Self-assembling resorcinarene capsules: solid and gas phase studies on encapsulation of small alkyl ammonium cations
Heidi
Mansikkamäki
a,
Maija
Nissinen
a,
Christoph A.
Schalley
b and
Kari
Rissanen
*a
aDepartment of Chemistry, University of Jyväskylä, PO Box 35, FIN-40014, University of Jyväskylä, Finland. E-mail: hemansik@st.jyu.fi, majoni@cc.jyu.fi, kari.rissanen@jyu.fi; Fax: +358 14 260 2501; Tel: +358 14 260 2672
bKekulé-Institut für Organische Chemie und Biochemie der Universität, Gerhard-Domagk-Str. 1, D-53121 Bonn, Germany. E-mail: c.schalley@uni-bonn.de
First published on 22nd November 2002
Abstract
The self-assembling process of unsubstituted resorcinarenes (1, 2 and 3) to dimeric capsules encapsulating small tetra-alkyl ammonium cations 4–7 has been studied in solid and gaseous states by X-ray crystallographic and mass spectrometric methods. Hydrogen bonding and cation-π interaction as well as the proper encapsulation in the empty cavity of the capsule appear to be the most important interactions in the capsule formation process. Competitive mass spectrometric studies clearly indicated preference of the tetramethyl ammonium cation (4) over tetraethyl ammonium cation (6) and especially tetrabutyl ammonium cation (7). The crystal structures of altogether eight dimeric capsules of resorcinarenes 1–3 with cations 4 and 5 were determined. In the solid state, the alkyl chain length of the host affects the crystal packing significantly. However, ethyl resorcinarene (2) is the only host, which binds the spherical halide anion (Cl− or Br−) in between the lower rim alkyl chains.
Introduction
Recently, supramolecular reversible encapsulation and self-assembling molecular aggregates with molecular recognition properties have attracted worldwide interest.1 These objectives are met with resorcinarenes which are versatile and easily available host compounds for complexation studies in solution, in the gas phase and in the solid state.2 One of the most interesting and elegant features of unsubstituted alkyl resorcinarenes is their capability to form dimeric3 and hexameric4 molecular capsules/containers encapsulating neutral and/or charged guests inside the cavity.The studies of the complexation of alkyl ammonium cations, e.g. biologically significant acetylcholine,5a by resorcinarenes and their tetraphenolate derivatives have demonstrated the complexation of suitably sized ammonium cations.3,5,6 Our recent interest is focused on the self-assembling hydrogen-bonded capsule formation process and recognition properties of unsubstituted alkyl resorcinarenes by complexation and encapsulation of ammonium cations. The complexation owes mainly to the electrostatic attraction between the π-basic cavity of the resorcinarene capsule and the size of the positively charged cation.
Most of the early studies manifest monomeric 1∶1 complexation, while 2∶1 complexation to dimeric capsules encapsulating alkyl ammonium cations has been observed in solid state studies.3 These studies emphasize the significance of the cation size for successful encapsulation, while the effect of the counterion and solvent is small.3,6 Recently, we have observed an interesting simultaneous encapsulation of diquats concomitant with binding of spherical anions such as Cl− and Br− as a bridge between the lower rim ethyl chains of ethyl resorcinarene (2).3d
In the previous studies,3a–c the dimeric capsule was reported to be eclipsed, yet our most recent study3d revealed the formation of a staggered capsule. Gas phase studies6 have indicated the possibility of obtaining directly hydrogen-bonded capsules without solvent or anion mediation, an interesting feature, which we believe to be achievable also in solid state when suitable crystallisation conditions are found.
The mass spectrometric examination of non-covalently bound supramolecular aggregates7 is not always straightforward. In particular, hydrogen-bonded aggregates often cause problems due to the use of competitive or even protic solvents or matrices. Nevertheless, several successful examples for such systems have been reported, among them rosette-type complexes,8 molecular boxes,9 hydrogen-bonded ferrocene oligomers,10 and self-assembling capsules.11 As a bottom line of all these studies, it is still a challenge to the imagination of the researcher to find suitable conditions for ionisation. If non-competitive conditions have to be used, it is advantageous to pre-generate the ions of interest in solution or the matrix before ionisation. This can be done by covalent addition of a crown ether–alkali metal ion complex8a or by attachment of an anion to the hydrogen bonding system8b,c as long as it does not destroy or alter the binding between the subunits too much. Other possibilities include sandwich complexes of Ag+ ions9 or the oxidation of ferrocene units by addition of iodine to the sample just before the ESI-MS experiment.10 In the case of capsules, which accept guests inside their cavity, this guest, e.g. a quaternary ammonium ion can of course be charged.11 Calixarene- and resorcinarene-based hydrogen-bonded capsules and the corresponding monomeric complexes have been the subject of several previous studies,6,12 some of which belong to the earliest examples for true gas-phase complexation studies.13
Based on the previously obtained results in solid and gaseous states, we wanted to study the factors influencing the ammonium cation induced self-assembly to dimeric capsules by investigating systematically the significance of the cation and anion size. The alkyl chain length was varied to survey the anion binding properties of the lower rim and packing of the capsules in the crystal lattice. The solid state studies were performed to find out the detailed structures of the capsules. In the mass spectral studies, the data obtained for the capsules with singly charged guest ions will be discussed; particular focus will be on the determination of structural features of these compounds.
Results and discussion
X-Ray crystallography and solid state capsules
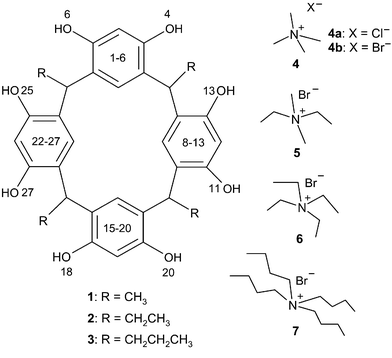 | ||
| Scheme 1 Structural formulae and relevant crystallographic numbering of resorcinarenes. | ||
| Solvent | 1 | 2 | 3 | |
|---|---|---|---|---|
| a Preliminary results. | ||||
| 4a | MeOH | Powder precipitate | 4 +@22·Cl−·6MeOH·H2O | 4 +@32·Cl−·4MeOH |
| EtOH | Capsulea | 4 +@22·Cl−·4.5H2O | 4 +@32·Cl−·4EtOH | |
| 4b | MeOH | 4 +@12·Br−·4MeOH·3H2O | 4 +@22·Br−·8MeOH | Powder precipitate |
| EtOH | Capsulea | Capsulea | 4 +@32·Br−·4EtOH | |
| 5 | MeOH | Powder precipitate | Capsulea | 5 +@32·Br·MeOH·0.5H2O |
| EtOH | Capsulea | Capsulea | Capsulea | |
In the capsule, 4+@12·Br−·4MeOH·3H2O, two molecules of 1 are linked via MeOH molecules, water molecules and bromide (disordered over two positions with one of the water molecules) and, as expected, the TMA cation (4+) is located inside the cavity formed by the resorcinarene hosts (Fig. 1). The distances of the methyl groups of the cation to the centroids of the aromatic groups of the resorcinarene host (3.41 and 3.62 Å) indicate C–H⋯π interaction between the host and the guest. The resorcinarene molecules adopt a slightly distorted cone conformation that is flattened on the sides where the Br− anion is mediating the capsule (Table 2). The capsule is staggered and slightly tighter, i.e. the capsule halves are slightly closer to each other than in our previous study with diquats,3d due to the smaller size of the cation, the distance between the planes defined by the methine bridges of the hosts being 8.36 Å. Survey of the crystal packing reveals perpendicular columns formed by sequential capsules (Fig. 2).
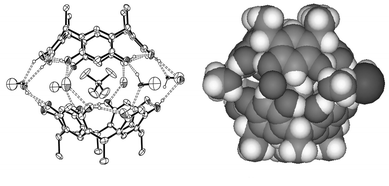 | ||
| Fig. 1 Ortep16 and CPK plots of the X-ray structure of hydrogen-bonded solvent–anion mediated resorcinarene 1 capsule 4+@12·Br−·4MeOH·3H2O. Non-hydrogen bonding molecules have been omitted for clarity from the Ortep plot. | ||
| Capsule | 4 +@12Br−·4MeOH·3H2O | 4 +@22·Cl−·6MeOH·H2O | 4 +@22·Cl−·4.5H2O | 4 +@22·Br−·8MeOH | 4 +@32·Cl−·4MeOH | 4 +@32·Cl−·4EtOH | 4 +@32·Br−·4EtOH | 5 +@32·Br−·4MeOH·0.5H2O |
|---|---|---|---|---|---|---|---|---|
| a Defined as a distance between the planes formed by the methine bridges of the resorcinarenes. b The angle is measured as a torsion angle between lower rim methine carbon of the upper host, centroid of the plane defined by methine carbons of the upper host, corresponding centroid of the lower host and the closest methine carbon of the lower host. c Asymmetric unit contains two unequal halves of the capsule. | ||||||||
| Capsule mediating hydrogen bonds to solvents/Å | 2.62–3.12 | 2.64–2.86 | 2.69–2.91 | 2.67–2.69 | 2.68–2.88 | 2.66–2.74 | 2.66–2.75 | 2.60–2.72 |
| Capsule mediating hydrogen bonds to anions/Å | 3.10 | 2.86 | 3.04 | — | 3.07–3.79 | 3.11–3.14 | 3.23, 3.25 | 3.23 |
| Distance between the capsule halves/Åa | 8.36 | 8.77, 8.80c | 8.35 | 8.86 | 8.34 | 8.53 | 8.59 | 8.98 |
| Distance between the centroids of opposite aromatic rings of a resorcinarene molecule/Å | 6.62/7.11 | 6.62/7.07 | 6.71/6.99 | 6.87/6.87 | 6.88/6.90 | 6.77/6.99 | 6.79/6.97 | 6.86/6.95 |
| 6.61/7.07c | ||||||||
| C–H⋯π interactions between cation and resorcinarenes/Å | 3.40–4.47 | 3.50–3.77 | 3.33–4.22 | 3.36–3.86 | 3.45–4.03 | 3.16–4.04 | 3.35–4.04 | 3.64–3.96 |
| Torsion angle between the capsule halves/°b | 40.4 | 42.8 | 40.0 | 0.0 | 34.8 | 0.1 | 0.1 | 0.1 |
| Number of intra/inter molecular hydrogen bonds | 4/16 | 3/16 | 4/13 | 4/12 | 4/12 | 4/12 | 4/12 | 4/13 |
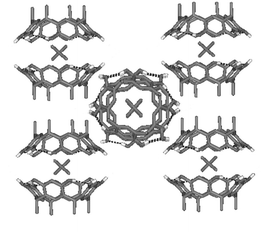 | ||
| Fig. 2 Crystal packing of 4+@12·Br−·4MeOH·3H2O showing the perpendicular columns formed by sequential resorcinarene capsules. Solvent molecules, anions and non-hydrogen bonding hydrogens are omitted for clarity. | ||
Preliminary crystallisation studies indicate encapsulation of both cations 4+ and 5+ in dimeric 1 in aqueous ethanol solution,15 but owing to the poor quality and instability of crystals formed, good enough X-ray data could not be obtained from either of them.
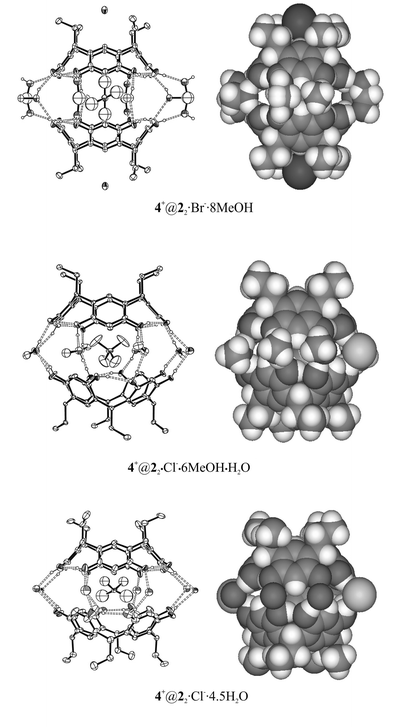 | ||
| Fig. 3 X-ray structures of the resorcinarene capsules of 2. 4+@22·Br−·8MeOH is mediated via methanol molecules, 4+@22·Cl−·6MeOH·H2O is methanol–anion mediated and 4+@22·Cl−·4.5H2O water–anion mediated. In capsule 4+@22·Cl−·6MeOH·H2O, the anion is simultaneously found in between the ethyl chains (see Fig. 2 in ref. 3d). Non-hydrogen-bonded hydrogen atoms are omitted for clarity from Ortep15 pictures. Only one of the crystallographically independent capsule halves is shown in the picture of 4+@22·Cl−·6MeOH·H2O. | ||
The two capsules 4+@22·Cl−·6MeOH·H2O and 4+@22·Br−·8MeOH obtained from methanol resemble closely the earlier results of the pair of capsules of 2 with larger diquats 1,4-diazoniabicyclo[2.2.2]octane dichloride and 1,4-dimethyl-1,4-diazoniabicyclo[2.2.2]octane dibromide,3d the first one being staggered and anion–solvent mediated and the latter one being eclipsed and only methanol mediated. However, in this case the cation is the same in both structures, thus the structure of the capsule cannot be explained by the size of the cation, but most probably the staggered structure relates to the capsule-mediating role of the anion. The role of the anions in crystal packing is also similar to the previously observed diquat capsules, i.e. the anions of 4+@22·Cl−·6MeOH·H2O and 4+@22·Br−·8MeOH are found in between the lower rim ethyl chains of the resorcinarene host and in the former capsule the anion is simultaneously mediating the capsule halves (Fig. 3 and ref. 3d).
When 2 was crystallised with 4a from aqueous ethanol, capsule 4+@22·Cl−·4.5H2O mediated by 4.5 water molecules and two chloride anions resulted, which means that on average 13 intracapsular hydrogen bonds are formed. No ethanol is present in the crystal, although only a trace of water was used in the crystallisation. The orientation of the capsule is eclipsed, which most probably relates to the small size of the mediating solvents and the capsule-mediating role of chloride.
In both staggered capsules 4+@22·Cl−·6MeOH·H2O and 4+@22·Cl−·4.5H2O, the conformation of the resorcinarenes is slightly distorted, while the conformation of resorcinarenes in eclipsed capsule 4+@22·Br−·8MeOH is near perfect (Table 2). The reason for this is the fact that the capsule mediating solvents consist only of methanol molecules, while the chlorides mediating the dimerisation cause a distortion from the perfect conformation in staggered capsules.
In all three capsules of 2, the cation is tightly embedded inside the capsule. Complex-stabilising C–H⋯π interactions between aromatic parts of the host and the guest are evident on the basis of the closest distances between the methyl groups of the cation and the centroids of the aromatic resorcinol groups (3.33–3.86 Å).
All three capsules have a different type of packing pattern. The packing of capsule 4+@22·Br−·8MeOH builds up of parallel columns formed by sequential capsules, the interstice of which is filled by a hydrogen-bonded bromide–methanol network (Fig. 4).
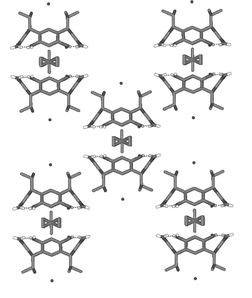 | ||
| Fig. 4 Crystal packing pattern of 4+@22·Br−·8MeOH. Non-hydrogen bonding hydrogens and disordered solvent molecules filling the interstice are excluded for clarity. | ||
Capsule 4+@22·Cl−·6MeOH·H2O reveals a similar crystal packing as the capsule of 2 with 1,4-diazoniabicyclo[2.2.2]octane dichloride reported by us previously.3d In both capsules, the chloride anions are mediating the mutual binding of the two capsule halves and are simultaneously located between the lower rim ethyl chains of the adjacent perpendicularly positioned capsules. Capsule 4+@22·Cl−·4.5H2O has a similar type of crystal packing when compared to the capsules of propyl resorcinarene (3) (Fig. 6), and is discussed in the following paragraph.
 | ||
| Fig. 5 X-ray structures of resorcinarene 3 capsules have great similarity to each other. All capsules are solvent–anion mediated. Non-hydrogen-bonded atoms have been omitted for clarity from Ortep plots. | ||
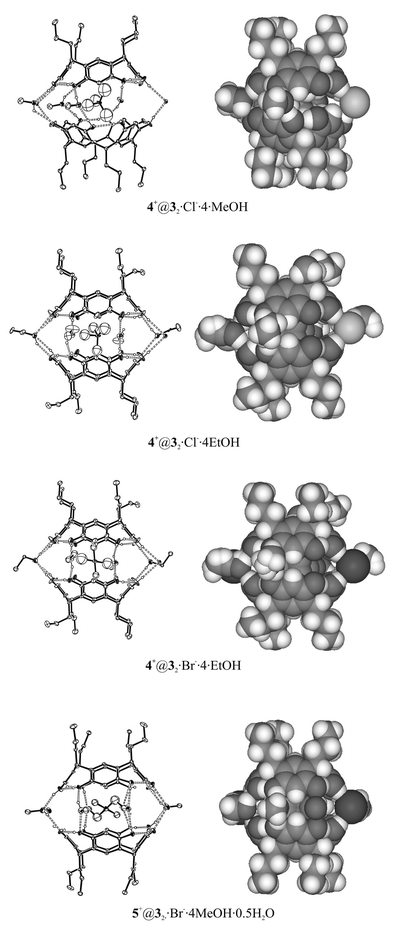
Three capsules out of four (4+@32·Cl−·4EtOH, 4+@32·Br−·4EtOH and 5+@32·Br−·4MeOH·0.5H2O) formed from the host 3 possess an eclipsed orientation and have similar unit cell dimensions, 4+@32·Cl−·4EtOH and 4+@32·Br−·4EtOH being isomorphous. The only capsule in staggered orientation is 4+@32·Cl−·4MeOH. The reason for favoured eclipsed orientation is in 4+@32·Cl−·4EtOH and 4+@32·Br−·4EtOH the larger size of the capsule mediating ethanol molecules compared to methanol or water and in 5+@32·Br−·4MeOH·0.5H2O the larger size of the cation inside the capsule.
Surprisingly, in the capsule 4+@32·Cl−·4MeOH with staggered orientation the resorcinarene molecules possess the most symmetrical conformation despite the distorting effect of the mediating anions described above, while the conformation of the resorcinarenes is slightly distorted in eclipsed oriented capsules of 3 (Table 2). As expected the staggered capsule is the tightest one, i.e. the distance between the planes defined by the methine bridges is 8.34 Å, and the loosest is 5+@32·Br−·4MeOH·0.5H2O with the larger size cation (8.98 Å).
The disordered cations are embedded inside the capsules of 3 and cation 5+, which attracts interest due to its larger size, lies in a horizontal position inside the cavity of the capsule. The horizontal position is similar to the position of 1,4-dimethyl-1,4-diazoniabicyclo[2.2.2]octane3d and tetraethyl ammonium cation3a observed earlier. C–H⋯π and cation-π interactions play a noticeable role also in these capsules, since the closest distances between carbon atoms of the cations and the electron rich face of the aromatic rings of the host vary from 3.35 to 4.04 Å.
An exceptional feature of the structures obtained from 2 is the location of the anions at the interstices between adjacent capsules rather than between the lower rim alkyl chains. At these positions, they simultaneously are capable of linking the two capsule halves together (Fig. 6).
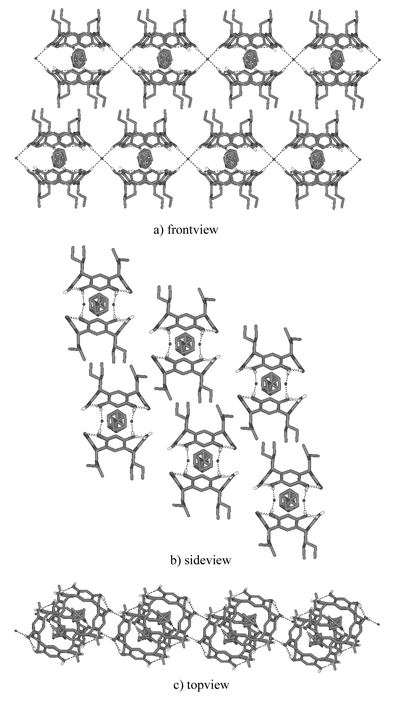 | ||
| Fig. 6 Crystal packing of structure 4+@32·Br−·4EtOH. Adjacent capsules are hydrogen-bonded to each other via capsule mediating anions (a and c). Hydrophilic and hydrophobic layers are formed due to the propyl chains of the resorcinarene that start to govern the crystal packing (b). Non-hydrogen bonding hydrogens and solvent molecules are omitted for clarity. | ||
All four capsules of 3 and one capsule of 2 (4+@22·Cl−·4.5H2O) have similar crystal packing patterns. The packing of 4+@32·Br−·4EtOH is shown in Fig. 6 and discussed here in more detail to represent all of the structures mentioned above. The adjacent molecular capsules in 4+@32·Br−·4EtOH are connected via capsule mediating anions to form side to side hydrogen-bonded continuous chains. The capsule ribbons next to each other are placed so that the aliphatic alkyl chains of resorcinarenes are pointing towards the alkyl chains of the adjacent resorcinarene. This leads to the formation of hydrophobic and hydrophilic layers in the crystal.
Mass spectrometric investigation of singly charged capsules
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =1 amu and are in perfect agreement with those calculated for these complexes based on natural isotope abundances (see inset). This confirms the elemental composition of the complex as well as its singly charged nature. Some other ions appearing in the spectra as low-intensity signals are mostly due to salt clustering. To our experience, acetonitrile is the best solvent choice. Test experiments with acetone or even more unpolar solvents caused solubility problems with the guest salts which could probably be overcome if more weakly bonding anions were used such as BF4− or PF6−. Protic solvents like methanol, however, generate significant amounts of protonated ions unnecessarily obscuring the spectra with additional signals.
=1 amu and are in perfect agreement with those calculated for these complexes based on natural isotope abundances (see inset). This confirms the elemental composition of the complex as well as its singly charged nature. Some other ions appearing in the spectra as low-intensity signals are mostly due to salt clustering. To our experience, acetonitrile is the best solvent choice. Test experiments with acetone or even more unpolar solvents caused solubility problems with the guest salts which could probably be overcome if more weakly bonding anions were used such as BF4− or PF6−. Protic solvents like methanol, however, generate significant amounts of protonated ions unnecessarily obscuring the spectra with additional signals.
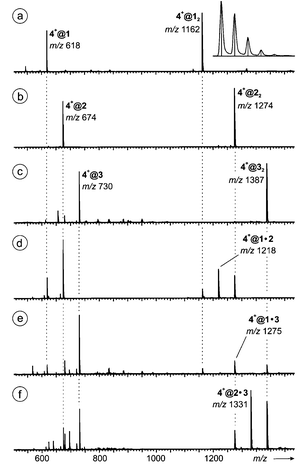 | ||
| Fig. 7 ESI mass spectra of 50 μM acetonitrile solutions of (a) 1, (b) 2, and (c) 3 with equimolar amounts of salt tetramethyl ammonium bromide 4+Br− providing the guest ion. Spectra (d)–(f) were obtained from mixtures of (d) 1 and 2, (e) 1 and 3, and (f) 2 and 3, 25 μM in each host monomer and 50 μM in 4+Br−. The inset in spectrum (a) shows the recorded isotope pattern (curve) for 4+@22 in comparison to that calculated on the basis of natural isotope abundances (vertical lines). | ||
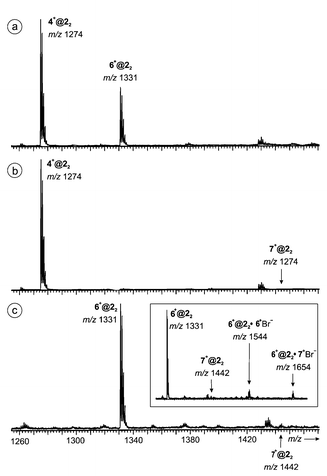 | ||
| Fig. 8 Competition of guests (a) 4+Br− and 5+Br−, (b) 4+Br− and 6+Br−, and (c) 6+Br− and 7+Br− for host 22. Note that only the region is shown in which the dimer–guest complexes are observed. All sample solutions were 50 μM with respect to the host monomer and 100 μM with respect to each guest salt. Higher excesses of the salts result in reduced signal intensities for the desired dimer–guest complexes. Instead, salt clusters become more prominent. The inset in the bottom spectrum shows two salt adducts to 6+@22. Ion pairs of both salts present in the mixture are attached to this ion with similar abundance (6+@22·6+Br−versus6+@22·7+Br−), while there is a strong preference for 6+@22 over 7+@22. | ||
Indeed, the spectrum of an equimolar mixture of tetramethyl and tetraethyl ammonium bromides 4+Br− and 6+Br− with resorcinarene 2 gave intense signals for both dimer–guest complexes with a 2∶1 preference for 4+@22 over 6+@22. Thus, the expected preference for ammonium ions smaller than those utilized in the crystallographic work is indeed observed. Similar experiments with either one of these two salts competing for complexation with tetrabutyl ammonium bromide 7+Br− only gave rise to signals for dimer–guest complexes containing the smaller cation. Signals for 7+@22 almost completely vanish within the noise.
Even more surprising is the formation of two salt adducts with low, but almost equal intensity (inset in Fig. 9): 6+@22·6+Br− (m/z 1544) and 6+@22·7+Br− (m/z 1654). The third expected salt-adduct 7+@22·7+Br− bearing two large tetrabutyl ammonium cations is again not observed. These findings strongly point to the existence of hydrogen-bonded capsular ions carrying the 6+ guest ion inside their cavity, while the additional ion pair is attached to the outer surface. While there is a strong preference for the smaller cation in the dimer–guest complexes, no such preference is seen when an additional ion pair is attached outside. The size selectivity, although only qualitatively examined here, strongly points to the formation of capsules that remain intact on their way into the gas phase.
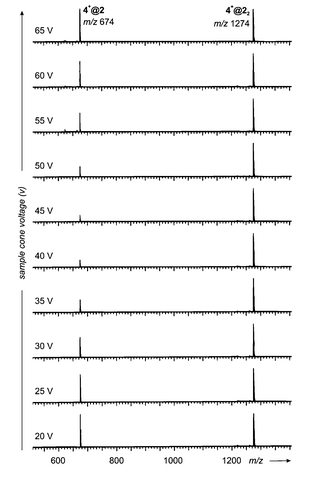 | ||
| Fig. 9 In-source fragmentation behaviour of 4+@2 and 4+@22 and its dependence on the sample cone voltage. The sample cone voltage rises from 20 (bottom) to 65 V (top) in steps of 5 V. Reproducibly, the signal for the monomer–guest complex 4+@2 first decreases in intensity relative to the corresponding dimer–guest complex, but at higher sample cone voltages increases again. | ||
Fig. 9 summarizes the results from these experiments. Starting at a sample cone voltage of 20 V, in the spectrum obtained from a solution of 4+Br− and 1, the monomer–guest complex 4+@2 and the capsule 4+@22 appear with almost the same abundance. Upon stepwise increase of the voltage to 45 V with increments of 5 V, the monomer–guest complex decreases significantly in intensity relative to its dimeric counterpart. Finally, an estimated 1∶5 ratio is obtained. Further increase of the sample cone voltage, however, did not lead to a further decrease in 4+@2 intensity. Rather, the intensity of this ion increases and at 65 V reaches again the 1∶1 ratio of the beginning. These results can be rationalized by assuming that the monomer–guest complex fragments into the “naked” cation and a neutral resorcinarene at lower energy as compared to the dimer–guest complex. That would explain the marked decrease in its intensity. At lower collision energies, first the monomer complex would be destroyed. Upon increase of the collision energy, at a certain point enough energy is provided for the decomposition of the dimer–guest complex into a neutral resorcinarene and the corresponding monomer–guest complex. This would regenerate the latter and thus also explain why its intensity increases at even higher voltages. If this scenario holds true, it means that the cation is bound more tightly to the resorcinarene dimer than to the monomer. This is only in line with a structure where the cation is inside a cavity and protected against release by additional forces, which presumably come from the seam of hydrogen bonding between the two resorcinarenes. A very similar behaviour has been observed for hydrogen-bonded calixarene capsules before and led to the same conclusions.11b Note, however, that the observed fragmentation of the dimer–guest complex to yield the monomer–guest complex is probably not the fragmentation process of lowest energy. More likely, the release of the guest cation, i.e. the loss of the complete capsule, has a lower barrier but the collisions in the ion source region may well open reaction channels higher than the lowest-energy fragmentation pathways.
Finally, it should be noted that capsules are not only formed as positive ions. The negative ion spectrum obtained with 1 and tetramethyl ammonium as the guest salt provides an interesting additional aspect (Fig. 10). Ions are observed that correspond to doubly deprotonated resorcinarenes binding tightly an ammonium ion in a 4+[@1−2H+] complex at m/z 616. Accordingly, the dimer 4+[@1−H+]2 (m/z 1160) is also formed with quite considerable abundance. The net charge of these complexes is −1. Weaker in intensity, but still clearly visible is a signal for a singly-deprotonated dimer 4+Br−@[12−H+] (m/z 1242) which receives its negative charge from an attached bromide ion. A further member in this series is 4+Br−2@12 at m/z 1323. In this complex both resorcinarenes are neutral. The positive charge of the ammonium ion is overcompensated by complexation of two bromide ions. Although we do not have any structural information from the mass spectrometric experiments, these complexes are of particular interest, because partial deprotonation of the resorcinarenes may well increase the strength of the hydrogen bonds connecting them and thus stabilize a capsule significantly as compared to its neutral relatives.
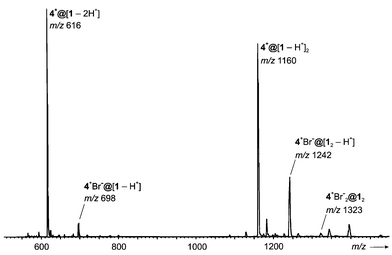 | ||
| Fig. 10 Negative ion ESI mass spectrum of a 50 μM solution of 1 containing tetramethyl ammonium bromide as the guest salt (50 μM). | ||
Conclusions
In conclusion, a total of eight crystal structures of dimeric molecular capsules formed by resorcinarenes 1, 2 and 3 with small ammonium cations 4+ and 5+ in aqueous methanol and ethanol solutions were determined. All capsules are mediated by hydrogen-bonded networks via solvent and in most cases also via spherical counter anions. C–H⋯π and cation-π interactions probably act as a capsule forming driving force since several weak interactions between the aromatic parts of the host and the carbons of the guest are evident on the basis of their closest distances.The only host capable of binding the spherical anion at the lower rim alkyl chain is 2, which indicates that the methyl group of 1 is too short and bulky for anion binding while propyl chains of 3 already start to pack as intervening hydrophobic layers typical to longer alkyl chain resorcinarenes such as C11.18
Half of the capsules possess staggered structures, which supposedly should be tighter, i.e. capsule halves should be drawn closer together. However, staggered capsule 4@22·Cl−·6MeOH·H2O is significantly looser than other staggered capsules and, as well, eclipsed capsule 4@32·Cl−·4EtOH is significantly tighter than other eclipsed capsules (Table 2). Thus, no direct conclusion about the spatial orientation of the capsule halves and the tightness of the capsule can be drawn. It seems that the interplay of cations, anions and solvents used gives rather unpredictable results with no straightforward pattern – except for the relatively easy capsule formation in all cases. Still, it is reasonable to assume that by using bigger cations, such as the tetraethyl ammonium cation (6) and 1,4-dimethyl-1,4-diazoniabicyclo[2.2.2]octane,3d and bulkier solvents, such as ethanol, the formation of an eclipsed oriented capsule is favoured.
With mass spectrometric experiments, we provide several different pieces of evidence which speak in favour of capsule formation not only in the solid state, but also in solution and even in the gas phase: size selectivity, heterodimer formation, and fragmentation behaviour. Of course, structural evidence coming from mass spectrometric experiments is necessarily indirect and somewhat limited, but on the other hand, these experiments also demonstrate the power of this technique beyond mere determination of accurate molecular masses. One might speculate what the binding forces are which hold the capsular structure together and make it stable enough to be intactly transferred into the gas phase. Likely, the seam of hydrogen bonds is not the only force “at work”. Cation-π and C–H⋯π interactions are known to stabilize such assemblies significantly.6,11,12
Our future tasks include using even smaller cations than TMA and searching for different crystallisation conditions to obtain perhaps even tighter capsules. NMR competitive and titration experiments are underway to complement these studies in solution.
Experimental
Crystal structures
Suitable single crystals for X-ray analysis were obtained by slow evaporation of the aqueous methanol or ethanol solutions (Table 1).The X-ray crystallographic data for all complexes were recorded with a Nonius Kappa CCD diffractometer. Graphite monochromatised MoKα radiation [λ(MoKα)=0.71073 Å] and temperature of 173.0±0.1 K were used in all cases. The CCD data were processed with Denzo-SMN v0.95.37318a and all structures were solved by direct methods (SHELXS-9718b) and refined on F2 by full-matrix least-squares techniques (SHELXL-9718c). The hydrogen atoms were calculated to their idealised positions with isotropic temperature factors (1.2 or 1.5 times the carbon temperature factor) and refined as riding atoms. Hydrogen atoms for any water molecules were not determined.
Carbon atoms of the cations of 4+@22·Cl−·4.5H2O, 4+@22·Br−·8MeOH, 4+@32·Cl−·4EtOH, 4+@32·Br−·4EtOH, 5+@32·Br−·4MeOH·0.5H2O and 4+@22·Cl−·6MeOH·H2O and disordered solvent molecules for structures 4+@32·Br−·4EtOH, 4+@22·Cl−·4.5H2O, 4+@22·Cl−·6MeOH·H2O and 4+@32·Cl−·4EtOH were treated isotropically. The cations of 4+@22·Cl−·4.5H2O, 4+@22·Br−·8MeOH, 4+@32·Cl−·4EtOH, 4+@32·Br−·4EtOH and 5+@32·Br−·4MeOH·0.5H2O are disordered and no hydrogen atoms were determined for these cations. Bromide–chloride and one of the water molecules are disordered between themselves in 4+@12·Br−·4MeOH·3H2O and in 4+@22·Cl−·6MeOH·H2O. However, in the later one the disorder is treated solely as chloride to keep the refinement stabile.
Residual electron density below 1.30 e Å−3 was found in 4+@22·Br−·8MeOH close to the Br− atom, in 4+@22·Cl−·4.5H2O close to the cation and in 5+@22·Cl−·4MeOH·0.5H2O close to disordered methanol carbon.
In structures 4+@22·Br−·8MeOH and 5+@32·Br−·4MeOH·0.5H2O one geometrical restraint was used to keep the disordered cation chemically reasonable. See Table 3 for detailed crystallographic data.
| Capsule | 4 +@12·Br−·4MeOH·3H2O | 4 +@22·Cl−·6MeOH·H2O | 4 +@22·Cl−·4.5H2O | 4 +@22·Br−·8MeOH | 4 +@32·Cl−·4MeOH | 4 +@32·Cl−·4EtOH | 4 +@32·Br−·4EtOH | 5 +@32·Br−·4MeOH·0.5H2O |
|---|---|---|---|---|---|---|---|---|
|
a
I>2σ(I).
b For data I>2σ(I).
|
||||||||
| Formula | 2C32H32O8·C4H12N+Br−·4CH3OH·3H2O | 2C36H40O8·C4H12N+Cl−·6CH3OH·H2O | 2C36H40O8·C4H12N+Cl−·4.5H2O | 2C36H40O8·C4H12N+Br−·8CH3OH | 2C40H48O8·C4H12N+Cl−·4CH3OH | 2C40H48O8·C4H12N+Cl−·4C2H5OH | 2C40H48O8·C4H12N+Br−·4C2H5OH | 2C40H48O8·C6H18N+Br−·4 C2H5OH·0.5H2O |
| M | 1407.4 | 1521.3 | 1392.0 | 1611.8 | 1551.3 | 1607.4 | 1651.9 | 1634.9 |
| a/Å | 14.2645(7) | 27.4410(4) | 13.6265(3) | 14.2230(7) | 22.4960(4) | 13.3702(4) | 13.3833(2) | 13.2301(2) |
| b/Å | 14.2675 | 27.4283(6) | 14.2716(2) | 14.2230 | 13.7567(3) | 13.7036(4) | 13.7536(2) | 13.5456(3) |
| c/Å | 18.4532(9) | 26.0750(5) | 20.8343(4) | 21.547(2) | 27.7823(6) | 14.4145(7) | 14.4512(3) | 14.7790(4) |
| α/° | 90 | 90 | 90 | 90 | 90 | 106.713(2) | 106.6801(5) | 105.8011(6) |
| β/° | 90 | 121.702(2) | 90 | 90 | 100.656(1) | 90.504(2) | 91.0839(5) | 90.7846(7) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 117.936(2) | 117.2219(5) | 118.409(1) |
| V/Å3 | 3754.8(3) | 16697.3(5) |
4051.7(1) | 4358.9(4) | 8449.5(3) | 2202.3(1) | 2230.4(1) | 2209.2(1) |
| Z | 2 | 8 | 2 | 2 | 4 | 1 | 1 | 1 |
| Crystal system | Tetragonal | Monoclinic | Orthorhombic | Tetragonal | Monoclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2/c
2/c |
C2/c | Pnnn | I4/m | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| D c/Mg m−3 | 1.245 | 1.196 | 1.141 | 1.228 | 1.219 | 1.212 | 1.230 | 1.229 |
| μ/mm−1 | 0.619 | 0.116 | 0.114 | 0.543 | 0.115 | 0.113 | 0.530 | 0.534 |
| Reflections measured/unique | 13197/3284 |
19652/13270 |
11402/3237 |
9057/1978 | 17904/7253 |
12026/7692 |
12083/7741 |
12471/7769 |
| Refl. used in refinementa | 2233 | 5670 | 2223 | 1333 | 3703 | 4028 | 5376 | 5162 |
| R int | 0.073 | 0.069 | 0.063 | 0.063 | 0.108 | 0.047 | 0.039 | 0.044 |
| R/Rwb | 0.088/0.223 | 0.092/0.207 | 0.108/0.295 | 0.094/0.267 | 0.073/0.191 | 0.079/0.200 | 0.065/0.168 | 0.072/0.175 |
| No. of parameters | 217 | 1007 | 232 | 136 | 503 | 548 | 531 | 525 |
| GooF | 1.068 | 1.040 | 1.062 | 1.101 | 0.998 | 1.021 | 1.027 | 1.040 |
CCDC reference numbers 196091–196098.
See http://www.rsc.org/suppdata/nj/b2/b207875a/ for crystallographic data in CIF or other electronic format.
LCT ESI-TOF mass spectra
The mass spectrometric experiments were performed with a Micromass LCT ESI-TOF instrument equipped with a Z geometry electrospray ion source. For both positive and negative ion spectra, the samples were introduced into the source as acetonitrile solutions of the monomers 1–3 (50 μM) and the salts 4+Br− − 7+Br− (50 μM) at flow rates of 15 μL min−1. A constant spray and highest intensities were achieved with a capillary voltage of 3700 V at a source temperature of 80°C and a desolvation temperature of 120°C. Other selected source parameters were as follows: sample cone voltage: 20–30 V, extraction cone voltage: 3–6 V, flow of cone gas: 10 L h−1, flow of desolvation gas: 150 L h−1. Other parameters did not influence the ion intensities much. The sample cone voltage was optimized at a somewhat higher value of 50 V for negative ion spectra. For the examination of heterodimer formation, two of the sample solutions were mixed in a 1∶1 ratio and then subjected to the same experiments. Competition experiments with two different guests were performed with solutions containing the capsule monomers (50 μM) and each guest at 100 μM concentrations, i.e. in fourfold excess relative to the dimeric capsules. Since only qualitative data were required, no corrections with respect to ESI response factors were done.19 It is assumed that these are similar for the capsules under study when only the guest cation is exchanged. In particular, this can be expected to be true for capsules, where the guest ion is located inside a host, which is the same for all ions in such a competition experiment. Multiple scans (50–200) were recorded and averaged for each spectrum in order to improve the signal-to-noise ratio.
Since the instrument does not permit MS/MS experiments, the fragmentation behaviour of the samples was examined by in-source fragmentation induced by collisions with the gas molecules present in the ion source. For this purpose, the ions were accelerated to different kinetic energies through tuning the sample cone voltage accordingly. At high voltages, the ions undergo collisions at higher velocities and usually pronounced fragmentation is observed.
Acknowledgements
We thank M.Sc. M. Luostarinen, Dr. D. Falàbu and Ph.Lic. J. Ropponen for providing compounds 1, 2, and 5, respectively. The financial support of The Graduate School of Bioorganic Chemistry (H. M.) and Academy of Finland (project no. 100319; M. N.) are gratefully acknowledged. C.A.S. is grateful for fellowship support by the Fonds der Chemischen Industrie (Liebig fellowship) and the German Academic Exchange Service (DAAD). Part of the work has been funded by the Deutsche Forschungsgemeinschaft.References
- (a) M. Morgan Conn and J. Rebek, Jr., Chem. Rev., 1997, 97, 1647 CrossRef CAS; (b) F. Hof, S. L. Craig, C. Nuckolls and J. Rebek, Jr., Angew. Chem., Int. Ed., 2002, 41, 1488 CrossRef CAS.
- P. Timmerman, W. Verboom and D. N. Reinhouldt, Tetrahedron, 1996, 52, 2663 CrossRef CAS.
- (a) K. Murayama and K. Aoki, Chem. Commun., 1998, 607 RSC; (b) A. Shivanyuk, K. Rissanen and E. Kolehmainen, Chem. Commun., 2000, 1107 RSC; (c) A. Shivanyuk and J. Rebek, Jr., Chem. Commun., 2001, 2374 RSC; (d) H. Mansikkamäki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902–1903 RSC.
- (a) L. R. MacGillvray and J. L. Atwood, Nature, 1997, 389, 469 CrossRef CAS; (b) A. Shivanyuk and J. Rebek, Jr., Chem. Commun., 2001, 2424 RSC; (c) A. Shivanyuk and J. Rebek, Jr., Proc. Natl. Acad. Sci. USA, 2001, 98, 2374 CrossRef; (d) T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257 CrossRef CAS.
- (a) K. Murayama and K. Aoki, Chem. Commun., 1997, 119 RSC; (b) H.-J. Schneider, D. Güttes and U. Schneider, Angew. Chem., Int. Ed. Engl., 1986, 25, 647 CrossRef; (c) H.-J. Schneider, D. Güttes and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6449 CrossRef CAS; (d) U. Schneider and H.-J. Schneider, Chem. Ber., 1994, 127, 2455 CAS.
- M. Mäkinen, M. Nissinen, K. Rissanen and P. Vainiotalo, J. Am. Soc. Mass Spectrom. Search PubMed , submitted.
- For reviews on the application of mass spectrometry to different aspects in host–guest and supramolecular chemistry, see (a) J. S. Brodbelt and C.-C. Liou, Pure Appl. Chem., 1993, 65, 409 CAS; (b) D. V. Dearden, H. Zhang, I.-H. Chu, P. Wong and Q. Chen, Pure Appl. Chem., 1993, 65, 423 CAS; (c) J. S. Brodbelt and D. V. Dearden, Mass Spectrometry in: Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle, J.-M. Lehn, and J. A. Ripmeester, Pergamon, Oxford, 1996, vol. 8, 567 Search PubMed; (d) M. Przybylski and M. O. Glocker, Angew. Chem., 1996, 108, 878; Angew. Chem., Int. Ed. Eng., 1996, 35, 806 Search PubMed; (e) J. S. Brodbelt, Int. J. Mass Spectrom., 2000, 200, 57 CrossRef CAS; (f) C. A. Schalley, Int. J. Mass Spectrom., 2000, 194, 11–39 CrossRef CAS; (g) C. B. Lebrilla, Acc. Chem. Res., 2001, 34, 653 CrossRef CAS; (h) C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253 CrossRef CAS.
- (a) K. C. Russell, E. Leize, A. Van Dorsselaer and J.-M. Lehn, Angew. Chem., 1995, 107, 244; K. C. Russell, E. Leize, A. Van Dorsselaer and J.-M. Lehn, Angew. Chem., Int. Ed. Eng., 1995, 34, 209 CrossRef CAS; (b) X. Cheng, Q. Gao, R. D. Smith, E. E. Simanek, M. Mammen and G. M. Whitesides, Rapid Commun. Mass. Spectrom., 1995, 9, 312 CAS; (c) X. Cheng, Q. Gao, R. D. Smith, E. E. Simanek, M. Mammen and G. M. Whitesides, J. Org. Chem., 1996, 61, 2204 CrossRef CAS.
- (a) P. Timmerman, R. H. Vreekamp, R. Hulst, W. Verboom, D. N. Reinhoudt, K. Rissanen, K. A. Udachin and J. Ripmeester, Chem. Eur. J., 1997, 3, 1823 CrossRef CAS; (b) K. A. Joliffe, M. Crego Calama, R. Fokkens, N. M. M. Nibbering, P. Timmerman and D. N. Reinhoudt, Angew. Chem., 1998, 110, 1294 CrossRef; K. A. Joliffe, M. Crego Calama, R. Fokkens, N. M. M. Nibbering, P. Timmerman and D. N. Reinhoudt, Angew. Chem., Int. Ed., 1998, 37, 1247 CrossRef CAS; (c) P. Timmerman, K. A. Joliffe, M. Crego Calama, J.-L. Weidmann, L. J. Prins, F. Cardullo, B. H. M. Snellink-Ruël, R. H. Fokkens, N. M. M. Nibbering and S. Shinkai, Chem. Eur. J., 2000, 6, 4104 CrossRef CAS.
- M. Scherer, J. L. Sessler, M. Moini, A. Gebauer and V. Lynch, Chem. Eur. J., 1998, 4, 152 CrossRef CAS.
-
(a) C. A. Schalley, J. M. Rivera, T. Martín, J. Santamaría, G. Siuzdak and J. Rebek, Jr., Eur. J. Org. Chem., 1999, 1325 CrossRef CAS;
(b) C. A. Schalley, R. K. Castellano, M. S. Brody, D. M. Rudkevich, G. Siuzdak and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 4568 CrossRef CAS;
(c) M. S. Brody, D. M. Rudkevich, C. A. Schalley and J. Rebek, Jr., Angew. Chem., 1999, 111, 1738 CrossRef; M. S. Brody, D. M. Rudkevich, C. A. Schalley and J. Rebek, Jr., Angew. Chem., Int. Ed., 1999, 38, 1640 CrossRef CAS;
(d) C. A. Schalley, T. Martín, U. Obst and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 2133 CrossRef CAS;
(e) A. Lützen, A. R. Renslo, C. A. Schalley, B. M. O'Leary and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 7455 CrossRef;
(f) B. M. O'Leary, T. Szabo, N. Svenstrup, C. A. Schalley, A. Lützen and J. Rebek, Jr., J. Am. Chem. Soc., 2001, 123, 11519 CrossRef CAS.
- (a) J. A. Bryant, M. T. Blanda, M. Vincenti and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2167 CrossRef CAS; (b) L. M. Nuwaysir, J. A. Castoro, C. L.-C. Yang and C. L. Wilkins, J. Am. Chem. Soc., 1992, 114, 5748 CrossRef CAS; (c) F. Inokuchi, Y. Shiomi, H. Kawabata, T. Sakaki and S. Shinkai, Chem. Lett., 1993, 1595 CAS; (d) F. Inokuchi, K. Araki and S. Shinkai, Chem. Lett., 1994, 1383 CAS; (e) F. Inokuchi, Y. Miyahara, T. Inazu and S. Shinkai, Angew. Chem., 1995, 107, 1459; F. Inokuchi, Y. Miyahara, T. Inazu and S. Shinkai, Angew. Chem., Int. Ed. Engl., 1994, 34, 1364 CrossRef; (f) P. S. H. Wong, X. Yu and D. V. Dearden, Inorg. Chim. Acta, 1996, 246, 259 CrossRef CAS; (g) R. Warmuth, Chem. Commun., 1998, 59 RSC; (h) A. Irico, M. Vincenti and E. Dalcanale, Chem. Eur. J., 2001, 7, 2034 CrossRef CAS; (i) M. C. Letzel, C. Agena and J. Mattay, Eur. J. Mass Spectrom., 2001, 7, 35 Search PubMed; (j) E. Ventola, K. Rissanen and P. Vainiotalo, Chem. Commun., 2002, 1110 RSC; (k) M. Mäkinen, P. Vainiotalo and K. Rissanen, J. Am. Soc. Mass Spectrom., 2002, 7, 851 CrossRef CAS.
-
(a) M. Vincenti, E. Dalcanale, P. Soncini and G. Guglielmetti, J. Am. Chem. Soc., 1990, 112, 445 CrossRef CAS;
(b) M. Vincenti, E. Pelizzetti, E. Dalcanale and P. Soncini, Pure Appl. Chem., 1993, 65, 1507 CAS;
(c) M. Vincenti, J. Mass Spectrom., 1995, 30, 925 CAS;
(d) M. Vincenti, C. Minero, E. Pelizzetti, A. Secchi and E. Dalcanale, Pure Appl. Chem., 1995, 67, 1075 CAS;
(e) M. Vincenti and E. Dalcanale, J. Chem. Soc., Perkin Trans. 2, 1995, 1069 RSC;
(f) A. Arduini, M. Cantoni, E. Graviani, A. Pochini, A. Secchi, A. R. Sicuri, R. Ungaro and M. Vincenti, Tetrahedron, 1995, 51, 599 CrossRef CAS;
(g) M. Vincenti, A. Irico and E. Dalcanale, Adv. Mass Spectrom., 1998, 14, 129 Search PubMed;
(h) J. M. J. Nuutinen, A. Irico, M. Vincenti, E. Dalcanale, J. H. M. Pakarinen and P. Vainiotalo, J. Am. Chem. Soc., 2000, 122, 10090 CrossRef CAS;
(i) A. Irico and M. Vincenti, Int. J. Mass Spectrom., 2002, 214, 23 CrossRef.
- M. C. Letzel, C. Agena and J. Mattay, J. Mass Spectrom., 2002, 37, 63 CrossRef CAS.
- H. Mansikkamäki, M. Nissinen and K. Rissanen, unpublished results.
- ORTEP3 for Windows – L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 Search PubMed.
- H. Adams, F. Davis and C. J. M. Stirling, Chem. Commun., 1994, 2527 RSC.
- (a) Z. Otwinowski and W. Minor in Processing of X-ray Diffraction Data Collected in Oscillation Mode, Methoods in Enzymology, vol. 276: Macromolecular Chrystallography, Part A, ed. C. W. Carter, Jr. and R. M. Sweet, Academic Press, New York, 1997, 307 Search PubMed; (b) G. M. Sheldrick, SHELXS-97, A program for Automatic Solution of Crystal Structures, University of Göttingen, Germany, 1997; (c) G. M. Sheldrick, SHELXS-97, A program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
- E. Leize, A. Jaffrezic and A. Van Dorsselaer, J. Mass Spectrom., 1996, 31, 53 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |