Chemoenzymatic total syntheses of the sesquiterpene (−)-patchoulenone†
Martin G. Banwell*a, David C. R. Hocklessa and Malcolm D. McLeodab
aResearch School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 0200, Australia. E-mail: mgb@rsc.anu.edu.au
bSchool of Chemistry F11, The University of Sydney, Sydney, NSW 2006, Australia
First published on 29th October 2002
Abstract
The bicyclo[5.3.1]undec-7(8)-en-3-one 6, which is prepared from the monochiral cis-1,2-dihydrocatechol 4, affords a mixture of products 7, 8 and 9 on exposure to protic acid. Each of compounds 8 and 9 rearranges to congener 7 on treatment with SnCl2 or upon sustained reaction with protic acid. Reaction of the last compound with hydrogen in the presence of palladium on carbon affords a mixture of the saturated diols 10 and 11 with the latter capable of elaboration to (−)-patchoulenone (1) in four simple steps. An alternate and more efficient route to compound 11 involved a radical cyclisation route wherein enone 6 was treated with SmI2 and thiophenol, the latter reagent being employed to ensure efficient reduction. The major product, 12, thus formed was then debenzylated to give the patchoulenone precursor 11. In an even more efficient route to the title sesquiterpene, the bicyclo[2.2.2]octenone 21 was reacted with isopropenyllithium to give the dienol 22, which engaged in an anionic oxy-Cope rearrangement to afford the bicyclo[5.3.1]undec-7(8)-enone 23 and for which an X-ray crystal structure determination has been carried out. Reductive cyclisation of the last compound using SmI2 in the presence of thiophenol then gave, in a stereoselective manner, diol monoether 24, which after subjection to debenzylation, oxidation and dehydration steps afforded (−)-patchoulenone (1).
(−)-Patchoulenone (1) is a prominent member of the cyperene class of sesquiterpenes and was first isolated in 1964 from Cyperus rotundus Linné (Cyperaceae), a plant common in Sudan, India, China, Thailand and Japan.1,2 The compound has also been identified as a constituent of, inter alia, the root bark of Uvaria narum Wall. (Annonaceae)3 and Piptostigma fugax.4 Despite a number of the source plants being used in traditional medicines, only a modest amount is known about the biological properties of (−)-patchoulenone. Thus, compound 1 shows2in vitro activity (EC50 1.08
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×10−4 M) against the malarial parasite Plasmodium falciparum, strong anti-fungal activity against Rhizoctonia solani and Saprolegnia asterophora,4 and significant toxicity in a brine shrimp bioassay.4 The isomeric sesquiterpene cyperotundone (2)5 and its deoxygenated counterpart cyperene (3)6 have been isolated from the same or related plant sources. Once again, there is little information available regarding the biological properties of these congeners.
×10−4 M) against the malarial parasite Plasmodium falciparum, strong anti-fungal activity against Rhizoctonia solani and Saprolegnia asterophora,4 and significant toxicity in a brine shrimp bioassay.4 The isomeric sesquiterpene cyperotundone (2)5 and its deoxygenated counterpart cyperene (3)6 have been isolated from the same or related plant sources. Once again, there is little information available regarding the biological properties of these congeners.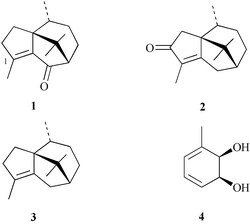
The 1,4,9,9-tetramethyl-2,4,5,6,7,8-hexahydro-3H-3a,7-methanoazulene framework associated with the cyperene-type sesquiterpenes has been the subject of a number of synthetic studies7 and the title compound has itself been synthesised by Hikino et al.,8 who used (+)-camphor as the starting material. The racemic modification of patchoulenone has also been prepared via the Lewis acid catalysed addition of a diazo ketone to a tethered olefin.9 We now report two distinct and chemoenzymatic total syntheses of (−)-patchoulenone that employ the monochiral cis-1,2-dihydrocatechol 4, obtained by microbial oxidation of toluene, as starting material.10
Results and discussion
Mechanistic and synthetic studies concerning a key transannular cyclisation reaction – construction of the patchoulenone framework
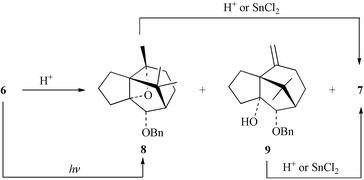
In connection with the development of synthetic approaches to taxoids, we have described11 the conversion of the dihydrocatechol 4 into the diene 5 and shown that the latter readily engages in an oxy-anionic Cope reaction to give the bicyclo[5.3.1]undec-7(8)-en-3-one 6 (Scheme 1). As has been observed in a closely related system,12 the carbon-carbon double bond and carbonyl group within enone 6 are in sufficiently close proximity that the compound exhibits a significant absorption (ε 2475) at λmax 245 nm. As a consequence of the proximity of these moieties, compound 6 engages in a rapid, acid-catalysed intramolecular Prins reaction13 to give isomer 7, which embodies the tricyclic framework associated with cyperene-type natural products such as 1–3. These observations prompted a detailed investigation of the conversion 6→7 with a view to optimising the yield of this process so that it could be exploited in the synthesis of (−)-patchoulenone (1). In this connection, a deuterochloroform solution of compound 6 containing traces of acid was observed, by 1H NMR techniques, to be consumed within 15 min at room temperature. Under these conditions a mixture of three products was obtained with the major ones being identified as the tricyclic alkene 7 and the oxetane 8
(Scheme 2).14 A trace of the isomeric system 9 was also detected. After 24 h compounds 8 and 9 had both been cleanly converted into isomer 7. That these conversions are acid-catalysed follows from the observation that a solution of compound 6 in base-washed deuterochloroform is completely stable and even after 24 h none of the above-mentioned rearrangement products could be detected. Furthermore, when a crystal of camphorsulfonic acid is added to the NMR sample the isomerisation process commences immediately, although the process is slower than that observed when HCl is the catalyst. These observations are consistent with the operation of an intramolecular Prins reaction and confirmation of the structure of oxetane 8 generated in these processes follows from the fact that this compound can be formed in high yield by subjecting precursor 6 to an intramolecular Paterno–Büchi reaction.15 Furthermore, when a pure sample of compound 8 produced in this way was treated with acidic deuterochloroform it was readily converted into alkene 7.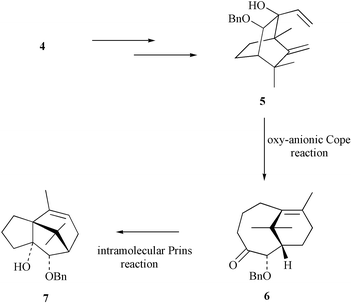 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
The close mechanistic relationship between the Prins reaction and the carbonyl-ene reaction, which can be catalysed by Lewis acids, prompted treatment of a solution of substrate 6 with tin(II) chloride (SnCl2)16 in deuterochloroform solution. Under such conditions the rapid formation of the ene product 7 occurs and small amounts of the oxetane 8, but not the isomeric alkene 9, can be detected by 1H NMR techniques (interestingly, independent subjection of compounds 8 and 9 to treatment with SnCl2 results in their rather slow – although still high yielding – conversion into isomer 7). These results suggest that compounds 8 and 9 are not intermediates in the Lewis acid catalysed carbonyl-ene reaction. Such results also suggest that the mechanisms associated with the Brønsted acid and Lewis acid catalysed conversions 6→7 are different. Thus, the former process is most likely a stepwise one while the latter is a concerted event. In a preparative sense the most effective method for acquiring useful quantities of compound 7 involved treating precursor 6 with catalytic amounts of SnCl2 in chloroform at 18°C for 1 h and under these conditions a 97% yield of product was obtained.
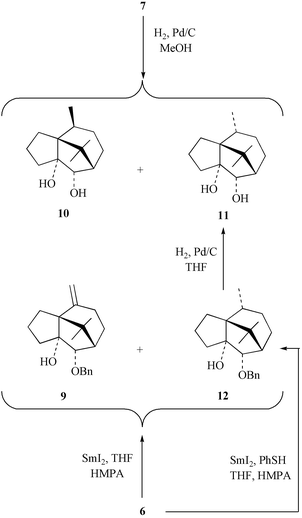
The synthesis of patchoulenone from compound 7 requires, inter alia, the diastereofacially selective hydrogenation of the double bond within the latter. In the event (Scheme 3), reaction of alkene 7 with dihydrogen in the presence of palladium on carbon afforded, as a consequence of hydrogenation of the alkene double bond and hydrogenolytic cleavage of the benzyl ether moiety, a ca. 1∶3 mixture of product 10 and the required epimer 11
(80% combined yield). These products could be separated from one another by flash chromatography and the structure of the major one follows from its conversion into patchoulenone. While the selectivity of this hydrogenation process is not especially high it compares rather favourably with related examples reported by Büchi,7 Hikino,8 and Erman.9 Nevertheless, we considered alternate means for producing compound 11 so as to achieve a more efficient process. In this connection the one-electron reduction of the carbonyl group within compound 6 would give a radical anion that might be expected to undergo a 5-exo-trig cyclisation onto the nearby double bond and the resulting tertiary radical might be further reduced and then protonated to give compound 12, the monobenzyl ether of the target compound 11. Since such reductive cyclisations have been effected with samarium(II) iodide,17 substrate 6 was reacted with this reagent (Scheme 3) in a mixture of THF and HMPA at 0°C. After 15 min all of the starting material had been consumed and a mixture of the target product 12
(39%) and the previously observed alkene 9
(54%) was obtained. On the basis that this mixture of products might derive, at least in part, from disproportionation of the above-mentioned tertiary radical,18–20 it was considered that the inclusion of a hydrogen donor source in the reducing medium might afford higher yields of the reductive cyclisation product 12. In the event, treatment of compound 6 with samarium(II) iodide under the same conditions as used before, save for the addition (or inclusion) of thiophenol (1 M),21,22 afforded the tricyclic alcohol 12
(71% yield), the structure of which follows from its hydrogenolytic cleavage to diol 11
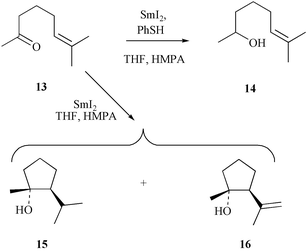
(95%). There was no sign of any product resulting from direct reduction (no cyclisation) of the carbonyl group within the starting ketone 6.23 This outcome is taken as testimony to the rapid rate at which the derived radical anion cyclises onto the proximate double bond since reaction of the model unsaturated ketone 1324
(Scheme 4) under the same conditions (i.e. SmI2 with thiophenol present) only gives the direct reduction product 14
(87%). Interestingly, reaction of the same model ketone with SmI2 alone gives equimolar quantities of the reductive cyclisation product 15 and its unsaturated counterpart 16
(63% combined yield). It is presumed that this mixture of products derives from disproportionation of the tertiary radical resulting from 5-exo-trig cyclisation of the initially formed radical anion.
 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
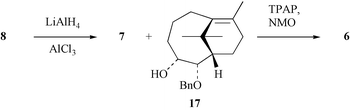
The reductive cleavage of photochemically generated oxetane 8 was also examined as an alternate means for preparing compound 12 (Scheme 5).15a,b However, when substrate 8 was treated with a mixture of lithium aluminium hydride (LAH) and aluminium trichloride, the product 17 (65%), which is isomeric with the hoped-for alcohol 12, was obtained along with small quantities (6%) of compound 7. Treatment of the major product of this reaction with the Ley–Griffith oxidant25 gave ketone 6 (85%) and this sequence of events is taken to imply that in the first step the oxetane 8 is isomerised to ketone 6 by the aluminium trichloride, which is, in turn, reduced to the observed alcohol by the LAH.
 | ||
| Scheme 5 | ||
Completion of the first synthesis of (−)-patchoulenone
The exploitation of compound 11 in the completion of our first synthesis of (−)-patchoulenone (1) is shown in Scheme 6. Thus, the diol was treated with the Swern reagent and the resulting acyloin 18 (91%) subjected to dehydration with thionyl chloride in pyridine. The ensuing enone 19 (68%) was reacted with the Gilman reagent derived from methyllithium and the commercially available copper(I) bromide/dimethyl sulfide complex.26 The enolate anion thus formed was trapped with trimethylsilyl chloride to give the silylenol ether 20. This compound was obtained as a single diastereoisomer (at C–1) but its instability prevented determination of the configuration at the newly created stereogenic centre. Dehydrogenation of compound 20 was most conveniently effected with DDQ and 2,6-lutidine27 and in this manner the title compound (1) was obtained (77% from 19) as a white crystalline solid. The physical and spectroscopic data obtained on this material match those reported1 for the natural product.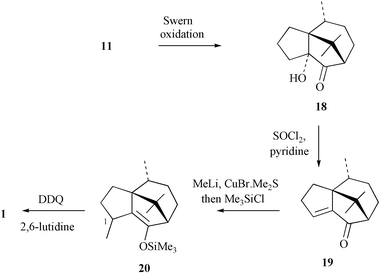 | ||
| Scheme 6 | ||
A second and more efficient synthesis of (−)-patchoulenone
The synthesis of (−)-patchoulenone outlined above is capable of improvement to the extent that the latter steps used for installing the C–1 methyl group could be avoided by incorporating this substituent at an earlier stage in the synthesis. To these ends, the previously reported11 ketone 21 (Scheme 7) was reacted with isopropenyllithium (generated in situ from 2-bromopropene and tert-butyllithium) and the nucleophilic addition product 22 thereby obtained in 89% yield. The illustrated stereochemistry associated with this product, which is expected on the basis of the blocking effect of the benzyloxy group within precursor 21, follows from its ready participation in a sodium hydride promoted anionic oxy-Cope rearrangement process to give, after protic work-up, the bicyclo[5.3.1]undec-7(8)-en-3-one 23 (76% from 21) as a white crystalline solid. The structure of the last compound was established by single-crystal X-ray analysis (Fig. 1 and Table 1), which reveals that the methyl group at C–4 is in the α-configuration and that the ketone and carbon-carbon double bond moieties are in a close spatial relationship to one another. As a consequence, and in keeping with the behaviour of its nor-analogue 6, compound 23 readily participated in a reductive cyclisation reaction when subjected to reaction with SmI2 in the presence of thiophenol. The product of this reaction, compound 24 (74%), was readily debenzylated with dihydrogen in the presence of palladium on carbon, to give diol 25 (97%), which was oxidised to acyloin 26 (66% at 79% conversion) with the Parikh–von Doering reagent. Dehydration of the last compound with thionyl chloride/pyridine then afforded (−)-patchoulenone (1) (72%), which was identical, in all respects, with the material obtained by the route described above.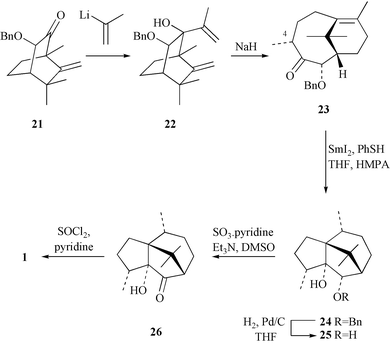 | ||
| Scheme 7 | ||
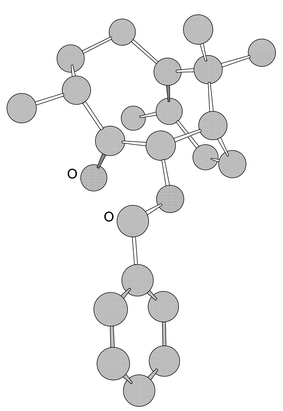 | ||
| Fig. 1 CS Chem3D ProTM drawing of compound 23 generated using data derived from an X-ray crystallographic study (hydrogen atoms omitted for clarity). | ||
| Formula | C22H30O2 |
| FW | 326.48 |
| Crystal system | monoclinic |
| Space group | P21 (#4) |
| a/Å | 10.017(1) |
| b/Å | 9.245(2) |
| c/Å | 11.062(1) |
| β/° | 112.073(7) |
| Z | 2 |
| T/°C | 23 |
| λ/Å | 1.54178 |
| μ/cm−1 | 5.18 |
| No. of reflections | 1621 |
| Unique reflections [I>3σ(I)] | 864 |
| S | 1.79 |
| R | 0.042 |
| wR | 0.033 |
Experimental
Unless otherwise specified,1H and 13C NMR spectra were recorded on a Varian Gemini 300 spectrometer using CDCl3 as solvent. Infrared spectra were recorded on either a Perkin–Elmer 683 or 1800 FTIR instrument while mass spectral analyses were conducted on a VG Micromass 7070F double-focussing spectrometer. Melting points were recorded on a Reichert hot-stage microscope and are uncorrected. Thin layer chromatographic analyses were carried out on aluminium-backed 0.2 mm thick silica gel 60 GF254 plates supplied by Merck while flash chromatographic purifications were conducted according to the method of Still et al.28 and using Merck silica gel 60 (230–400 mesh) as adsorbent. All solvents and common reagents were purified by established procedures.29Unless otherwise specified, all reactions were carried out under an atmosphere of dry nitrogen.
Synthetic studies
×20 mL). The combined organic extracts were washed with brine (1×20 mL), dried over magnesium sulfate and concentrated under reduced pressure. Purification by flash chromatography (15% ethyl acetate–petroleum ether) afforded 6 as colourless oil (196 mg, 90%); [α]D
−96.0 (c 1.3, CHCl3); Rf 0.37 (15% ethyl acetate–petroleum ether); λmax
(ε)
(EtOH) 245 (2475), 205 (13460), 201 (14230); (CH3CN) 237 (3125), 204 (sh, 15620) nm; νmax 1692 (s), 1639 (w), 1606 (w) cm−1; δH
(300 MHz, CDCl3) 7.34–7.27 (5H, m), 4.49 (1H, d, J 12.0 Hz), 4.26 (1H, d, J 12.0 Hz), 4.24 (1H, d, J 4.3 Hz), 2.40–2.00 (8H, m), 1.82–1.72 (3H, m), 1.44 (3H, s), 1.37 (3H, s), 1.07 (3H, s); δC
(75 MHz, CDCl3) 210.8, 138.1, 137.9, 134.5, 128.3, 127.8, 127.6, 81.2, 71.0, 56.0, 42.8, 36.3, 29.0, 27.2, 26.6, 26.5, 25.9, 20.4, 15.9; m/z 312 (M+, 7%), 294 (24), 221 (17), 204 (34), 188 (15), 175 (22), 165 (21), 152 (29), 135 (35), 121 (23), 109 (25), 91 (100). Found (HRMS): M+, 312.2084; C21H28O2 requires 312.2089.°C for 1 h, then quenched with sodium bicarbonate solution (10 mL of a saturated aqueous solution) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried over magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Purification by flash chromatography (10% ethyl acetate–petroleum ether) afforded 7 as colourless oil (73 mg, 97%), [α]D
−32.0 (c 2.0, CHCl3); Anal. found: C, 80.82; H, 8.84; C21H28O2 requires C, 80.73; H, 9.03%; Rf 0.45 (10% ethyl acetate–petroleum ether); νmax 3547 (m), 1606 (w), 1500 (m) cm−1; δH
(300 MHz, CDCl3) 7.39–7.28 (5H, m), 5.41 (1H, s), 4.73 (1H, d, J 11.8 Hz), 4.42 (1H, d, J 11.8 Hz), 3.92 (1H, d, J 6.5 Hz), 2.99 (1H, br s), 2.46 (1H, d, J 15.8 Hz), 2.12–1.73 (7H, m), 1.69 (3H, s), 1.58 (1H, m), 1.01 (3H, s), 1.00 (3H, s); δC
(75 MHz, CDCl3) 140.5, 138.7, 128.2, 127.5, 127.4, 121.1, 91.5, 83.8, 72.3, 65.2, 50.4, 42.1, 39.2, 27.5, 26.7, 25.6, 25.1, 23.3, 21.7; m/z 312 (M+, 17%), 294 (34), 221 (22), 204 (59), 189 (19), 175 (30), 147 (33), 135 (65), 121 (34), 101 (31), 91 (100). Found (HRMS): M+ 312.2086; C21H28O2 requires 312.2089.°C, at which point thin layer chromatography revealed the complete conversion of 8 into 7. Accordingly, the reaction mixture was quenched with sodium bicarbonate (10 mL of a saturated aqueous solution) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried over magnesium sulfate and concentrated under reduced pressure to give 7 as a colourless oil.°C. The reaction mixture was quenched with sodium bicarbonate solution (10 mL of a saturated aqueous solution) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried over magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Subjection of this material to flash chromatography (10% ethyl acetate–petroleum ether) afforded 7
(7.2 mg, 90%) as a colourless oil.°C. The mixture was then filtered to remove the catalyst and concentrated under reduced pressure. Subjection of the residue to column chromatography (25% ethyl acetate–petroleum ether, 2 elutions) gave 10
(12.6 mg, 21%) as a colourless oil; [α]D
+37.2 (c 0.7, CHCl3); Rf 0.22 (25% ethyl acetate–petroleum ether); νmax 3331 (br, s) cm−1; δH
(300 MHz, CDCl3) 4.24 (1H, app t, J 5.9 Hz), 2.97 (1H, d, J 6.0 Hz), 2.89 (1H, s), 2.21 (1H, m), 2.10–1.60 (9H, m), 1.50 (1H, m), 1.26–1.18 (1H, m), 1.08 (3H, d, J 7.8 Hz), 1.08 (3H, s), 0.91 (3H, s); δC
(75 MHz, CDCl3) 88.4, 76.6, 61.6, 51.6, 46.0, 40.1, 33.8, 31.2, 28.1, 25.6, 25.2, 24.4, 21.7, 19.4; m/z 224 (M+, 13%), 206 (46), 194 (50), 191 (40), 179 (24), 173 (27), 163 (42), 150 (45), 137 (49), 135 (43), 125 (73), 122 (61), 111 (100). Found (HRMS): M+ 224.1772; C14H24O2 requires 224.1776.A second fraction afforded 11 (35.5 mg, 59%) as a colourless oil; [α]D −21.4 (c 0.6, CHCl3); Rf 0.27 (25% ethyl acetate–petroleum ether); νmax 3312 (br s) cm−1; δH (300 MHz, CDCl3) 4.19 (1H, app t, J 6.4 Hz), 3.01 (1H, d, J 6.8 Hz), 2.44 (1H, s), 2.09–1.40 (12H, m), 1.11 (3H, d, J 6.8 Hz), 0.92 (6H, s); δC (75 MHz, CDCl3) 90.0, 75.4, 61.0, 52.0, 45.6, 40.5, 34.4, 28.1, 27.9, 27.2, 26.9, 22.8, 20.8, 18.5; m/z 224 (M+, 6%), 206 (27), 194 (20), 191 (16), 163 (14), 150 (19), 137 (18), 135 (22), 125 (100), 122 (40), 111 (80). Found (HRMS): M+ 224.1777; C14H24O2 requires 224.1776.
°C was treated dropwise with samarium(II) iodide (1.00 mL of a 0.1 M solution in THF, 0.100 mmol). The completion of the reaction was indicated by the appearance of a moderately persistent purple colour. The reaction mixture was then poured into water (10 mL) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried over magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Subjection of this material to flash chromatography (12% ether–petroleum ether, 2 elutions) gave 9
(10.8 mg, 54%) as a colourless oil; [α]D
+65 (c 0.4, CHCl3); Rf 0.35 (10% ethyl acetate–petroleum ether); νmax 3530 (m), 3331 (m), 1635 (s), 1606 (w) cm−1; δH
(300 MHz, CDCl3) 7.40–7.31 (5H, m), 4.78 (1H, s), 4.68 (1H, m), 4.64 (2H, m), 4.05 (1H, d, J 6.1 Hz), 3.26 (1H, s), 2.57 (1H, m), 2.24–1.53 (10H, m), 0.96 (3H, s), 0.83 (3H, s); δC
(75 MHz, CDCl3) 151.4, 138.1, 128.4, 127.7, 127.4, 107.5, 87.4, 84.3, 72.7, 67.4, 50.3, 46.0, 40.4, 29.7, 26.8, 26.7, 24.8, 23.3, 21.6; m/z 312 (M+, 3%), 294 (39), 221 (24), 204 (71), 203 (68), 185 (37), 175 (41), 161 (39), 147 (23), 135 (37), 133 (40), 119 (32), 105 (36), 91 (100). Found (HRMS): M+ 312.2096; C21H28O2 requires 312.2089.The second fraction afforded 12
(7.8 mg, 39%), mp 53–54°C; [α]D
+17.9 (c 0.9, CHCl3); Rf 0.48 (10% ethyl acetate–petroleum ether); νmax 3549 (s), 1605 (m) cm−1; δH
(300 MHz, CDCl3) 7.37–7.29 (5H, m), 4.60 (2H, m), 3.95 (1H, d, J 5.9 Hz), 3.32 (1H, s), 2.08–1.33 (12H, m), 1.14 (3H, d, J 6.8 Hz), 0.93 (3H, s), 0.91 (3H, s); δC
(75 MHz, CDCl3) 138.2, 128.4, 127.7, 127.4, 89.4, 83.1, 72.6, 60.9, 50.5, 45.7, 40.6, 34.7, 28.2, 28.1, 27.1, 27.0, 22.8, 21.5, 18.4; m/z 223 (M+−C7H7, 100%), 205 (22) 149 (13), 135 (35), 125 (15), 121 (12), 111 (18), 109 (16), 91 (71). Found (HRMS): M+−C7H7 223.1699; C14H23O2 requires 223.1698.
°C was treated dropwise with thiophenol (0.131 mL, 1.28 mmol), followed by samarium(II) iodide (0.96 mL of a 0.1 M solution in THF, 0.096 mmol). The completion of the reaction was indicated by the appearance of a persistent purple colour. The reaction mixture was then poured into potassium hydroxide (10 mL of a 0.5 M solution) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with potassium hydroxide (10 mL of a 0.5 M solution), then brine (1×10 mL), before being dried over magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Subjection of this material to flash chromatography (10% ethyl acetate–petroleum ether) gave 12
(7.1 mg, 71%) as a colourless oil. This material was identical, in all respects, with a sample of 12 obtained as described immediately above.°C (ice bath) and hydrochloric acid (20 mL of a 1 M aqueous solution) was added in one portion. Stirring was continued for 4 h after which sodium hydroxide (15 mL of a 1 M aqueous solution) was added, giving pH 7 (indicator paper). The mixture was extracted with diethyl ether (3×50 mL), the combined organic extract washed with brine (2×25 mL), dried over magnesium sulfate, concentrated under reduced pressure, then filtered through a short pad of silica (25% ethyl acetate–petroleum ether) to give tetrahydro-2-hydroxy-2H-pyran30
(5.77 g, 52%), bp 70°C (10 mmHg, Kugelrohr), as a colourless oil; Rf 0.21 (25% ethyl acetate–petroleum ether); νmax 3377 (br s) cm−1; δH
(300 MHz, CDCl3) 4.89 (1H, m), 4.25 (1H, d, J 4.9 Hz), 4.02 (1H, m), 3.54 (1H, m), 1.86–1.77 (2H, m), 1.54–1.49 (4H, m); δC
(75 MHz, CDCl3) 94.5, 63.9, 31.9, 25.2, 20.3; m/z 102 (M+, 35%), 101 (M+–H, 36), 85 (62), 84 (56), 83 (39), 74 (20), 69 (14), 56 (100).THF (86 mL) was added to a mixture of isopropyltriphenylphosphonium bromide (8.15 g, 0.0212 mol) and potassium hydride (656 mg, 0.0164 mol) and the resulting suspension was stirred at 18°C. A solution of tetrahydro-2-hydroxy-2H-pyran (2.11 g, 0.0207 mol) in THF (5 mL+5 mL washing) was added by cannula, resulting in the evolution of gas. The ensuing mixture was heated at reflux for 2 h after which time a pale pink colour developed. The cooled reaction mixture was diluted with water (50 mL) and extracted with diethyl ether (3×50 mL). The combined organic extract was washed with brine (2×25 mL), dried over sodium sulfate and concentrated under reduced pressure to give a pale yellow oil. Purification by flash chromatography (25% ethyl acetate–petroleum ether) afforded 6-methylhept-5-en-1-ol31
(2.12 g, 80%) as a colourless oil; Rf 0.36 (25% ethyl acetate–petroleum ether); νmax 3330 (br s), 1673 (w) cm−1; δH
(300 MHz, CDCl3) 5.12 (1H, m), 3.63 (2H, app q, J 6.1 Hz), 2.00 (2H, m), 1.84 (1H, t, J 5.2 Hz), 1.69 (3H, s), 1.60 (3H, s), 1.56 (2H, m), 1.40 (2H, m); δC
(75 MHz, CDCl3) 131.7, 124.4, 62.9, 32.4, 27.8, 26.0, 25.7, 17.7; m/z 128 (M+, 47%), 110 (14), 95 (54), 85 (12), 82 (100), 69 (87). Found (HRMS): M+ 128.1205; C8H16O requires 128.1201.
A magnetically stirred solution of 6-methylhept-5-en-1-ol (0.379 g, 2.96 mmol) in dichloromethane (6 mL) was treated with 4-methylmorpholine N-oxide (1.04 g, 8.86 mmol), powdered 4 Å molecular sieves (1.50 g) and tetrapropylammonium perruthenate (52 mg, 0.148 mmol). The reaction mixture was stirred for 1 h at 18°C, then filtered through silica (dichloromethane) and concentrated under reduced pressure to give a brown oil. Purification by flash chromatography (10% ethyl acetate–petroleum ether) afforded 6-methylhept-5-enal31
(0.370 g, 99%) as a colourless oil; Rf 0.44 (10% ethyl acetate–petroleum ether); νmax 1727 (s) cm−1; δH
(300 MHz, CDCl3) 9.76 (1H, t, J 1.8 Hz), 5.08 (1H, m), 2.42 (2H, td, J 7.3, 1.8 Hz), 2.03 (2H, q, J 7.3 Hz), 1.69 (3H, s), 1.67 [2H, m, (obscured)], 1.60 (3H, s); δC
(75 MHz, CDCl3) 202.9, 132.8, 123.3, 43.4, 27.3, 25.7, 22.3, 17.7; m/z
(GCMS) 126 (M+, 7%), 108 (M+−H2O, 5), 93 (M+−H2O−CH3, 3), 82 (100), 69 (30), 67 (80), 55 (28).
Methylmagnesium chloride (1.81 mL of a 3 M solution in THF, 5.43 mmol) was added, dropwise, to a magnetically stirred solution of 6-methylhept-5-enal (0.229 g, 1.81 mmol) in diethyl ether (9 mL) maintained at 0°C (ice bath) under a nitrogen atmosphere. The resulting mixture was then carefully quenched with water (20 mL) and extracted with diethyl ether (3×20 mL). The combined organic extract was washed with brine (1×20 mL), dried over sodium sulfate and concentrated under reduced pressure to give a pale yellow oil. Purification by flash chromatography (25% ethyl acetate–petroleum ether) afforded 7-methyloct-6-en-2-ol32
(14; 188 mg, 73%) as a colourless oil; Rf 0.38 (25% ethyl acetate–petroleum ether); νmax 3355 (br s) cm−1; δH
(300 MHz, CDCl3) 5.11 (1H, m), 3.79 (1H, m), 1.99 (2H, m), 1.69 (3H, s), 1.61 [1H, m (partially obscured)], 1.60 (3H, s), 1.50–1.32 (4H, m), 1.18 (3H, d, J 6.2 Hz); δC
(75 MHz, CDCl3) 131.7, 124.5, 68.1, 39.0, 28.0, 26.1, 25.8, 23.5, 17.7; m/z 142 (M+, 27%), 109 (21), 95 (31), 82 (100), 71 (38), 69 (42), 67 (47), 55 (31). Found (HRMS): M+ 142.1358; C9H18O requires 142.1358.
A magnetically stirred solution of 7-methyloct-6-en-2-ol (153 mg, 1.07 mmol) in dichloromethane (4.3 mL) was treated with 4-methylmorpholine N-oxide (379 mg, 3.23 mmol), powdered 4 Åmolecular sieves (0.500 g) and tetrapropylammonium perruthenate (19 mg, 0.0541 mmol). The ensuing mixture was stirred for 4 h at 18°C, then filtered through silica (dichloromethane) and concentrated under reduced pressure to give a brown oil. Purification by flash chromatography (10% ethyl acetate–petroleum ether) afforded 7-methyloct-6-en-2-one (13;33 144 mg, 95%) as a colourless oil; Rf 0.63 (25% ethyl acetate–petroleum ether); νmax 1717 (s) cm−1; δH
(300 MHz, CDCl3) 5.08 (1H, m), 2.44 (2H, t, J 7.45), 2.14 (3H, s), 1.99 (2H, m), 1.69 (3H, s), 1.66–1.56 [2H, m (obscured)], 1.59 (3H, s); δC
(75 MHz, CDCl3) 209.4, 132.4, 123.7, 43.2, 29.9, 27.3, 25.7, 24.0, 17.7; m/z 140 (M+, 13%), 82 (100), 67 (54).
°C (ice bath), was treated with thiophenol (0.146 mL, 1.43 mmol), followed by the dropwise addition of samarium(II) iodide (3.8 mL of a 0.1 M solution in THF, 0.380 mmol). The completion of the reaction was indicated by the persistence of a purple colour. The reaction mixture was then poured into potassium hydroxide (10 mL of a 0.5 M aqueous solution) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), then dried with magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Purification by flash chromatography (20% ethyl acetate–petroleum ether) gave 7-methyloct-6-en-2-ol (14; 18 mg, 87%) as a colourless oil and identical, in all respects, with the material obtained by the method described immediately above.°C (ice bath) under a nitrogen atmosphere. The completion of the reaction was indicated by the persistence of a purple colour. The reaction mixture was then poured into water (10 mL) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL) and concentrated under reduced pressure to give a colourless oil. Separation of the crude mixture by column chromatography (12% ethyl acetate–petroleum ether, 2 elutions) gave compound 1534
(6.2 mg, 31%); Rf 0.39 (20% ethyl acetate–petroleum ether); νmax 3375 (br m) cm−1; δH
(300 MHz, CDCl3) 1.93 (1H, m), 1.75–1.13 (8H, m), 1.19 (3H, s), 1.03 (3H, d, J 6.4 Hz), 0.89 (3H, d, J 6.5 Hz); δC
(75 MHz, CDCl3) 80.7, 56.4, 43.2, 29.4, 28.7, 22.4, 22.3, 22.0, 19.6; m/z
(GCMS) 142 (M+, 3%), 127 (M+−Me, 7), 124 (M+−H2O, 10), 109 (M+−H2O−CH3, 42), 95 (10), 82 (100), 71 (95), 67 (29), 58 (42).A second fraction afforded compound 16
(6.3 mg, 32%); Rf 0.36 (20% ethyl acetate–petroleum ether); νmax 3374 (br s), 3082 (m), 1644 (m) cm−1; δH
(300 MHz, CDCl3) 4.87 (1H, s), 4.74 (1H, s), 2.44 (1H, app t, J 8.9 Hz), 1.88 (1H, m), 1.81 (3H, s), 1.77–1.60 (6H, m), 1.14 (3H, s); δC
(75 MHz, CDCl3) 145.6, 111.5, 80.9, 57.3, 41.3, 28.1, 24.2, 23.0, 20.7; m/z
(GCMS) 140 (M+, 1%), 122 (M+−H2O−CH3, 8), 107 (5), 82 (100), 71 (25), 67 (41), 55 (23). Found (HRMS): M+ 140.1197; C9H16O requires 140.1201.
°C. The resulting mixture was treated with a solution of 8
(10.9 mg, 0.0349 mmol) in THF (0.25 mL+0.25 mL washing), then heated at 70°C for 8 h. After this time the reaction mixture was cooled to 0°C and quenched by the dropwise addition of ethyl acetate (0.5 mL), then tartaric acid (10 mL of a 1 M aqueous solution). Stirring was continued until clear layers resulted. The mixture was then extracted with diethyl ether (3×10 mL), the combined organic extract washed with brine (1×10 mL), dried over magnesium sulfate, then concentrated under reduced pressure. Purification by flash chromatography (15% ethyl acetate–petroleum ether) afforded 17
(7.1 mg, 65%) as a colourless oil, [α]D
−91 (c 0.2 CHCl3); Rf 0.43 (15% ethyl acetate–petroleum ether); νmax 3538 (m), 3461 (m) cm−1; δH
(300 MHz, CDCl3) 7.37–7.24 (5H, m), 4.54 (2H, m), 4.19 (1H, app t, J 10.4 Hz), 3.42 (1H, dd, J 5.0, 1.8 Hz), 2.41–2.28 (3H, m), 2.19–1.98 (4H, m), 1.89–1.80 (2H, m), 1.76 (3H, s), 1.63 (1H, m), 1.51 (1H, m), 1.43 (1H, d, exch), 1.16 (3H, s), 1.08 (3H, s); δC
(75 MHz, CDCl3) 142.0, 139.0, 128.2, 128.1, 128.0, 127.6, 127.4, 84.3, 72.0, 70.9, 49.0, 36.5, 31.2, 28.5, 28.0, 25.4, 22.7, 20.7, 17.1; m/z 314 (M+, 8%), 223 (13), 205 (20), 188 (16), 173 (25), 163 (19), 145 (47), 133 (23), 123 (25), 107 (36), 91 (100). Found (HRMS): M+ 314.2246; C21H30O2 requires 314.2246.°C and then filtered through silica (dichloromethane washing) and concentrated under reduced pressure to give a brown oil. Purification by flash chromatography (10% ethyl acetate–petroleum ether) afforded 6
(5.9 mg, 85%) as a colourless oil that was identical, in all respects, with authentic material.°C was treated with oxalyl chloride (49 μL, 0.559 mmol) and the resulting mixture stirred at −78°C for 0.5 h. A solution of diol 11
(41.8 mg, 0.186 mmol) in dichloromethane (0.5 mL+0.5 mL washing) was then added by cannula and the resulting mixture stirred at −78°C for 0.5 h. Triethylamine (156 µL, 1.12 mmol) was added and the mixture stirred at −78°C for 10 min, then allowed to warm to 18°C over 20 min. The reaction mixture was then poured into water (10 mL) and extracted with dichloromethane (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried with magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Subjection of this material to flash chromatography (10% ethyl acetate–petroleum ether) gave 18
(37.9 mg, 91%) as a colourless oil; [α]D
−0.19 (c 1.0, CHCl3); Rf 0.23 (10% ethyl acetate–petroleum ether); νmax 3456 (br s), 1736 (s) cm−1; δH
(300 MHz, CDCl3) 2.25 (1H, m), 2.26–1.55 (12H, m), 1.14 (3H, d, J 7.0 Hz), 1.04 (3H, s), 0.95 (3H, s); δC
(75 MHz, CDCl3) 225.0, 91.5, 60.8, 59.2, 42.2, 38.7, 33.4, 28.7, 27.5, 27.2, 26.3, 25.3, 20.6, 18.1; m/z 222 (M+, 0.5%), 194 (48), 179 (24), 161 (11), 151 (15), 137 (31), 125 (53), 124 (51), 111 (100). Found (HRMS): M+ 222.1619; C14H22O2 requires 222.1620.°C for 1 h, then poured onto ice (10 g) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried with magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Subjection of this material to flash chromatography (10% ethyl acetate–petroleum ether) gave 19
(13.0 mg, 68%) as a colourless oil; [α]D
−150 (c 0.5, CHCl3); Rf 0.37 (10% ethyl acetate–petroleum ether); νmax 1705 (s), 1639 (m) cm−1; δH
(300 MHz, CDCl3) 6.49 (1H, dd, J 3.6, 2.1 Hz), 2.78–2.55 (2H, m), 2.22 (1H, m), 2.11 (1H, app t, J 3.5 Hz), 2.04–1.88 (2H, m), 1.79–1.72 (2H, m), 1.59 (1H, m), 1.14 (1H, m), 1.05 (3H, s), 0.89 (3H, s), 0.86 (3H, d, J 6.5 Hz); δC
(75 MHz, CDCl3) 206.8, 148.4, 132.7, 63.1, 62.8, 40.9, 37.8, 34.8, 28.0, 27.3, 26.5, 26.2, 18.9, 17.7; m/z 204 (M+, 100%), 189 (17), 161 (48), 149 (38), 133 (49), 122 (30), 105 (30), 91 (52). Found (HRMS): M+ 204.1512; C14H20O requires 204.1514.°C then treated with methyllithium (445 µL of a 1.10 M solution in diethyl ether, 0.489 mmol). The initially yellow solution faded to colourless within 5 min, after which it was warmed to −40°C for 0.5 h. The solution was then re-cooled to −78°C and hexamethylphosphoramide (85 µL, 0.489 mmol) was added, followed by 19
(10 mg, 0.0489 mmol) in THF (0.5 mL+0.5 mL washing), giving a bright yellow solution that was immediately treated with trimethylsilyl chloride (62 µL, 0.489 mmol), resulting in rapid decolourisation. The reaction mixture was stirred for 0.5 h at −78°C, then quenched at this temperature with pH 7.4 buffer (10 mL) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried with magnesium sulfate and concentrated under reduced pressure to give enol ether 20 as a clear, colourless oil. This material was dissolved in dichloromethane (2 mL) and 2,6-lutidine (37 µL, 0.318 mmol) was added, followed by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (44.4 mg, 0.196 mmol). After 5 min the reaction mixture was poured into sodium bicarbonate (10 mL of a saturated aqueous solution) and extracted with dichloromethane (2×10 mL). The combined organic extract was washed with brine (1×10 mL), dried with magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Purification by flash chromatography (10% ethyl acetate–petroleum ether) afforded (−)-patchoulenone (1) as colourless crystals (8.2 mg, 77%), mp 50–51°C (lit.1 52.5°C); [α]D
−101 (c 0.4, CHCl3)
[lit.1
−97.1 (c 8.0)]; Rf 0.36 (10% ethyl acetate–petroleum ether); νmax 1710 (s), 1660 (s) cm−1; δH
(300 MHz, CDCl3) 2.83 (1H, m), 2.49 (1H, dd, J 18.2, 10.2 Hz), 2.23 (1H, m), 2.12 (3H, s), 2.11 (1H, m, partially obscured), 2.05–1.88 (2H, m), 1.82–1.57 (3H, m), 1.21 (1H, m), 1.67 (3H, s), 0.94 (3H, s), 0.89 (3H, d, J 6.5 Hz); δC
(75 MHz, CDCl3) 207.4, 148.6, 139.6, 63.6, 63.0, 43.4, 41.4, 34.6, 28.0, 26.4, 26.2, 25.9, 19.0, 17.9, 15.2; m/z 218 (M+, 100), 203 (43), 189 (14), 175 (78), 161 (40), 147 (50), 133 (29), 105 (30), 91 (31). Found (HRMS): M+ 218.1670; C15H22O requires 218.1671.°C under a nitrogen atmosphere, was charged with tert-butyllithium (6.07 mL of a 1.7 M solution in pentane, 0.0103 mol) and the resulting mixture stirred at −78°C for 15 min. Ketone 21
(294 mg, 1.03 mmol) in diethyl ether (1.0 mL+1.0 mL washing) was then added by cannula and the reaction mixture was stirred for 5 min at −78°C before being warmed to 18°C, then quenched with water (20 mL) and extracted with diethyl ether (3×20 mL). The combined organic extract was washed with brine (1×20 mL), dried with magnesium sulfate and concentrated under reduced pressure. Subjection of the residue to flash chromatography (10% ethyl acetate–petroleum ether) afforded 22
(0.299 g, 89%) as a colourless oil. This material was immediately dissolved in THF (5.0 mL+4.0 mL washing) and the resulting solution was added to a flask containing sodium hydride (41.8 mg, 1.74 mmol). The ensuing mixture was heated at reflux for 3 h, then cooled, quenched with water (20 mL) and extracted with diethyl ether (3×20 mL). The combined organic extract was washed with brine (20 mL), dried over magnesium sulfate and concentrated under reduced pressure. Subjection of the residue to flash chromatography (10% ethyl acetate–petroleum ether) afforded 23
[257 mg, 86%
(76% from 21)] as a colourless oil, mp 100–102°C; [α]D
−60 (c 1.0, CHCl3); Rf 0.36 (10% ethyl acetate–petroleum ether); λmax
(ε)
(EtOH) 252 (2108), 207 (11468); (CH3CN) 248 (2650), 204 (sh, 16151); (hexane) 244 (2748), 196 (18655) nm; νmax 1691 (s), 1609 (w), 1585 (w) cm−1; δH
(300 MHz, CDCl3) 7.33–7.24 (5H, m), 4.48 (1H, d, J 12.0 Hz), 4.26 (1H, d, J 5.4 Hz), 4.18 (1H, d, J 12.0 Hz), 2.70 (1H, m), 2.40–2.27 (3H, m), 2.21–1.95 (3H, m), 1.91–1.70 (2H, m), 1.54 (1H, m), 1.44 (3H, s), 1.42 (3H, s), 1.07 (3H, s), 0.92 (3H, d, J 6.7 Hz); δC
(75 MHz, CDCl3) 216.2, 138.3, 136.1, 135.0, 128.3, 127.5, 84.5, 71.0, 55.9, 45.0, 36.3, 36.2, 29.9, 28.3, 27.3, 25.4, 20.5, 18.2, 15.7 (one signal overlapping or obscured); m/z 326 (M+, 11%), 308 (15), 235 (18), 218 (76), 189 (18), 165 (30), 149 (48), 137 (43), 123 (17), 109 (31), 91 (100). Found (HRMS): M+ 326.2246; C22H30O2 requires 326.2246.°C, was treated with thiophenol (0.314 mL, 3.06 mmol) and then, dropwise, samarium(II) iodide (3.8 mL of a 0.1 M solution in THF, 0.380 mmol). The completion of the reaction was indicated by the persistence of a purple colour. The reaction mixture was then poured into potassium hydroxide (1×10 mL of a 0.5 M solution) and extracted with diethyl ether (3×10 mL). The combined organic extract was washed with potassium hydroxide (10 mL of a 0.5 M solution) and brine (1×10 mL), then dried with magnesium sulfate and concentrated under reduced pressure to give a colourless oil. Subjection of this material to flash chromatography (10% diethyl ether–petroleum ether) gave 24
(37 mg, 74%) as a colourless oil; [α]D
+21.2 (c 2.1, CHCl3); Rf 0.47 (5% diethyl ether–petroleum ether); νmax 3530 (s), 1606 (w) cm−1; δH
(300 MHz, CDCl3) 7.38–7.29 (5H, m), 4.63–4.54 (2H, m), 3.85 (1H, d, J 5.9 Hz), 2.96 (1H, s), 2.09–2.06 (2H, m), 1.87–1.48 (8H, m), 1.33 (1H, m), 1.13 (3H, d, J 6.6 Hz), 0.94 (3H, s), 0.93 (3H, s), 0.89 (3H, d, J 6.4 Hz); δC
(75 MHz, CDCl3) 138.2, 128.4, 127.7, 127.5, 88.9, 81.7, 72.3, 60.9, 50.3, 46.2, 40.6, 35.5, 35.4, 28.5, 28.1, 25.8, 23.3, 21.6, 18.5, 12.6; m/z 328 (M+, 0.4%), 237 (94), 219 (31), 207 (20), 201 (11), 177 (10), 163 (25), 149 (52), 139 (29), 125 (31), 123 (25), 121 (25), 109 (31), 91 (100). Found (HRMS): M+ 328.2401; C22H32O2 requires 328.2402.°C the reaction mixture was filtered to remove the catalyst, concentrated under reduced pressure and the residue filtered through a short plug of silica (15% ethyl acetate–petroleum ether) to afford 25
(24.8 mg, 97%) as a crystalline solid, mp 168–169°C (sealed tube); [α]D
−17.8 (c 0.5, CHCl3); Rf 0.38 (15% ethyl acetate–petroleum ether); νmax 3349 (br s) cm−1; δH
(300 MHz, CDCl3) 4.13 (1H, dd, J 7.3, 6.0 Hz), 2.43 (1H, d, J 7.4 Hz), 2.18 (1H, s), 2.07–1.39 (11H, m), 1.11 (3H, d, J 6.8 Hz), 0.97 (3H, s), 0.92 (3H, s), 0.92 (3H, d, J 6.6 Hz); δC
(75 MHz, CDCl3) 89.1, 74.4, 60.9, 52.3, 46.0, 40.5, 35.4, 35.2, 28.3, 28.0, 25.8, 23.2, 20.7, 18.6, 12.3; m/z 238 (M+, 25%), 220 (27), 208 (19), 180 (15), 152 (39), 139 (100), 125 (55), 122 (30), 107 (18). Found (HRMS): M+ 238.1937; C15H26O2 requires 238.1933.°C for 24 h, then at 35°C for 24 h and then diluted with water (10 mL) and extracted with hexane (3×10 mL). The combined organic extract was washed with brine (1×10 mL), dried with magnesium sulfate and concentrated under reduced pressure to afford a light yellow oil. Subjection of this material to flash chromatography (10% ethyl acetate–petroleum ether) afforded 25
(3.4 mg, 21% recovery) and ketone 26
(8.3 mg, 66% at 79% conversion) as colourless crystals, mp 107–108°C (sealed tube); [α]D
+11.2 (c 0.6, CHCl3); Rf 0.53 (15% ethyl acetate–petroleum ether); νmax 3522 (s), 3434 (s), 1728 (s) cm−1; δH
(300 MHz, CDCl3) 2.29–2.17 (3H, m), 1.99–1.55 (9H, m), 1.12 (3H, d, J 6.7 Hz), 1.03–1.01 (9H, m); δC
(75 MHz, CDCl3) 224.9, 89.9, 60.9, 59.8, 43.2, 38.9, 35.1, 34.2, 29.7, 27.6, 26.3, 25.3, 21.1, 18.0, 13.1; m/z 236 (M+, 0.7%), 208 (78), 193 (22), 165 (11), 151 (37), 139 (63), 138 (60), 125 (100), 110 (48). Found (HRMS): M+ 236.1773; C15H24O2 requires 236.1776.°C and stirred at this temperature for 22 h. The ensuing mixture was cooled to 18°C, diluted with diethyl ether (10 mL), then poured into water (10 mL). The aqueous phase was separated and re-extracted with ether (2×10 mL). The combined organic extract was washed with brine (1×10 mL), dried with magnesium sulfate and concentrated under reduced pressure to afford a light yellow oil. Subjection of this material to flash chromatography (8% ethyl acetate–petroleum ether) afforded (−)-patchoulenone (1; 5.5 mg, 72%) as colourless crystals. This material was identical, in all respects, with the sample obtained as described earlier.Crystal data and refinement details for compound 23
Crystallographic data for the title compound are given in Table 1. Intensity data were collected on a Rigaku AFC6R diffractometer using the ω−2θ scan technique to a maximum 2θ value of 120°. Scans of (1.20+0.30 tanθ)° were made at a speed of 16° min−1
(in omega). The weak reflections [I<10.0σ(I)] were rescanned (maximum of four scans) and the counts were accumulated to ensure good counting statistics. Stationary background counts were recorded on each side of the reflection. The ratio of peak counting time to background counting time was 2∶1.Three representative reflections were measured after every 150 reflections, which revealed that no decay correction was required. An empirical absorption correction based on azimuthal scans was applied, which resulted in transmission factors ranging from 0.94 to 1.00. The data were corrected for Lorentz and polarisation effects.
The structure was solved by direct methods35 and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included at geometrically determined positions, which were periodically recalculated but were not refined. The final cycle of full-matrix least-squares refinement was based on 864 observed reflections [I>3.00σ(I)] and 217 variable parameters. The maximum and minimum peaks on the final difference Fourier map correspond to 0.13 and −0.11 e
Å−3, respectively. All calculations were performed using the teXsan36 crystallographic software package of Molecular Structure Corporation.
CCDC reference number 188927. See http://www.rsc.org/suppdata/nj/b2/b206372g/ for crystallographic files in CIF or other electronic format.
Acknowledgements
We thank the Institute of Advanced Studies for financial support, including the provision of a post-Doctoral Fellowship to MM, and Drs G. Whited and L. Kwart [Genencor International Inc. (Palo Alto)] for providing generous supplies of the cis-1,2-dihydrocatechol 4. Helpful discussions with Professor A. L. J. Beckwith (ANU) are gratefully acknowledged.References
- B. Trivedi, O. Motl, V. Herout and F. Sorm, Collect. Czech. Chem. Commun., 1964, 29, 1675 CAS.
- C. Thebtaranonth, Y. Thebtaranonth, S. Wanauppathamkul and Y. Yuthavong, Phytochemistry, 1995, 40, 125 CrossRef CAS.
- A. Hisham, V. Wray, L. Pieters, M. Claeys, R. Dommisse and A. Vlietinck, Magn. Reson. Chem., 1992, 30, 295 CAS.
- H. Achenbach and A. Schwinn, Phytochemistry, 1995, 38, 1037 CrossRef CAS.
- H. Hikino, K. Aota and T. Takemoto, Chem. Pharm. Bull., 1966, 14, 890 CAS.
- B. Trivedi, O. Motl, J. Smoliková and F. Sorm, Tetrahedron Lett., 1964, 1197 CrossRef CAS.
- G. Büchi, W. D. MacLeod, Jr. and J. Padilla O., J. Am. Chem. Soc., 1964, 86, 4438 CrossRef and refererences cited there in.
- H. Hikino, K. Ito, K. Aota and T. Takemoto, Chem. Pharm. Bull., 1968, 16, 43 CAS.
- W. F. Erman and L. C. Stone, J. Am. Chem. Soc., 1971, 93, 2821 CrossRef CAS.
- For reviews on the applications of cis-1,2-dihydrocatechols in synthesis see: (a) D. A. Widdowson, D. W. Ribbons and S. D. Thomas, Janssen Chim. Acta, 1990, 8, 3 Search PubMed; (b) H. A. J. Carless, Tetrahedron: Asymmetry, 1992, 3, 795 CrossRef CAS; (c) S. M. Brown and T. Hudlicky, in Organic Synthesis: Theory and Applications, ed. T. Hudlicky, JAI, Greenwich, CT, 1993, vol. 2, p. 113 Search PubMed; (d) T. Hudlicky and J. W. Reed, in Advances in Asymmetric Synthesis, ed. A. Hassner, JAI, Greenwich, CT, 1995, vol. 1, p. 271 Search PubMed; (e) T. Hudlicky and A. J. Thorpe, Chem. Commun., 1996, 1993 RSC; (f) T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35 Search PubMed.
- M. G. Banwell, P. Darmos, D. C. R. Hockless and M. D. McLeod, Synlett, 1998, 897 CAS.
- R. A. Holton and A. D. Williams, J. Org. Chem., 1988, 53, 5981 CrossRef CAS.
- For reviews on the Prins and related carbonyl-ene reactions see: (a) B. B. Snider, in Comprehensive Organic Synthesis, eds. B. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 2, p. 527 Search PubMed; (b) D. R. Adams and S. P. Bhatnagar, Synthesis, 1977, 661 CrossRef CAS. For some recent articles concerning the mechanism of the carbonyl-ene cyclisation reaction see: (c) J. A. Marshall and M. W. Andersen, J. Org. Chem., 1992, 57, 5851 CrossRef CAS; (d) D. C. Braddock, K. K. Hii and J. M. Brown, Angew Chem., Int. Ed., 1998, 37, 1720 CrossRef CAS.
- Oxetane formation has been observed during the course of an intermolecular carbonyl-ene reaction: H. Kwart and M. Brechbiel, J. Org. Chem., 1982, 47, 5409. Search PubMed We are not aware of any examples of oxetane formation being observed during the course of a Prins reaction.
- For related examples of intramolecular Paterno-Büchi reactions see: (a) J. Meinwald and A. T. Hamner, J. Chem Soc., Chem. Commun., 1969, 1302 RSC; (b) G. L. Lange and M. Bosch, Tetrahedron Lett., 1971, 315 CrossRef CAS; (c) C. A. Dvorak, C. Dufour, S. Iwasa and V. H. Rawal, J. Org. Chem., 1998, 63, 5302 CrossRef CAS.
- For other examples of the carbonyl-ene reaction promoted by the mild Lewis acid SnCl2 see: G. Foster and P. Johnson, Br. Pat. 1205397 (Cl.c 07c), 1970(Chem. Abs., 1970, 73, 131809a).
- G. A. Molander and C. Kenny, J. Am. Chem. Soc., 1989, 111, 8236. For a useful recent review of this type of reduction/cyclisation sequence see: G. A. Molander and C. R. Harris, Chem. Rev., 1996, 96, 307 Search PubMed.
- Curran et al.18a have observed that the SmI2-mediated reduction of tertiary radicals to the corresponding anion/alkyl samarium species is an inefficient process with the result that the rather long-lived radical can engage in disproportionation reactions. The rate constant for dispropotionation of the t-butyl radical in solution is ca. 108 M−1 s−118b while the rate constant for reduction of a δ,ω-unsaturated tertiary radical by SmI2 has been estimated, by Curran and his co-workers,18a as being less than 104 M−1 s−1. (a) D. P. Curran, T. L. Fevig, C. P. Jasperse and M. J. Totleben, Synlett, 1992, 943 CrossRef CAS; (b) M. J. Gibian and R. C. Corley, Chem. Rev., 1973, 73, 441 CrossRef CAS.
- The formation of compound 9 from isomer 6 cannot be due to a SmI3-promoted carbonyl-ene reaction since treatment of the latter material with SmI3 (generated from SmI2 and di-iodomethane) in THF–HMPA does not lead to any reaction.
- In principle, tris-(trimethylsilyl)silylhydride20a and Bu3SnH20b promoted reductive cyclisation of compound 6 should afford target 12 but these processes proved to be very inefficient. (a) K. J. Kulicke, Synlett, 1992, 91; (b) E. J. Enholm, Tetrahedron Lett., 1989, 37, 4939 CrossRef.
- The rate constant for hydrogen atom transfer from thiophenol to the t-butyl radical is estimated to be ca. 108 M−1 s−1: J. A. Franz, B. A. Bushau and M. S. Alnajjar, J. Am. Chem. Soc., 1989, 111, 268 Search PubMed.
- Ogawa et al. have exploited thiophenol in H-atom transfer to a radical generated under SRN1 conditions and involving SmI2 as a reducing agent: A. Ogawa, S. Ohya and T. Hirao, Chem. Lett., 1997, 275 Search PubMed.
- Only trace amounts of diphenyldisulfide are observed in the reaction mixture due to its ready reductive cleavage by SmI2: X. Jia, Y. Zhang and X. Zhou, Synth. Commun., 1994, 24, 387 Search PubMed.
- Ketone 13 was prepared by the route detailed in the Experimental.
- S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639 CrossRef.
- H. O. House, C.-Y. Chu, J. M. Wilkins and M. J. Umen, J. Org. Chem., 1975, 40, 1460 CrossRef CAS.
- (a) I. Fleming and I. Paterson, Synthesis, 1979, 736 CrossRef CAS; (b) T. L. Fevig, R. L. Elliott and D. P. Curran, J. Am. Chem. Soc., 1988, 110, 5064 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- D. D. Perrin and W. L. F. Amarego, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 3rd edn., 1988 Search PubMed.
- G. F. Woods Jr., Org. Synth. Coll. Vol., 1955, 3, 470 Search PubMed.
- K. S. Shrestha, K. Honda, M. Asami and S. Inoue, Bull. Chem. Soc. Jpn., 1999, 72, 73 CrossRef CAS.
- J. K. Crandall and M. Mualla, Tetrahedron Lett., 1986, 27, 2243 CrossRef CAS.
- J.-G. Jun, D. W. Lee and B. P. Mundy, Synth. Commun., 1998, 28, 2499 CAS.
- T. Shono, I. Nishiguchi, H. Ohmizu and M. Mitani, J. Am. Chem. Soc., 1978, 100, 545 CrossRef CAS.
- A. Altomare, M. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- teXsan: Single Crystal Structure Analysis Software, v. 1.8, Molecular Structure Corporation, The Woodlands, TX, 77381, USA, 1997.
Footnote |
| † Aspects of this work have been reported in preliminary form: M. Banwell and M. McLeod, Chem. Commun., 1998, 1851. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |