Monocyclopentadienyl complexes of niobium, tantalum and tungsten containing heterodifunctional P,O ligands†
Xavier
Morise
*a,
Malcolm L. H.
Green
*b,
Pierre
Braunstein
*a,
Leigh H.
Rees
b and
Ino C.
Vei
b
aLaboratoire de Chimie de Coordination, UMR CNRS 7513, Université Louis Pasteur, 4, rue Blaise Pascal, 67070, Strasbourg Cedex, France. E-mail: morise@chimie.u-strasbg.fr; braunst@chimie.u-strasbg.fr.
bInorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK OX1 3QR. E-mail: Malcolm.Green@chemistry.oxford.ac.uk.
First published on 13th September 2002
Abstract
The reactions of P,O type ligands with the half-sandwich complexes [(η-C5R5)MCl4]
(R5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =H5, Me5, iPrH4; M=Nb, Ta, W) have been investigated. Monodentate P-adducts were obtained with the β-amidophosphine Ph2PCH2C(O)NPh2, whereas in the case of the keto ligand Ph2PCH2C(O)Ph a spontaneous HCl elimination occurred to give direct access to the corresponding phosphinoenolate complexes. The crystal structures of [(η-C5H5)Nb
=H5, Me5, iPrH4; M=Nb, Ta, W) have been investigated. Monodentate P-adducts were obtained with the β-amidophosphine Ph2PCH2C(O)NPh2, whereas in the case of the keto ligand Ph2PCH2C(O)Ph a spontaneous HCl elimination occurred to give direct access to the corresponding phosphinoenolate complexes. The crystal structures of [(η-C5H5)Nb![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) Cl3{PPh2CH
Cl3{PPh2CH![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) C(O
C(O![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) )Ph}], [(η-C5H5)TaCl3{PPh2CHC(O)Ph}] and [(η-C5Me5)TaCl3{PPh2CHC(O)Ph}] have been determined. Interestingly, the acetamido derived phosphine Ph2PNHC(O)Me afforded O-adducts, which is an unusual bonding mode for a P,O ligand.
)Ph}], [(η-C5H5)TaCl3{PPh2CHC(O)Ph}] and [(η-C5Me5)TaCl3{PPh2CHC(O)Ph}] have been determined. Interestingly, the acetamido derived phosphine Ph2PNHC(O)Me afforded O-adducts, which is an unusual bonding mode for a P,O ligand.
Introduction
Bifunctional P,O ligands, which combine a soft phosphine moiety and a hard oxygen function, such as ester, amide or ketone, remain the subject of numerous studies, owing to the various properties and/or applications of their transition-metal complexes.1–4 In metal complexes with these P,O molecules both P-monocoordination and P,O chelation are commonly found. The P,O chelate ligands give stable metal complexes and due to the different properties of the P and O ligand atoms they can strongly influence the stereoelectronic properties at the metal centre.3 Furthermore, in late transition metal complexes, the greater lability of the O-ligand atom favours fluxional processes resulting from reversible O-atom dissociation and recoordination.1–4 The O-atom dissociation in these hemilabile ligands creates a vacant coordination site, and conversely weak O-association can give rise to stabilisation of an otherwise coordinatively unsaturated species. For this reason complexes with P,O ligands have been studied for their catalytic properties,5–13 especially in reactions involving the activation of small molecules.14–19In contrast to the wealth of late transition metal complexes synthesized, which contain a β-keto-, ester- or amidophosphine ligand, there have been only a few studies on the early transition-metal chemistry of these ligands and they have mainly focused on molybdenum. Thus, the half-sandwich complexes of the β-amidophosphine Ph2PCH2C(O)NPh21, [(η-C5H5)MoCl2{1-κ2P,O}] or [(η-C5Me5)MoCl2{1-κ2P,O}][BF4],20 and of the keto-functionalised N-pyrrolylphosphine Ph2PNHC4H3{C(O)Me}-2 (PpyrO), [(η-C5H5)MoCl(CO)2{(PpyrO)-κP}] and [(η-C5H5)Mo(CO)2{(PpyrO)-κ2P,O}][BF4],21 have been reported. More recently, we have described the synthesis of η-arene-molybdenum complexes, such as [(η-C6H5R)Mo(η-C3H5){Ph2PXHC(O)R′-κ2P,O}][PF6]
(R=H, Me; R′=Ph, NPh2, Me; X=N, C).22,23 In these complexes, the P,O ligands act either as P-monodentate phosphine or as a P,O-chelate, whereas O-monocoordination, which may have been anticipated owing to the relatively hard nature of molybdenum vs. late transition metals, was not observed. Although ligands with carbonyl functions (ketones,24–26 amides,27,28 acids/esters29,30) are known to coordinate readily to groups 5 and 6 metals, there has been no example reported, to the best of our knowledge, when a phosphine donor is also present in the ligand. This also applies to somewhat related ligands, such as R2PNHP(O)R2,31 or even when the P atom is not available for coordination as in R3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NC(O)R′.32,33 We will present here a rare example of a P,O phosphine acting as a O-monodentate ligand in half-sandwich Nb, Ta and W complexes.
NC(O)R′.32,33 We will present here a rare example of a P,O phosphine acting as a O-monodentate ligand in half-sandwich Nb, Ta and W complexes.
Anionic phosphino-enolates, formed from ketophosphines, are also a well-developed group of P,O ligands.34–49 Interest in this chemistry partly stems from the fact that nickel phosphino-enolate complexes catalyze the oligomerisation of ethylene in α-olefins (Shell Higher Olefin Process).34,40,50,51 Substitutional modification of the phosphino-enolate ligands can modify catalyst activity and the chain length distribution.49–52 Numerous phosphino-enolate complexes of group 8–10 metals have been synthesized and their reactivity studied.35–39,41–46,49,53–58 For example, Rh(I) complexes in this class are efficient catalysts for the transfer dehydrogenation of alkanes to alkenes.59
As for their neutral counterparts, the early transition metal chemitry of anionic P,O ligands has been little investigated. The η-arene-molybdenum complexes [(η-C6H5R)Mo(η-C3H5){Ph2PXC(O)Ph-κ2P,O}] or [(η-C6H5R)Mo{Ph2PXC(O)Ph-κ2P,O}2] (R=H, Me; R′=Ph, NPh2, Me; X=N, C) represent, to the best of our knowledge, rare examples of early transition metal complexes, containing a chelating phosphinoenolate ligand.22,23 Very recently, Edwards et al. reported the compound [(η-C5H5)2Ti{OC(O)CH2PPh2}2], prepared by treatment of [(η-C5H5)2TiCl2] with the anion Ph2PCH2C(O)O−.60 The titanocene and zirconocene complexes of a camphor-derived phosphino-enolate have also been described.61 Further zirconocene phosphino-enolates derived from the α-ketophosphine Ph2PC(O)R (R=Me, Et) have been prepared by Floriani et al.62,63
Here, we report the first investigations into the coordination chemistry of P,O ligands, namely Ph2PCH2C(O)NPh21, Ph2PCH2C(O)Ph 2 and Ph2PNHC(O)Me 3, with the half-metallocene complexes [(η-C5R5)MCl4]
4
(a: R5=H5, M=Nb; b: R5=H5, M=Ta; c: R5=Me5, M=Ta; d: R5=iPrH4, M=W). The latter were chosen because they represent versatile precursors for new materials, the design of highly efficient catalysts for organic transformations and polymerization reactions64–68 or for W![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) W triply bonded complexes (4d),69 which have a remarkably rich chemistry.70 We will show that under similar reaction conditions, the P,O ligands 1–3 lead to complexes in which they adopt either a P-monodentate, an anionic P,O-chelating or, quite unexpectedly, a neutral O-monodentate coordination mode.
W triply bonded complexes (4d),69 which have a remarkably rich chemistry.70 We will show that under similar reaction conditions, the P,O ligands 1–3 lead to complexes in which they adopt either a P-monodentate, an anionic P,O-chelating or, quite unexpectedly, a neutral O-monodentate coordination mode.
Results and discussion
Half-sandwich complexes [(η-C5R5)MCl4] 4a–d have Lewis acid character and form mono-adducts [(η-C5R5)MCl4(L)] with neutral ligands such as L=amines, phosphines or phosphites.71 Early studies led to the conclusion that adducts of 4a, [(η-C5H5)NbCl4(L)] where L=a tertiary phosphine or phosphite, were labile in contrast to their tantalum analogues.72–74 The niobium adducts [(η-C5H4Me)NbCl4(L)]
(L=PMe3, PEt3 or P(OMe)3) slowly decompose in solution.75 Recently, Poli et al. showed that adducts of 4a with PMenPh3−n
(n=3, 2 or 1) could be isolated provided that no excess of the phosphine, which may cause reduction side-products, was present.76 The adducts of 4c, [(η-C5H4iPr)WCl4(L)] where L=vinylPH2, allylPH2 or PMe3, are stable.77,78
Formation of P-monoadducts
Treatment of [(η-C5H5)NbCl4] 4a with one mol. equiv. of 1 gave the P-monoadduct [(η-C5H5)NbCl4{PPh2CH2C(O)NPh2}] 5a as a brown solid. The proposed structure is supported by the presence, in the 31P{1H} NMR spectrum (CD2Cl2), of a very broad peak at ca.δ 33.1 (w1/2>2500 Hz), owing to the quadrupolar broadening due to Nb (spin 9/2), and by the occurrence of a νCO vibration at 1658 cm−1 in the IR spectrum, which indicates non-coordination of the amide moiety (free ligand: 1657 cm−1).42 In the 1H NMR spectrum (CD2Cl2), the η-C5H5 and PCH2 protons are observed at δ 6.89 (singlet) and 3.88 (doublet, 2J(P,H)=7 Hz), respectively. The absence of coupling between the 31P nucleus and the Cp protons was also noted for [(η-C5H5)NbCl4(PMenPh3−n)]
(n=3, 2 or 1).76 Complexes [(η-C5H5)TaCl4{PPh2CH2C(O)NPh2}]
5b and [(η-C5H4iPr)WCl4{PPh2CH2C(O)NPh2}]
5c were prepared from 4b,c and 1. The spectroscopic data of the tantalum complex 5b mirror those of 5a. The paramagnetic tungsten complex 5c was characterized by IR spectroscopy (νCO 1662 cm−1), mass spectroscopy (m/z: 793 [M−Cl]) and elemental analysis. Complexes 5a–c were stable in solution for several hours and in the solid state for a few days, in the absence of oxygen. Contrary to the situation reported for the Mo complexes mentioned in the introduction,20,22,23 there was no observed tendency for chelation for 1 in complexes 5a–c.
Formation of P,O-enolate complexes
Surprisingly, attempts to form the P-monodentate analogues of 5a–c from [(η-C5H5)NbCl4] 4a and the β-ketophosphine Ph2PCH2C(O)Ph 2 failed. Instead, the reaction followed a different pathway, which resulted in the formation of a deep red solution from which HCl fumes emanated (the acidic nature of the gas was confirmed with pH-paper). After work up, brick-red [(η-C5H5)NbCl3{PPh2CHC(O)Ph}] 6a was isolated (Scheme 1). The 1H NMR spectrum of 6a contains a sharp singlet at δ 6.95 (Cp) and a doublet at δ 6.25 [J(P,H)=3.5 Hz], which is ascribed to the enolate proton. There were also signals arising from the aromatic hydrogens. The 31P{1H} NMR spectrum (CD2Cl2) shows a very broad hump at ca.δ 36.5, as a result of the P–Nb coupling (w1/2>2500 Hz). In the IR spectrum, a νCC+νCO vibration is observed at 1547 cm−1
(vs 1670 cm−1 for the νCO vibration of uncoordinated 2).35 This value lies within the range of those already reported for the same phosphino-enolate moiety in other metal complexes.35,37–39,41,43–46,49 The proposed structure of 6a was confirmed by X-ray diffraction (vide infra). Note that 6a was also obtained as the sole product when an excess of the ketophosphine ligand was used (2 to 4 mol. equiv.), no side-reaction being observed.
 | ||
| Scheme 1
Reaction conditions: Room temperature; solvent: M=Nb: DME; M=Ta: CH2Cl2; M=W: THF (1), toluene (2) or CH2Cl2/toluene 1∶1 (3). | ||
Spontaneous elimination of HCl was also observed when 2 was reacted with 4b–d, thus leading to the formation of the phosphino-enolate derivatives 6b–d, respectively (Scheme 1). The spectroscopic data of the tantalum complex 6b mirror those of 6a. In the case of the Cp* complex 6d, the 31P{1H} resonance occurs at slightly higher field (δ 31.3), whereas, in the 1H NMR spectrum, the enolate and η-C5Me5 protons are observed at δ 6.08 [J(P,H)=3.6 Hz] and 2.38 (singlet), respectively. The paramagnetic complex 6c was characterized by IR spectroscopy (νCC+νCO 1542 cm−1), mass spectroscopy (m/z: 701 [M]) and elemental analysis. It was isolated as a burgundy solid, whereas phosphine adducts of the type [(η-C5H4iPr)WCl4(PR3)] range from yellow ochre to brown.77,78 The X-ray structures of the tantalum complexes 6b,d have been determined (vide infra).
To the best of our knowledge, such a spontaneous and facile HCl elimination in the absence of base has not been described for transition metal complexes containing 2 or any similar ligands. Yet, thermal activation or treatment with I2 of Ru clusters of the type [Ru3(CO)12−nLn]
(L=2, n=1–3) have led to phosphino-enolate Ru complexes,43,44 whereas a Rh(III) phosphino-enolate complex has been obtained by deprotonation of a co-ordinated ketophosphine 2 by an excess of this ligand present in the reaction mixture, the latter being a slow process (ca. 1 week).48 Although the mechanism of the formation of 6 has not been established, we suggest, on the basis of the formation of complexes 5, that P-coordination of 2 at the metal centre occurs first. This would result in an enhanced acidity of the PCH2 protons and thus favour HCl elimination. This is consistent with our recent observation of an H/D exchange involving the methylene protons of 2 in the Mo(II) complexes [(η-C6H5R)Mo(η-C3H5){Ph2PCH2C(O)Ph}][PF6] (R=H, Me).23 The dehydrochlorination may also be facilitated by intra- and/or inter molecular H⋯Cl interactions involving the methylene protons, a bonding situation which has been encountered in the crystal structure of the Mo(II) complex [(η-C6H5Me)Mo(η-C3H5)Cl{Ph2PCH2C(O)Ph}].23
The complexes 6 exhibit remarkable stability both in solution and in the solid state. In the latter case they can be kept for weeks in the air. Note that when 6 was treated with an excess of PMe3, no opening or complete displacement of the P,O chelate was observed. The straightforward formation of these phosphino-enolate complexes, under the same reaction conditions as those used for the preparation of 5a–c, emphasizes a marked difference of behaviour between the β -ketophosphine 2 and its β-amido counterpart 1. This is related to the more acidic character of the CH2 protons in 2, owing to the different electronic influence of its Ph substituent vs. the NPh2 group in 1.
Formation of O-monoadducts
The reaction of 4a–c with the acetamido-phosphine Ph2PNHC(O)Me 3 led to the new complexes 7a–c in which the P,O ligand is O-monocoordinated and this is an unsual coordination mode for such a P,O ligand (Scheme 1). This proposed stucture is supported by the νCO vibration at 1646 cm−1
(7a,b) or 1651 cm−1
(7c), to be compared to 1715 cm−1 for the free ligand.79 The fact that the P atom does not coordinate to the metal centre is suggested by the 31P{1H} NMR resonances of 7a
(δ 33.1) and 7b
(δ 40.6), which occur in the same region as that of 3
(δ 31.1). These values suggest that the electronic properties of the phosphorus centre are slightly more affected by O-coordination of the ligand in the case of M=Ta (7b) than in the case of M=Nb (7a). Furthermore, the slightly broadened 31P singlet (w1/2=65 Hz) observed for 7a contrasts with the very broad signals (w1/2>2500 Hz) observed for 5a and 6a, in which the P atoms are coordinated to Nb. The 1H NMR spectra of 7a,b show all the expected signals, including those of the NH protons at ca.δ 9 (free ligand: δ 6.15).79 Complexes 7 are stable for a few days in the solid state, in the absence of air and moisture. They represent rare examples of transition metal derivatives in which a neutral functionalized phosphine is coordinated to the metal via the alternative donor atom instead of the P atom. Precedents include N- and O-adducts of MnI2 of 2-(diphenylphosphino)pyridine and 2, respectively.80 In the case of phosphino-enolate zirconocene and titanocene complexes, O-monocoordination of the anionic P,O ligands results from the formation of a covalent M–O bond.61–63 Previous investigations on the coordination properties of 3 concerned the synthesis of neutral and cationic Pd,79 Mo22 and Rh complexes,81 for which O-monocoordination was never encountered. Comparative studies showed that 3 is a better P,O-chelate than the keto- and amidophosphines 1 and 2, respectively.79
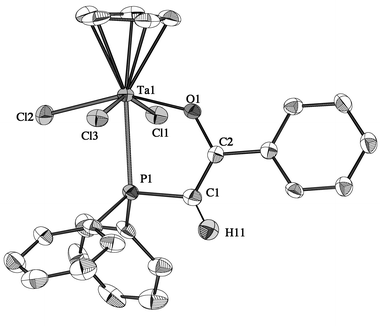
Crystal structures of 6a,b,d
Crystal data and data collection parameters are given in Table 1. There are two crystallographically different but very similar molecules in the asymmetric unit of 6b whose selected bond lengths and angles are given in Table 2 together with those of complexes 6a,d. ORTEP views of 6a, 6b and 6d are pictured in Fig. 1, 2 and 3, respectively. If one considers the Cp centroid of the cyclopentadienyl ligands as an apex of the metal coordination polyhedron, then the three complexes exhibit a distorted pseudooctahedral geometry, with the three chlorine atoms and the oxygen atom lying in the equatorial plane and the P atom trans to the cyclopentadienyl ring.82 The Cpcent–M–P(1) angles range from 175.76° (6a) to 178.48° (6d) and are slightly more obtuse than those found in the ylide complex [(η-C5Me5)TaCl4(CH2PMePh2)]
(173°)83 or in [(η-C5H5)NbCl4(PMePh2)]
(174.6°),76 the only phosphine adduct of [(η-C5H5)NbCl4] that has been structurally characterized to date. The chlorine and oxygen atoms are bent away from the cyclopentadienyl ring towards the P ligand. The molecular structures show M–Cl [2.3873(9)–2.446(1)
Å] bond lengths, Cpcent–M–Cl [103.35–105.28°] and Cl–M–Cl [87.08(4)–90.31(3)°
cis; 150.95(3)–152.37(8)°
trans] bond angles which are consistent with those reported for related structures.76,83–85 The M–O(1) bond lengths are similar to those found in other transition metal complexes with the same phosphino-enolate ligand35–39,41,43–46,49 or in [(η-C5Me5)TaCl3{OC(SiMe3)NNCPh2}] [1.98(1)
Å].85 In the case of [(η-C5Me5)TaCl3{PPh2CHC(O)Ph}] 6d
[2.024(2)
Å] it is slightly longer than in the other complexes 6a,b
[1.977(2)–1.983(3)
Å]. The M–P(1) bond lengths of ca. 2.66 Å are shorter than those found in [(η-C5H4Me)TaCl4{PH2(C6H2iPr3)}]
[2.710 Å]86 or in [(η-C5H5)NbCl4(PMePh2)]
[2.7844(9)
Å]76 and similar to those reported for [Nb(O)Cl3(PMe3)3]
[2.640(3)
Å].87 The other bond lengths and angles of the phosphino-enolate and cyclopentadienyl ligands are within the expected range.
| 6a | 6b | 6d | |
|---|---|---|---|
| Formula | C25H21Cl3OPNb·0.5C7H8 | C25H21Cl3OPTa | C30H31Cl3OPTa |
| FW | 613.75 | 655.72 | 725.86 |
| Colour | Red | Yellow | Yellow |
| Crystal system | Monoclinic | Triclinic | Trigonal |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| T/K | 150(2) | 150(2) | 150(2) |
| a/Å | 7.876(1) | 8.4611(1) | 42.5519(5) |
| b/Å | 26.392(1) | 16.6441(3) | 42.5519(5) |
| c/Å | 12.650(1) | 16.8443(3) | 8.7407(5) |
| α/° | 90 | 92.6314(8) | 90 |
| β/° | 97.01(1) | 99.3326(9) | 90 |
| γ/° | 90 | 94.1166(11) | 120 |
| V/Å3 | 2609.8 | 2330.7 | 13706.1 |
| Z | 4 | 4 | 18 |
| μ/mm−1 | 0.851 | 5.145 | 3.945 |
| Unique reflections | 5001 | 10582 |
6943 |
| Reflections | 4100 with I>3σ(I) |
8244 with I>2σ(I) |
5794 with I>3σ(I) |
| R | 0.0515 | 0.0314 | 0.0281 |
| R w | 0.0543 | 0.0366 | 0.0298 |
| 6a | 6b (molecule 1) | 6b (molecule 2) | 6d | |
|---|---|---|---|---|
| M–P1 | 2.667(1) | 2.670(1) | 2.674(1) | 2.6596(9) |
| Cpcent–M | 2.132 | 2.127 | 2.142 | 2.168 |
| M–Cl1 | 2.4186(9) | 2.415(1) | 2.410(1) | 2.4209(8) |
| M–Cl2 | 2.4057(9) | 2.4131(11) | 2.402(1) | 2.3873(9) |
| M–Cl3 | 2.446(1) | 2.422(1) | 2.413(1) | 2.3934(8) |
| M–O1 | 1.977(2) | 1.983(3) | 1.982(3) | 2.023(2) |
| P1–C1 | 1.779(4) | 1.779(5) | 1.792(4) | 1.773(3) |
| C1–C2 | 1.345(5) | 1.352(6) | 1.342(6) | 1.349(5) |
| C2–O1 | 1.346(4) | 1.349(5) | 1.352(3) | 1.342(4) |
| Cpcent–M–P1 | 175.76 | 175.80 | 177.40 | 178.48 |
| Cpcent–M–Cl1 | 104.02 | 103.59 | 105.28 | 104.39 |
| Cpcent–M–Cl2 | 104.24 | 104.05 | 104.60 | 103.99 |
| Cpcent–M–Cl3 | 103.44 | 104.02 | 103.35 | 103.81 |
| Cpcent–M–O1 | 101.83 | 103.17 | 101.78 | 103.82 |
| Cl1–M–Cl2 | 88.92(3) | 88.25(4) | 87.41(4) | 89.49(3) |
| Cl1–M–Cl3 | 152.37(3) | 151.26(3) | 152.35(4) | 150.95(3) |
| Cl1–M–O1 | 87.60(8) | 85.21(9) | 85.50(9) | 82.04(7) |
| Cl2–M–Cl3 | 87.26(3) | 87.08(4) | 87.16(4) | 90.31(3) |
| Cl2–M–O1 | 153.80(8) | 153.56(8) | 152.76(9) | 152.12(7) |
| Cl3–M–O1 | 83.921(8) | 86.47(9) | 87.04(9) | 84.70(7) |
| P1–M–O1 | 73.97(7) | 74.10(8) | 74.23(8) | 74.83(7) |
| P1–C1–C2 | 115.4(3) | 115.3(3) | 114.7(3) | 116.0(3) |
| C1–C2–O1 | 121.3(3) | 121.5(4) | 122.3(4) | 122.6(3) |
| C2–O1–M | 131.8(2) | 131.6(2) | 131.2(3) | 129.2(2) |
 | ||
| Fig. 1 Molecular structure of 6a. Hydrogen atoms, except H(11), and the solvent molecule (toluene) are omitted for clarity. | ||
 | ||
| Fig. 2 Molecular structure of 6b. Only one of the two independent molecules of the unit cell is shown. Hydrogen atoms, except H(11), are omitted for clarity. | ||
 | ||
| Fig. 3 Molecular structure of 6d. Hydrogen atoms, except H(11), are omitted for clarity. | ||
Conclusion
We have described here the first investigations on the chemistry of half-sandwich complexes of the type [(η-C5R5)MCl4] (M=Nb, Ta or W) with β-carbonylphosphines. Depending on the nature of the P,O ligand three different coordination modes have been observed. With Ph2PCH2C(O)NPh21 P-monoadducts were obtained, whereas Ph2PNHC(O)Me 3 led to O-monoadducts, which is an unsual coordination mode for P,O ligands. The reactions with the β-ketophosphine Ph2PCH2C(O)Ph 2 followed another pathway. In this case, no phosphine adduct was isolated, instead HCl elimination readily occurred giving the corresponding phosphino-enolate complexes. To the best of our knowledge, such a facile formation of transition metal phosphino-enolate complexes, in the absence of a base, has not been reported previously. The crystal structures of the Nb and Ta complexes 6a,b,d have been determined. The striking difference between 1 and 3 in bonding cannot be of steric origin since P-coordination of 3 would have led to a very similar steric situation at the metal centre. Therefore, the bonding difference must be of electronic origin, which has not been quantified at the moment and deserves further investigations.
Experimental
Reagents and physical measurements
All manipulations of air- and/or moisture sensitive materials were performed under an inert atmosphere of argon using standard Schlenk line techniques, or in an inert atmosphere dry box containing dinitrogen. Solvents were dried over the appropriate drying agent and distilled under nitrogen. Deuterated solvents were dried over the appropriate drying agent and vacuum distilled prior to use. The compounds [(η-C5H5)NbCl4],75 [(η-C5H5)TaCl4],75 [(η-C5Me5)TaCl4],88 [(η-C5H4iPr)WCl4],89 Ph2PCH2C(O)Ph,35 Ph2PCH2C(O)NPh242 and Ph2PNHC(O)Me79 were prepared according to previously published methods. NMR spectra were recorded on a Varian Mercury 300 (1H, 13C, and 31P at 300.17, 75.48 and 121.51 MHz, respectively) spectrometer. They were referenced internally using the residual protio solvent (1H) and solvent (13C) resonances and measured relative to tetramethylsilane (1H and 13C; δ 0 ppm). 31P NMR was referenced externally to 85% H3PO4 (δ 0 ppm). Elemental analyses were provided by the microanalytical department, Inorganic Chemistry Laboratory, University of Oxford or by the Service de Microanalyses, Université Louis Pasteur, Strasbourg. Infra-red spectra were recorded on a Perkin Elmer 1600 Series FTIR or a Bruker IFS66 FTIR spectrometer. Mass spectra were recorded at the Inorganic Chemistry Laboratory, on a Macromass LC Tof spectrometer.Synthesis
=7 Hz, PCH2); 6.90–7.79 (m, 25H, Cp and aromatics). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 33.1 (vbr, w1/2>2500 Hz). IR (CH2Cl2): 1658 cm−1
(νCO). Anal. calc. for C31H27Cl4NNbOP: C, 53.55; H, 3.91; N, 2.01. Found: C, 53.81; H, 4.06; N, 1.95%.
=5.5 Hz, PCH2); 6.77 (s, 5H, Cp); 6.95–7.97 (m, 20H, aromatics). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 27.9. IR (CH2Cl2): 1658 cm−1
(νCO). Anal. calc. for C31H27Cl4NOPTa: C, 47.54; H, 3.47; N, 1.79. Found: C, 47.69; H, 3.38; N, 1.92%.
O). MS m/z: 793 (M+−Cl showing the expected isotopic pattern), 758 (M+−2Cl). Anal. calc. for C34H33Cl4OPW: C, 49.30; H, 4.02; N, 1.69. Found: C, 48.87; H, 4.27; N, 1.53%.
![[upper bond 1 start]](https://www.rsc.org/images/entities/b_char_e010.gif) Cl3{PPh2CH
Cl3{PPh2CH![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/b_char_e0f1.gif) C(O
C(O![[upper bond 1 end]](https://www.rsc.org/images/entities/b_char_e011.gif) )Ph}] 6a.
Solid [(η-C5H5)NbCl4]
(0.400 g, 1.33 mmol) and Ph2PCH2C(O)Ph (0.405 g, 1.33 mmol) were placed in a Schlenk flask and DME (30 mL) was added at ambient temperature. Rapidly, the solution turned deep red, and HCl emanated from it. The mixture was then stirred for 2 h and the volatiles were removed under reduced pressure. The red-brown residue was washed with Et2O (2×20 mL) and pentane (2×20 mL) and dried in vacuo affording 6a as a red-brown solid (0.704 g, yield 93%). 1H NMR (CD2Cl2, 300.18 MHz): δ 6.25 (d, 1H, 2JPH=3.5 Hz, PCH); 6.95 (s, 5H, Cp); 7.24–7.88 (m, 15H, aromatics). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 36.5 (vbr, w1/2>2500 Hz). IR (Nujol): 1547 cm−1
(νCC+νCO). Anal. calc. for C25H21Cl3NbOP: C, 52.90; H, 3.72. Found: C, 53.21; H, 3.82%.
)Ph}] 6a.
Solid [(η-C5H5)NbCl4]
(0.400 g, 1.33 mmol) and Ph2PCH2C(O)Ph (0.405 g, 1.33 mmol) were placed in a Schlenk flask and DME (30 mL) was added at ambient temperature. Rapidly, the solution turned deep red, and HCl emanated from it. The mixture was then stirred for 2 h and the volatiles were removed under reduced pressure. The red-brown residue was washed with Et2O (2×20 mL) and pentane (2×20 mL) and dried in vacuo affording 6a as a red-brown solid (0.704 g, yield 93%). 1H NMR (CD2Cl2, 300.18 MHz): δ 6.25 (d, 1H, 2JPH=3.5 Hz, PCH); 6.95 (s, 5H, Cp); 7.24–7.88 (m, 15H, aromatics). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 36.5 (vbr, w1/2>2500 Hz). IR (Nujol): 1547 cm−1
(νCC+νCO). Anal. calc. for C25H21Cl3NbOP: C, 52.90; H, 3.72. Found: C, 53.21; H, 3.82%.
X-Ray quality single crystals were grown by slow diffusion of Et2O into a toluene solution of 6a.
Cl3{PPh2CHC(O)Ph}] 6b.
Solid [(η-C5H5)TaCl4]
(0.400 g, 1.03 mmol) and Ph2PCH2C(O)Ph (0.313 g, 1.03 mmol) were placed in a Schlenk flask and CH2Cl2
(20 mL) was added at room temperature. The mixture was stirred for 3 h, during which its colour changed from yellow to orange. The volatiles were then removed under reduced pressure. The dark yellow residue was extracted with toluene (15 mL) at 60°C. The resulting clear yellow solution was evaporated under reduced pressure affording 6b as a yellow powder (0.563 g, yield 83%). 1H NMR (CDCl3, 300.18 MHz): δ 6.34 (d, 1H, 2JPH=3.0 Hz, PCH); 6.72 (s, 5H, Cp); 7.20–7.91 (m, 15H, aromatics). 31P{1H} NMR (CDCl3, 121.5 MHz): δ 37.2. IR (Nujol): 1552 cm−1
(νCC+νCO). Anal. calc. for C25H21Cl3OPTa: C, 45.79; H, 3.63. Found: C, 46.01; H, 3.46%.
X-Ray quality single crystals were grown by slow diffusion of petroleum ether (bp 40–60 °C) into a CDCl3 solution of 6b.
Cl3{PPh2CHC(O)Ph}] 6c.
Solid [(η-C5H4iPr)WCl4]
(0.600 g, 1.38 mmol) and Ph2PCH2C(O)Ph (0.420 g, 1.38 mmol) were placed in a Schlenk flask and toluene (15 mL) was added at room temperature. The mixture was stirred overnight, affording a claret solution. The volatiles were then removed under reduced pressure and the residue was washed with Et2O (15mL) and pentane (2×10 mL). It afforded 6c as a purple solid, which was dried in vacuo
(0.786 g, yield 81%). IR: 1539 cm−1
(Nujol), 1542 cm−1
(KBr)
(νCC+νCO). MS m/z: 701 (M+, showing the expected isotopic pattern), 666 (M+−Cl). Anal. calc. for C28H27Cl3OPW: C, 48.00; H, 3.88. Found: C, 47.83; H, 3.72%.
Cl3{PPh2CHC(O)Ph}] 6d.
This complex was prepared in a similar manner to 6b from [(η-C5Me5)TaCl4]
(0.400 g, 0.873 mmol) and Ph2PCH2C(O)Ph (0.266 g, 0.873 mmol). It was obtained as a yellow solid (0.544 g, yield 86%). 1H NMR (CDCl3, 300.18 MHz): δ 2.38 (s, 15H, Me); 6.08 (d, 1H, 2JPH=3.6 Hz, PCH); 6.90–7.90 (m, 15H, aromatics). 31P{1H} NMR (CDCl3, 121.5 MHz): δ 36.5 (vbr, w1/2>2500 Hz). IR (Nujol): 1552 cm−1
(νCC+νCO). Anal. calc. for C30H31Cl3OPTa: C, 49.64; H, 4.30. Found: C, 49.87; H, 4.18%.
X-Ray quality single crystals were grown by slow diffusion of Et2O into a CDCl3 solution of 6d.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C(Me)NHPPh2}]
7a.
This complex was prepared in a similar manner to 5a from [(η-C5H5)NbCl4]
(0.500 g, 1.67 mmol) and Ph2PNHC(O)Me (0.354 g, 1.67 mmol) in DME (20 mL). It was obtained as an orange-pink solid (0.751 g, yield 88%). 1H NMR (CD2Cl2, 300.18 MHz): δ 2.16 (s, 3H, Me); 7.18 (s, 5H, Cp); 7.22–7.95 (m, 10H, aromatics); 9.64 (br s, 1H, NH). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 33.1 (w1/2=65 Hz). IR (CH2Cl2): 1646 cm−1
(νCO). Anal. calc. for C19H19Cl4NNbOP: C, 42.02; H, 3.53; N, 2.58. Found: C, 42.26; H, 3.81; N, 2.61%.
C(Me)NHPPh2}]
7b.
This complex was prepared in a similar manner to 5b from [(η-C5H5)TaCl4]
(0.150 g, 0.38 mmol) and Ph2PNHC(O)Me (0.080 g, 0.38 mmol) in CH2Cl2
(10 mL). It was obtained as a yellow-brown solid (0.185 g, yield 80%). 1H NMR (CDCl3, 300.18 MHz): δ 2.61 (s, 3H, Me); 6.91 (s, 5H, Cp); 6.80–8.02 (m, 10H, aromatics); 8.85 (br s, 1H, NH). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 40.6. IR (CDCl3): 1646 cm−1
(νCO). Anal. calc. for C19H19Cl4NOPTa: C, 36.16; H, 3.03; N, 2.22. Found: C, 36.28; H, 3.05; N, 2.26%.
C(Me)NHPPh2}]
7c.
This complex was prepared in a similar manner to 5c from [(η-C5H4iPr)WCl4]
(1.000 g, 2.30 mmol) and Ph2PNHC(O)Me (0.490 g, 2.30 mmol) in a 1∶1 CH2Cl2–toluene mixture (30 mL). It was obtained as a green-brown solid (1.212 g, yield 81%). IR (Nujol): 1651 cm−1
(νCO). MS m/z: 641 (M+−Cl, showing the expected isotopic pattern), 606 (M+−2Cl). Anal. calc. for C22H25Cl4NOPW: C, 39.08; H, 3.73; N, 2.07. Found: C, 38.70; H, 3.78; N, 2.03%.
C(Me)NHPPh2}]
7a.
This complex was prepared in a similar manner to 5a from [(η-C5H5)NbCl4]
(0.500 g, 1.67 mmol) and Ph2PNHC(O)Me (0.354 g, 1.67 mmol) in DME (20 mL). It was obtained as an orange-pink solid (0.751 g, yield 88%). 1H NMR (CD2Cl2, 300.18 MHz): δ 2.16 (s, 3H, Me); 7.18 (s, 5H, Cp); 7.22–7.95 (m, 10H, aromatics); 9.64 (br s, 1H, NH). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 33.1 (w1/2=65 Hz). IR (CH2Cl2): 1646 cm−1
(νCO). Anal. calc. for C19H19Cl4NNbOP: C, 42.02; H, 3.53; N, 2.58. Found: C, 42.26; H, 3.81; N, 2.61%.
C(Me)NHPPh2}]
7b.
This complex was prepared in a similar manner to 5b from [(η-C5H5)TaCl4]
(0.150 g, 0.38 mmol) and Ph2PNHC(O)Me (0.080 g, 0.38 mmol) in CH2Cl2
(10 mL). It was obtained as a yellow-brown solid (0.185 g, yield 80%). 1H NMR (CDCl3, 300.18 MHz): δ 2.61 (s, 3H, Me); 6.91 (s, 5H, Cp); 6.80–8.02 (m, 10H, aromatics); 8.85 (br s, 1H, NH). 31P{1H} NMR (CD2Cl2, 121.5 MHz): δ 40.6. IR (CDCl3): 1646 cm−1
(νCO). Anal. calc. for C19H19Cl4NOPTa: C, 36.16; H, 3.03; N, 2.22. Found: C, 36.28; H, 3.05; N, 2.26%.
C(Me)NHPPh2}]
7c.
This complex was prepared in a similar manner to 5c from [(η-C5H4iPr)WCl4]
(1.000 g, 2.30 mmol) and Ph2PNHC(O)Me (0.490 g, 2.30 mmol) in a 1∶1 CH2Cl2–toluene mixture (30 mL). It was obtained as a green-brown solid (1.212 g, yield 81%). IR (Nujol): 1651 cm−1
(νCO). MS m/z: 641 (M+−Cl, showing the expected isotopic pattern), 606 (M+−2Cl). Anal. calc. for C22H25Cl4NOPW: C, 39.08; H, 3.73; N, 2.07. Found: C, 38.70; H, 3.78; N, 2.03%.
Crystallography
Data were collected on an Enraf-Nonius DIP2000 Image Plate diffractometer (6a) and on an Enraf-Nonius KappaCCD diffractometer (6b,d) (graphite-monochromated MoKα radiation λ=0.71073 Å). Intensity data were processed using the DENZO-SMN package90 running on a Silicon Graphics Indy computer. The structures were solved using the direct-methods program SIR-92.91 Subsequent full-matrix least-squares refinements were carried out using the SHELXL-9392
(6a) or the CRYSTALS93 programs. The structure of 6b consists of two “pseudo-dimers” representing two slightly different mononuclear complexes, referred to as molecules 1 and 2 in Table 2. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were positioned geometrically after each cycle of refinement. A 3-term Chebychev polynomial weighting scheme was applied. Selected crystal data and refinement details are given in Table 1.
CCDC reference numbers 190864–190866.
See http://www.rsc.org/suppdata/nj/b2/b202983a/ for crystallographic data in CIF or other electronic format.
Acknowledgements
For providing financial support, we would like to thank the Centre National de la Recherche Scientifique/Royal Society cooperation programme (XM), the RSC for a travel grant (XM, JGA No. 0103 306), the AG Leventis Foundation (IV) and the EPSRC (LHR). We thank Dr. David Watkin and Andrew Cowley (Crystallography Laboratory, Oxford) for their generous assistance with the crystal structure determinations.References
- A. Bader and E. Lindner, Coord. Chem. Rev., 1991, 108, 27 CrossRef CAS and references cited therein.
- E. Lindner, S. Pautz and M. Haustein, Coord. Chem. Rev., 1996, 155, 145 CrossRef CAS.
- C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233 Search PubMed and references cited therein.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680 CrossRef CAS.
- R. W. Wegman, A. G. Abatjoglou and A. M. Harrison, J. Chem. Soc., Chem. Commun., 1987, 1891 RSC.
- G. J. P. Britovsek, W. Keim, S. Mecking, D. Sainz and T. Wagner, J. Chem. Soc., Chem. Commun., 1993, 1632 RSC.
- E. Lindner, Q. Wang, H. A. Mayer, A. Bader, H. Kühbauch and P. Wegner, Organometallics, 1993, 12, 3291 CrossRef CAS.
- M. Alvarez, N. Lugan and R. Mathieu, J. Organomet. Chem., 1994, 468, 249 CrossRef CAS.
- C. Abu-Gnim and I. Amer, J. Chem. Soc., Chem. Commun., 1994, 115 RSC.
- H. Yang, M. Alvarez, N. Lugan and R. Mathieu, J. Chem. Soc., Chem. Commun., 1995, 1721 RSC.
- G. J. P. Britovsek, K. J. Cavell and W. Keim, J. Mol. Catal., 1996, 110, 77 Search PubMed.
- S. Mecking and W. Keim, Organometallics, 1996, 15, 2650 CrossRef CAS.
- H. Yang, M. Alvarez-Gressier, N. Lugan and R. Mathieu, Organometallics, 1997, 16, 1401 CrossRef CAS.
- P. Braunstein, D. Matt, Y. Dusausoy, J. Fischer, A. Mitschler and L. Ricard, J. Am. Chem. Soc., 1981, 103 CAS.
- J. I. Dulebohn, S. C. Haefner, K. A. Berglund and K. R. Dunbar, Chem. Mater., 1992, 4, 506 CrossRef CAS.
- K. R. Dunbar, Comments Inorg. Chem., 1992, 13, 313 Search PubMed.
- H. Werner, M. Schulz and B. Windmüller, Organometallics, 1995, 14, 3659 CrossRef CAS.
- E. Lindner, B. Keppleler, H. A. Mayer, K. Gierling, R. Fawzi and M. Steinmann, J. Organomet. Chem., 1996, 526, 175 CrossRef.
- H. Werner, J. Bank, B. Windmüller, O. Gevert and W. Wolfsberger, Helv. Chim. Acta, 2001, 84, 3163 CrossRef CAS.
- J.-M. Camus, D. Morales, J. Andrieu, P. Richard, R. Poli, P. Braunstein and F. Naud, J. Chem. Soc., Dalton Trans., 2000, 2577 RSC.
- C. D. Andrews, A. D. Burrows, J. M. Lynam, M. F. Mahon and M. T. Palmer, New J. Chem., 2001, 25, 824 RSC.
- N. G. Jones, M. L. H. Green, I. Vei, X. Morise and P. Braunstein, J. Chem. Soc., Dalton Trans., 2002, 1487 RSC.
- N. G. Jones, M. L. H. Green, I. C. Vei, L. H. Rees, S. I. Pascu, D. Watkin, A. Cowley, X. Morise and P. Braunstein, J. Chem. Soc., Dalton Trans., 2002 Search PubMed , in the press.
- J. A. Canich, F. A. Cotton and S. A. Duraj, Inorg. Chim. Acta, 1989, 41, 156.
- S. Gambarotta, M. Pasquali, C. Floriani, A. Chiesi-Villa and C. Guastini, Inorg. Chem., 1981, 20, 1173 CrossRef CAS.
- F. E. Kuhn, A. D. Lopes, A. M. Santos, E. Herdtweck, J. J. Haider, C. C. Romao and A. G. Santos, J. Mol. Catal. A: Chem., 2000, 151, 147 CrossRef CAS.
- D. J. McCabe, E. N. Duesler and R. T. Paine, Inorg. Chem., 1987, 26, 2300 CrossRef CAS.
- V. S. Sergienko, N. A. Ovchinnikova, M. A. Porai-Koshits and M. A. Glushkova, Koord. Khim., 1986, 12, 1650 Search PubMed.
- D. G. L. Holt, L. F. Larkworthy, D. C. Povey, G. W. Smith and G. J. Leigh, J. Chem. Soc., Dalton Trans., 1990, 3229 RSC.
- F. A. Cotton and G. W. Rice, Inorg. Chem., 1978, 17, 2004 CrossRef CAS.
- P. Bhattacharyya, A. M. Z. Slawin, M. B. Smith and J. D. Woollins, Inorg. Chem., 1996, 35, 3675 CrossRef CAS.
- L. R. Falvello, M. M. Garcia, I. Lazaro, R. Navarro and E. P. Urriolabeitia, New J. Chem., 1999, 227 RSC.
- R. Navarro and E. P. Urriolabeitia, J. Chem. Soc., Dalton Trans., 1999, 4111 RSC.
- W. Keim, A. Behr, B. Gruber, B. Hoffmann, F. H. Kowaldt, U. Kürschner, B. Limbäcker and F. P. Sistig, Organometallics, 1986, 5, 2356 CrossRef CAS and references cited therein.
- S.-E. Bouaoud, P. Braunstein, D. Grandjean, D. Matt and D. Nobel, Inorg. Chem., 1986, 25, 3765 CrossRef CAS.
- F. Balegroune, P. Braunstein, D. Grandjean, D. Matt and D. Nobel, Inorg. Chem., 1988, 27, 3320 CrossRef CAS.
- S.-E. Bouaoud, P. Braunstein, D. Grandjean, D. Matt and D. Nobel, Inorg. Chem., 1988, 27, 2279 CrossRef CAS.
- P. Braunstein, D. Matt, D. Nobel, F. Balegroune, S.-E. Bouaoud, D. Grandjean and J. Fischer, J. Chem. Soc., Dalton Trans., 1988, 353 RSC.
- P. Braunstein, T. M. Gomes-Carneiro, D. Matt, F. Balegroune and D. Grandjean, Organometallics, 1989, 8, 1737 CrossRef CAS.
- W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235 CrossRef.
- P. Berno, P. Braunstein, C. Floriani, A. Chiesi-Villa and C. Guastini, Inorg. Chem., 1991, 30, 1407 CrossRef CAS.
- J. Andrieu, P. Braunstein and A. D. Burrows, J. Chem. Res. (S), 1993, 380 Search PubMed.
- P. Braunstein, S. Coco-Cea, M. I. Bruce, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1992, 2539 RSC.
- P. Braunstein, S. Coco-Cea, M. I. Bruce, B. W. Skelton and A. H. White, J. Organomet. Chem., 1992, 423, C38 CrossRef CAS.
- P. Braunstein, L. Douce, F. Balegroune, D. Grandjean, D. Bayeul, Y. Dusausoy and P. Zanello, New J. Chem., 1992, 16, 925 Search PubMed.
- P. Braunstein, D. J. Kelly, A. Tiripicchio and F. Ugozzoli, Inorg. Chem., 1993, 32, 4845 CrossRef CAS.
- P. Braunstein, Y. Chauvin, J. Nähring, Y. Dusausoy, D. Bayeul, A. Tiripicchio and F. Ugozzoli, J. Chem. Soc., Dalton Trans., 1995, 851 RSC.
- P. Braunstein, Y. Chauvin, J. Nähring, A. DeCian and J. Fischer, J. Chem. Soc., Dalton Trans., 1995, 863 RSC.
- P. Braunstein, Y. Chauvin, J. Fischer, H. Olivier, C. Strohmann and D. V. Toronto, New J. Chem., 2000, 24, 437 RSC.
- P. Braunstein, Y. Chauvin, S. Mercier, L. Saussine, A. DeCian and J. Fischer, J. Chem. Soc., Chem. Commun., 1994, 2203 RSC.
- J. Pietsch, P. Braunstein and Y. Chauvin, New J. Chem., 1998, 22, 467 RSC.
- K. A. O. Starzewski and J. Witte, Angew. Chem., Int. Ed. Engl., 1987, 26, 63 CrossRef.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1980, 299 RSC.
- J. Bank, P. Steinert, P. Windmüller, W. Wolfsberger and H. Werner, J. Chem. Soc., Dalton Trans., 1996, 1153 RSC.
- P. Crochet and B. Demerseman, Organometallics, 1995, 14, 2173 CrossRef CAS.
- J. Vicente, M.-T. Chicote, M. A. Beswick and M. C. Ramirez-de-Arellano, Inorg. Chem., 1996, 35, 6592 CrossRef CAS.
- P. Crochet, B. Demerseman, M. I. Vallejo, M. P. Gamasa, J. Gimeno, J. Borge and S. Garcia-Granda, Organometallics, 1997, 16, 5406 CrossRef CAS.
- L. R. Falvello, S. Fernandez, R. Navarro, I. Pascual and E. P. Urriolabeitia, J. Chem. Soc., Dalton Trans., 1997, 763 RSC.
- P. Braunstein, Y. Chauvin, J. Nähring, A. DeCian, J. Fischer, A. Tiripicchio and F. Ugozzoli, Organometallics, 1996, 15, 5551 CrossRef CAS.
- D. A. Edwards, M. F. Mahon and T. J. Paget, Polyhedron, 2000, 19, 757 CrossRef CAS.
- C. Mattheis, P. Braunstein and A. Fischer, J. Chem. Soc., Dalton Trans., 2001, 800 RSC.
- P. Veya, C. Floriani, A. Chiesi-Villa and C. Guastini, Organometallics, 1991, 10, 2991 CrossRef CAS.
- P. Veya, C. Floriani, A. Chiesi-Villa and C. Guastini, Organometallics, 1994, 13, 208 CrossRef CAS.
- K. Mashima, Y. Nakayama and A. Nakamura, Adv. Polym. Sci., 1997, 133, 1 Search PubMed.
- A. Togni and R. L. Halterman, Metallocenes, Synthesis, Reactivity, Application, vol. 1 and 2, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed.
- J. M. Decker, S. J. Geib and T. Y. Meyer, Organometallics, 1999, 18, 4417 CrossRef CAS.
- K. Mashima, Y. Matsuo, S. Nakamura and K. Tani, J. Organomet. Chem., 2000, 593–594, 69 CrossRef CAS.
- K. Mashima, Y. Matsuo and K. Tani, Angew. Chem., Int. Ed. Engl., 2001, 40, 960 CrossRef CAS .and references cited therein.
- M. L. H. Green, P. C. McGowan and P. Mountford, J. Chem. Soc., Dalton Trans., 1995, 1207 RSC.
- M. L. H. Green and P. Mountford, Chem. Rev., 1994, 94, 29.
- R. Poli, Chem. Rev., 1991, 91, 509 CrossRef CAS and references cited therein.
- R. J. Burt and J. G. Leigh, J. Organomet. Chem., 1978, 148, C19 CrossRef CAS.
- R. J. Burt, J. G. Leigh and D. L. Hughes, J. Chem. Soc., Dalton Trans., 1981, 793 RSC.
- R. D. Sanner, S. T. Caster and W. J. Bruton, J. Organomet. Chem., 1982, 240, 157 CrossRef CAS.
- M. J. Bunker, A. DeCian, M. L. H. Green, J. J. E. Moreau and N. Siganporia, J. Chem. Soc., Dalton Trans., 1980, 2155 RSC.
- J. C. Fettinger, D. W. Keogh and R. Poli, Inorg. Chem., 1995, 34, 2343 CrossRef CAS.
- X. Morise, M. L. H. Green, P. C. McGowan and S. J. Simpson, J. Chem. Soc., Dalton Trans., 1994, 871 RSC.
- M. L. H. Green, X. Morise, A. H. Hamilton and A. N. Chernaga, J. Organomet. Chem., 1995, 488, 73 CrossRef CAS.
- P. Braunstein, C. Frison, X. Morise and R. D. Adams, J. Chem. Soc., Dalton Trans., 2000, 2205 RSC.
- P. Braunstein, D. J. Kelly, A. Tiripicchio and F. Ugozzoli, Bull. Soc. Chim. Fr., 1995, 132, 1083 Search PubMed.
- P. Braunstein, B. H. Heaton, C. Jacob, L. Manzi and X. Morise, to be published.
- P. Kubácek, R. Hoffmann and Z. Havlas, Organometallics, 1982, 1, 180 CrossRef CAS.
- R. Fandos, M. Gómez, P. Royo, S. Garcia-Blanco, S. Martinez-Carrera and J. Sanz-Aparicio, Organometallics, 1987, 6, 1581 CrossRef CAS.
- M. V. Galakhov, M. Gómez, G. Jiménez, P. Royo, M. A. Pellinghelli and A. Tiripicchio, Organometallics, 1994, 14, 1901.
- J. Arnold, T. D. Tilley, A. L. Rheingold and S. J. Geib, J. Chem. Soc., Chem. Commun., 1987, 793 RSC.
- G. A. A. Hadi, K. Fromm, S. Blaurock, S. Jelonek and E. Hey-Hawkins, Polyhedron, 1997, 16, 721 CrossRef CAS.
- V. Gibson, T. P. Kee, R. M. Sorrell, A. P. Bashell and M. McPartlin, Polyhedron, 1988, 7, 2221 CrossRef CAS.
- W. A. Herrmann, W. Kalcher, H. Biersack, I. Bernal and M. Creswick, Chem. Ber., 1981, 114, 3558 Search PubMed.
- M. L. H. Green, J. D. Hubert and P. Mountford, J. Chem. Soc., Dalton Trans., 1990, 3793 RSC.
- Z. Otwinowski and W. Minor, Processing of X-ray Diffraction Data Collected in Oscillation Mode, in Methods Enzymol., eds. C. W. Carter and R. M. Sweet, Academic Press, 1997, vol. 276, p. 357 Search PubMed.
- A Program Solution of Crystal Structure by Direct Methods: A. Almatore, G. Cascarano, C. Giavocazzo and A. Guargliardi, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, SHELXL93, Program for Crystal Structure Refinement, 1993, Intitüt für Anorganische Chemie der Universität, Tammanstrasse 4, D-3400 Göttingen, Germany.
- D. J. Watkin, K. C. Prout, J. R. Carruthers, P. W. Betteridge and R. I. Cooper, CRYSTALS issue 11, Chemical Crystallography Laboratory, Oxford, UK, 2001.
Footnote |
| † Dedicated to Prof. P. Royo on the occasion of his 65th birthday, with our warmest congratulations. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |