.5 (t, 2JCF=22 Hz, CF2CH2CH2CH2).
p-Rf8(CH2)3C6H4CH(S(CH2)3Rf8)(CH2)2Rf8 (12)
A Schlenk flask was charged with 4 (1.00 g, 0.987 mmol), 11 (0.763 g, 1.54 mmol), ZnI2 (0.315 g, 0.986 mmol), and CF3C6H5 (15 mL), and placed in a 60°C bath. The solution was stirred. After 16 h, water (50 mL) was added. The mixture was extracted with ether (2×50 mL). The extract was dried (MgSO4), and the solvents were removed by rotary evaporation. The residue was placed on the top of a silica gel column packed with hexanes. The column was eluted with hexanes (removing excess 11) and then hexanes/ether (20∶1 v/v). The solvents were removed from the latter fractions by oil pump vacuum to give 12 as a white solid (0.929 g, 0.632 mmol, 64%), mp 48°C (capillary), 47.8°C (DSC). Calcd for C39H21F51S: C, 31.42; H, 1.42. Found: C, 31.42; H, 1.34%.
NMR (CDCl3): 1H 1.68–1.72 (m, CHCH2CH2CF2), 1.89–2.19 (m, CF2CH2CH2CH2, SCH2CH2CH2, SCHCH2CH2CF2), 2.32 and 2.42 (2 quint, 2JHH=13 Hz, 3JHH=7 Hz, SCHH′CH2CH2 and SCHH′CH2CH2), 2.69 (t, 3JHH=7 Hz, CF2CH2CH2CH2), 3.74 (t, 3JHH=7 Hz, CHCH2CH2CF2), 7.14 and 7.22 (2d, 3JHH=8 Hz, C6H4); 13C{1H} (partial) 19.9 (s, CHCH2CH2CF2), 21.6, 27.0 (2s, CF2CH2CH2CH2, SCH2CH2CH2), 29.1, 29.4, 30.1 (3t, 2JCF=22 Hz, CF2CH2), 30.2 (s, SCH2CH2CH2), 34.5 (s, CF2CH2CH2CH2), 48.6 (s, CHCH2CH2CF2), 127.7, 128.8, 139.0, l40.3 (4s, C6H4).
Thioether palladacycle acetate 13
Complex 12 (0.740 g, 0.496 mmol), Pd(OAc)2 (0.111 g, 0.496 mmol), and AcOH (15 mL) were reacted in a procedure analogous to that given for 7 (2 h). A similar workup (15 cm×2.5 cm Ø column) gave 13 as a dark yellow gum (0.695 g, 0.210 mmol, 84%). Calcd for C82H46F102S2O4Pd2: C, 29.75; H, 1.40; S, 1.94. Found: C, 30.54; H, 1.55; S, 1.80%. IR (cm−1, thin film) νCO20c,31 1567, 1424.
NMR (4∶1 v/v CDCl3/CF3C6F5; see text regarding isomers): 1H 1.28–2.66 (m, 14H), 2.12 (br s, CH3) 2.85–4.50 (m, CHSCHH′), 6.66–7.20 (m, C6H3); 13C{1H} (partial) 14.1, 20.3, 22.1, 24.2 (4br s, CH2), 29.6–30.9 (br t, 3CH2CF2), 30.1, 30.4 (2s, CH3, SCH2), 35.2 (br s, CH2C6H3), 58.3 (br s, SCH2), 181.2 (br s, CO).
Partition coefficients (Table 1)
The following are representative. A. A 10 mL vial was charged with 6 (0.0236 g, 0.0160 mmol), CF3C6F11 (2.000 mL), and toluene (2.000 mL), fitted with a mininert valve, and vigorously shaken (2 min). After 12 h (24°C), a 0.400 mL aliquot of each layer was added to a hexane solution of eicosane (2.000 mL, 0.0273 M). GC analysis (average of 7–8 injections) showed 0.00304 mmol of 6 in the CF3C6F11 aliquot and 0.000040 mmol in the toluene aliquot (98.7∶1.3; a 2.000/0.400 scale factor gives a mass balance of 0.0226 g, 96%). B. A 10 mL vial was charged with 7 (0.0104 g, 0.0031 mmol), CF3C6F11 (2.000 mL), and toluene (2.000 mL), fitted with a mininert valve, and vigorously shaken (2 min). After 2 h (24°C), a 0.250 mL aliquot of the fluorous phase and a 0.750 mL aliquot of the non-fluorous phase were removed. The solvents were evaporated and the residues dried by oil pump vacuum (1 h). Each residue was taken up in CF3C6H5/EtOH (9∶1 v/v; 0.500 mL) and analyzed by HPLC (average of 5 injections, 200×4 mm Nucleosil 100-5 column, UV/visible detector). The relative peak intensities were (after normalization to the aliquot volumes) 95.5∶4.5.
Heck and Suzuki reactions
The following are representative. Table 2: A Schlenk tube was sequentially charged with DMF (6 mL), phenyl iodide (0.560 mL, 5.02 mmol), methyl acrylate (0.565 mL, 6.28 mmol), Et3N (1.400 mL, 10.05 mmol), and a solution of 7 in CF3C6H5 (0.000229 M; 0.015 mL, 3.44×10−6 mmol), fitted with a condenser, and placed in a 140°C oil bath. The solution was vigorously stirred (14 h), removed from the bath to cool, and diluted with ether to 25.00 mL. An aliquot (0.500 mL) was added to a toluene solution of tridecane (0.250 mL, 0.100 M). GC analysis showed only trans-methyl cinnamate (100%, TON 1460000). The aliquot was recombined with the mother solution. A standard basic aqueous workup and flash chromatography on silica gel (9∶1 v/v hexanes/EtOAc) gave trans-methyl cinnamate in 98% yield (0.797 g, 4.914 mmol). Table 3: A Schlenk tube was sequentially charged with 7 (0.0014 g, 0.00043 mmol), DMF (4 mL), phenyl iodide (0.250 mL, 2.24 mmol), methyl acrylate (0.250 mL, 2.78 mmol), and Et3N (0.625 mL, 4.48 mmol), and placed in a 100°C oil bath. The solution was vigorously stirred (2 h), and removed from the bath to cool (0.5 h). Then C8F17Br (1 mL) was added to give a biphasic system. The upper DMF layer was removed by syringe, and the C8F17Br phase extracted with DMF (2 mL). The combined DMF extracts were diluted with ether to 20.00 mL. An aliquot (0.5 mL) was added to a toluene solution of tridecane (0.250 mL, 0.100 M). GC analysis showed only trans-methyl cinnamate (100%; TON 5100 (rounded digits included)). The C8F17Br phase was taken to dryness by oil pump vacuum (0.5 h). The tube was recharged with identical quantities of DMF and all reactants except 7. A second cycle was analogously conducted. Table 4: A Schlenk tube was sequentially charged with PhB(OH)2 (1.455 g, 7.534 mmol), K3PO4 (2.132 g, 10.05 mmol), toluene (13 mL), phenyl iodide (0.560 mL, 5.023 mmol), and a solution of 7 in CF3C6H5 (0.000229 M; 0.020 mL, 4.58×10−6 mmol), fitted with a condenser, and placed in a 130°C oil bath. The suspension was vigorously stirred (38 h), removed from the bath to cool, and diluted with ether to 25.00 mL. An aliquot (0.5 mL) was added to a solution of tridecane (0.250 mL, 0.100 M). GC analysis showed the partial consumption of phenyl iodide (65%) and formation of biphenyl (40%, TON 440000).
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG; GL 301/3-1) and Johnson Matthey PMC (palladium loan) for support, and Dr. Gerhard Frank (Lehrstuhl für Mikrocharakterisierung, Universität Erlangen-Nürnberg) for assistance with the TEM measurements. This work was conducted as part of a Ph.D. thesis defended in the Department of Chemistry at the University of Utah.References
- (a) I. T. Horváth, Acc. Chem. Res., 1998, 31, 641 CrossRef CAS; (b) E. de Wolf, G. van Koten and B.-J. Deelman, Chem. Soc. Rev., 1999, 28, 37 RSC; (c) M. Cavazzini, F. Montanari, G. Pozzi and S. Quici, J. Fluorine Chem., 1999, 94, 183 CrossRef CAS; (d) E. G. Hope and A. M. Stuart, J. Fluorine Chem., 1999, 100, 75 CrossRef CAS; (e) J. Yoshida and K. Itami, Chem. Rev., 2002, 102, 3693 CrossRef CAS.
- Survey of practical considerations and underlying physical principles: L. P. Barthel-Rosa and J. A. Gladysz, Coord. Chem. Rev., 1999, 190–192, 587 Search PubMed.
- J. A. Gladysz and D. P. Curran, Tetrahedron, 2002, 58, 3823 CrossRef CAS and following papers in this special issue devoted to “fluorous chemistry”.
- M. Wende, R. Meier and J. A. Gladysz, J. Am. Chem. Soc., 2001, 123, 11490 CrossRef CAS.
- (a) K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 9, 1371 CrossRef; (b) J. Xiang, A. Orita and J. Otera, Adv. Synth. Catal., 2002, 344, 84 Search PubMed.
- D. E. Bergbreiter, P. L. Osburn, A. Wilson and E. M. Sink, J. Am. Chem. Soc., 2000, 122, 9058 CrossRef CAS.
- (a) I. T. Horváth, G. Kiss, R. A. Cook, J. E. Bond, P. A. Stevens, J. Rábai and E. J. Mozeleski, J. Am. Chem. Soc., 1998, 120, 3133 CrossRef CAS; (b) D. F. Foster, D. Gudmunsen, D. J. Adams, A. M. Stuart, E. G. Hope, D. J. Cole-Hamilton, G. P. Schwarz and P. Pogorzelec, Tetrahedron, 2002, 58, 3901 CrossRef CAS.
- (a) D. Rutherford, J. J. J. Juliette, C. Rocaboy, I. T. Horváth and J. A. Gladysz, Catal. Today, 1998, 42, 381 CrossRef CAS; (b) B. Richter, A. L. Spek, G. van Koten and B.-J. Deelman, J. Am. Chem. Soc., 2000, 122, 3945 CrossRef CAS; (c) E. de Wolf, A. L. Spek, B. W. M. Kuipers, A. P. Philipse, J. D. Meeldijk, P. H. H. Bomans, P. M. Frederik, B.-J. Deelman and G. van Koten, Tetrahedron, 2002, 58, 3911 CrossRef CAS.
- J. J. J. Juliette, D. Rutherford, I. T. Horváth and J. A. Gladysz, J. Am. Chem. Soc., 1999, 121, 2696 CrossRef CAS.
- (a) L. V. Dinh and J. A. Gladysz, Tetrahedron Lett., 1999, 40, 8995 CrossRef CAS; (b) E. de Wolf, E. A. Speets, B.-J. Deelman and G. van Koten, Organometallics, 2001, 20, 3686 CrossRef CAS.
- (a) J.-M. Vincent, A. Rabion, V. K. Yachandra and R. H. Fish, Angew. Chem., Int. Ed. Engl., 1997, 36, 2346 CrossRef CAS; J.-M. Vincent, A. Rabion, V. K. Yachandra and R. H. Fish, Angew. Chem., 1997, 109, 2438; (b) S. Quici, M. Cavazzini, S. Ceragioli, F. Montanari and G. Pozzi, Tetrahedron Lett., 1999, 40, 3647 CrossRef; (c) G. Pozzi, M. Cavazzini, F. Cinato, F. Montanari and S. Quici, Eur. J. Org. Chem., 1999, 1947 CrossRef CAS; (d) T. Nishimura, Y. Maeda, N. Kakiuchi and S. Uemura, J. Chem. Soc., Perkin Trans. 1, 2000, 4301 RSC; (e) S. Colonna, N. Gaggero, F. Montanari, G. Pozzi and S. Quici, Eur. J. Org. Chem., 2001, 181 CrossRef CAS; (f) G. Ragagnin, B. Betzemeier, S. Quici and P. Knochel, Tetrahedron, 2002, 58, 3985 CrossRef CAS.
- Heck reactions: (a) J. Moineau, G. Pozzi, S. Quici and D. Sinou, Tetrahedron Lett., 1999, 40, 7683 CrossRef CAS; (b) L. Xu, W. Chen, J. F. Bickley, A. Steiner and J. Xiao, J. Organomet. Chem., 2000, 598, 409 CrossRef CAS; (c) Y. Nakamura, S. Takeuchi, S. Zhang, K. Okumura and Y. Ohgo, Tetrahedron Lett., 2002, 43, 3053 CrossRef CAS.
- Suzuki reactions: (a) S. Schneider and W. Bannwarth, Helv. Chim. Acta, 2001, 84, 735 CrossRef CAS; (b) C. Rocaboy and J. A. Gladysz, Tetrahedron, 2002, 58, 4007 CrossRef CAS.
- Stille, Sonogashira, and other reactions: (a) S. Schneider and W. Bannwarth, Angew. Chem., Int. Ed., 2000, 39, 4142 CrossRef CAS; S. Schneider and W. Bannwarth, Angew. Chem., 2000, 112, 4293 CrossRef; (b) C. Markert and W. Bannwarth, Helv. Chim. Acta, 2002, 6, 1877 CrossRef; (c) B. Betzemeier and P. Knochel, Angew. Chem., Int. Ed. Engl., 1997, 36, 2623 CrossRef CAS; B. Betzemeier and P. Knochel, Angew. Chem., 1997, 109, 2736; (d) S. Saito, Y. Chounan, T. Nogami, O. Ohmori and Y. Yamamoto, Chem. Lett., 2001, 444 CrossRef CAS.
- (a) Y. Tian, Q. C. Yang, T. C. W. Mak and K. S. Chan, Tetrahedron, 2002, 58, 3951 CrossRef CAS; (b) Y. Nakamura, S. Takeuchi, K. Okumura, Y. Ohgo and D. P. Curran, Tetrahedron, 2002, 58, 3963 CrossRef CAS; (c) see also H. Kleijn, E. Rijnberg, J. T. B. H. Jastrezebski and G. van Koten, Org. Lett., 1999, 1, 853 Search PubMed.
- (a) M. Cavazzini, S. Quici and G. Pozzi, Tetrahedron, 2002, 58, 3943 CrossRef CAS; (b) A. Endres and G. Maas, Tetrahedron, 2002, 58, 3999 CrossRef CAS; (c) K. Mikami, Y. Mikami, H. Matsuzawa, Y. Matsumoto, J. Nishikido, F. Yamamoto and H. Nakajima, Tetrahedron, 2002, 58, 4015 CrossRef CAS; (d) Q. Zhang, Z. Luo and D. P. Curran, J. Org. Chem., 2000, 65, 8866 CrossRef CAS; (e) R. Kling, D. Sinou, G. Pozzi, A. Choplin, F. Quignard, S. Busch, S. Kainz, D. Koch and W. Leitner, Tetrahedron Lett., 1998, 39, 9439 CrossRef CAS.
- (a) J. Dupont, M. Pfeffer and J. Spencer, Eur. J. Inorg. Chem., 2001, 1917 CrossRef CAS; (b) W. A. Herrmann, V. P. W. Böhm and C.-P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS.
- (a) A. Zapf and M. Beller, Chem. Eur. J., 2001, 7, 2908 CrossRef CAS; (b) V. P. W. Böhm and W. A. Herrmann, Chem. Eur. J., 2001, 7, 4191 CrossRef CAS.
- M. Ohff, A. Ohff and D. Milstein, Chem. Commun., 1999, 357 RSC.
- (a) A. S. Gruber, D. Zim, G. Ebeling, A. L. Monteiro and J. Dupont, Org. Lett., 2000, 2, 1287 CrossRef CAS; (b) D. Zim, A. S. Gruber, G. Ebeling, J. Dupont and A. L. Monteiro, Org. Lett., 2000, 2, 2881 CrossRef CAS; (c) J. Dupont, A. S. Gruber, G. S. Fonseca, A. L. Monteiro and G. Ebeling, Organometallics, 2001, 20, 171 CrossRef CAS.
- (a) D. G. Blackmond, T. Rosner and A. Pfaltz, Org. Proc. Res. Dev., 1999, 3, 275 Search PubMed; (b) additional N-donor palladacycles that are effective catalyst precursors: I. P. Beletskaya, A. N. Kashin, N. B. Karlstedt, A. V. Mitin, A. V. Cheprakov and G. M. Kazankov, J. Organomet. Chem., 2001, 622, 89 Search PubMed.
- Recent reviews: (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS; (b) N. J. Whitcombe, K. K. Hii and S. E. Gibson, Tetrahedron, 2001, 57, 7449 CrossRef CAS.
- Recent reviews: (a) A. Suzuki, J. Organomet. Chem., 2002, 653, 83 CrossRef CAS; (b) N. Miyaura, J. Organomet. Chem., 2002, 653, 54 CrossRef CAS.
- (a) C. Rocaboy, D. Rutherford, B. L. Bennett and J. A. Gladysz, J. Phys. Org. Chem., 2000, 13, 596 CrossRef CAS; (b) C. Rocaboy, F. Hampel and J. A. Gladysz, J. Org. Chem., 2002, 67, 6863 CrossRef CAS.
- C. Rocaboy and J. A. Gladysz, Org. Lett., 2002, 4, 1993 CrossRef CAS.
- T. Soós, B. L. Bennett, D. Rutherford, L. P. Barthel-Rosa and J. A. Gladysz, Organometallics, 2001, 20, 3079 CrossRef CAS.
- M. Rottländer, L. Boymond, L. Bérillon, A. Leprête, G. Varchi, S. Avolio, H. Laaziri, G. Quéguiner, A. Ricci, G. Cahiez and P. Knochel, Chem. Eur. J., 2000, 6, 767 CrossRef CAS.
- C. Rocaboy, W. Bauer and J. A. Gladysz, Eur. J. Org. Chem., 2000, 2621 CrossRef CAS.
- (a) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS; (b) M. Frigerio, M. Santagostino and S. Sputore, J. Org. Chem., 1999, 64, 4537 CrossRef CAS.
- H. K. Cammenga and M. Epple, Angew. Chem., Int. Ed. Engl., 1995, 34, 1171 CrossRef CAS; H. K. Cammenga and M. Epple, Angew. Chem., 1995, 107, 1284 CrossRef.
- H. Onoue and I. Moritani, J. Organomet. Chem., 1972, 43, 431 CrossRef.
- Y. Guindon, R. Frenette, R. Fortin and J. Rokach, J. Org. Chem., 1983, 48, 1357 CrossRef CAS.
- (a) C. Naud, P. Calas, H. Blancou and A. Commeyras, J. Fluorine Chem., 2000, 104, 173 CrossRef CAS; (b) N. Mureau, F. Guittard and S. Géribaldi, Tetrahedron Lett., 2000, 41, 2885 CrossRef CAS.
- F. Szonyi and A. Cambon, J. Fluorine Chem., 1989, 42, 59 CrossRef CAS.
- J. A. Gladysz, Pure Appl. Chem., 2001, 73, 1319 Search PubMed.
- (a) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef CAS; M. T. Reetz and E. Westermann, Angew. Chem., 2000, 112, 170 CrossRef , and earlier papers referenced therein; (b) J. Le Bars, U. Specht, J. S. Bradley and D. G. Blackmond, Langmuir, 1999, 15, 7621 CrossRef.
- (a) V. Chechik and R. M. Crooks, J. Am. Chem. Soc., 2000, 122, 1243 CrossRef CAS; (b) L. K. Yeung and R. M. Crooks, Nano Lett., 2001, 1, 14 CrossRef CAS; (c) R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181 CrossRef CAS.
- (a) M. Moreno-Mañas, R. Pleixats and S. Villarroya, Organometallics, 2001, 20, 4524 CrossRef CAS; (b) see also M. Moreno-Mañas, R. Pleixats and S. Villarroya, Chem. Commun., 2002, 60 Search PubMed.
- M. Nowotny, U. Hanefeld, H. van Koningsveld and T. Maschmeyer, Chem. Commun., 2000, 1877 RSC.
- (a) K. Köhler, R. G. Heidenreich, J. G. E. Krauter and J. Pietsch, Chem. Eur. J., 2002, 8, 622 CrossRef CAS; (b) F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, Chem. Eur. J., 2000, 6, 843 CrossRef CAS.
- Related data for the Suzuki reaction: (a) R. B. Bedford, C. S. J. Cazin, M. B. Hursthouse, M. E. Light, K. J. Pike and S. Wimperis, J. Organomet. Chem., 2001, 633, 173 CrossRef CAS; (b) S.-W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS; (c) C. Ramararo, S. V. Ley, S. C. Smith, I. M. Shirley and N. DeAlmeida, Chem. Commun., 2002, 1132 RSC.
- For incisive overviews of nanoparticle catalysts, see: (a) S. Özkar and R. G. Finke, J. Am. Chem. Soc., 2002, 124, 5796 CrossRef; (b) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS.
- (a) T. Rosner, J. Le Bars, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 1848 CrossRef CAS; (b) T. Rosner, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 4621 CrossRef CAS.
- L. Dinh and R. C. Costa, research in progress, Universität Erlangen-Nürnberg.
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Rieder, J. Am. Chem. Soc., 2001, 123, 10139 CAS.
- J. Rebek, Tetrahedron, 1979, 35, 723 CrossRef CAS.
- S. C. Watson and J. F. Eastham, J. Organomet. Chem., 1967, 9, 165 CrossRef CAS.
- The Z/E HCCH assignments were based upon well established trends in the 3JHH values (Z, ca. 11 Hz; E, ca. 16 Hz).
- The Z/E CN assignment is tentative.
- These NMR assignments were confirmed by COSY and HETCOR experiments.
- This signal is assigned to the palladated aryl carbon.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |