DOI:
10.1039/B207292K
(Paper)
New J. Chem., 2003,
27, 28-31
Anisotropic thermal expansion in 18-crown-6·2 H2O·2 HNO3
Received
(in Montpellier, France)
24th July 2002
, Accepted 14th October 2002
First published on 25th November 2002
Abstract
Reaction of aqueous HNO3 with 18-crown-6 results in the isolation of the neutral inclusion compound 18-crown-6·2 H2O·2 HNO3. Variable temperature single crystal X-ray and neutron diffraction data reveal that the triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) unit cell undergoes a highly anisotropic change in shape with temperature, with the crystallographic β angle changing smoothly from 66.56° to 69.03° between room temperature and 20 K. This arises from a change in shape of the crown ether in response to increases in hydrogen bond distances in the HNO3⋯H2O⋯crown ether chain.
) unit cell undergoes a highly anisotropic change in shape with temperature, with the crystallographic β angle changing smoothly from 66.56° to 69.03° between room temperature and 20 K. This arises from a change in shape of the crown ether in response to increases in hydrogen bond distances in the HNO3⋯H2O⋯crown ether chain.
Introduction
The hydrogen bonding interaction between water and nitric acid is of significant fundamental and applied interest, not least because of its importance in atmospheric chemistry.1 Nitric acid–water or oxonium–nitrate hydrogen bonded interactions also play a fundamental role in the remarkable anisotropic thermal expansion properties of layered materials such as guanidinium nitrate and monoguanidinium dioxonium trinitrate.2–5 Compounds showing anisotropic or negative thermal expansion are of considerable interest in materials applications, particularly the preparation of composite materials with zero thermal expansion.6–10 In this contribution we report the preparation and temperature-dependent structure of a new nitric acid hydrate involving hydrogen bonding to a crown ether. This contribution forms part of a growing body of work highlighting the utility of the crown ethers as hydrogen bond acceptors in the stabilization, isolation and study of reactive or transient inorganic species.11–16
Results and discussion
Crystallisation of 18-crown-6 from a 50% v/v solution of concentrated nitric acid and water results in the formation of large, deliquescent, colourless crystals of the hydrate 18-crown-6·2 H2O·2 HNO3
(1). The solid state IR spectrum of the compound as a nujol mull displays a broad, strong ν(OH) band at 3409 cm−1 with δ(OH) at 1657 cm−1, close to the values of conc. HNO3
(3410 and 1665 cm−1), although the latter bands are broader. The ν(NO3) bands occur at 1353, 1294 and 958 cm−1, again consistent with aqueous nitric acid, although the first two bands are much sharper and less intense than the free acid.
The compound was initially characterised by single crystal X-ray crystallography at 100 K, which revealed the heavy atom skeleton and suggested that the neutral formulation H2O⋯HNO3, rather than the zwitterionic H3O+⋯O3N− form, predominates, Fig. 1. The observed structure is consistent with theoretical studies that have shown that much stronger hydrogen bonds are formed between nitric acid and water when the former acts as the hydrogen bond donor.1 The water–crown ether O⋯O distances of 2.76–2.80 Å are typical of moderate strength interactions of their type (Table 1),17 whereas the much shorter nitric acid–water interaction, O(5)⋯O(4), is consistent with a strong hydrogen bond typical of highly acidic systems.14,15,17
 |
| | Fig. 1 X-Ray crystal structure of 1 at 100 K showing the three independent OH⋯O hydrogen bonds (CH hydrogen atoms omitted for clarity). | |
Table 1 Selected hydrogen bond distances (Å) and angles (deg.)a
| (a) X-Ray data (100 K) |
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
<(DHA) |
| O(4)–H(42)⋯O(3) |
0.855(18) |
1.922(18) |
2.7645(12) |
168.1(15) |
| O(4)–H(41)⋯O(2)#1 |
0.868(19) |
1.939(19) |
2.7950(13) |
168.7(17) |
| O(5)–H(51)⋯O(4) |
1.01(2) |
1.50(2) |
2.4817(13) |
163.4(17) |
| (b) Neutron data (20 K) |
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
<(DHA) |
| O(4)–H(42)⋯O(3) |
0.975(3) |
1.795(3) |
2.7540(16) |
167.0(2) |
| O(4)–H(41)⋯O(2)#1 |
0.970(3) |
1.823(3) |
2.7828(17) |
169.5(2) |
| O(5)–H(51)⋯O(4) |
1.063(2) |
1.433(2) |
2.4772(15) |
165.87(18) |
| (c) Neutron data (250 K) |
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
<(DHA) |
Symmetry transformation used to generate equivalent atoms: #1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −x,−y+1, −z+2; D=hydrogen bond donor atom, A=hydrogen bond acceptor atom. −x,−y+1, −z+2; D=hydrogen bond donor atom, A=hydrogen bond acceptor atom.
|
| O(4)–H(42)⋯O(3) |
0.946(11) |
1.861(8) |
2.794(5) |
168.4(5) |
| O(4)–H(41)⋯O(2)#1 |
0.954(11) |
1.881(8) |
2.825(5) |
169.3(5) |
| O(5)–H(51)⋯O(4) |
1.028(11) |
1.476(8) |
2.485(6) |
165.5(6) |
Measurement of the crystallographic unit cell dimensions of 1 at 100 K and at room temperature (296 K), just below the melting point of the crystals (320 K), showed a marked increase in the unit cell dimensions a, b and c with increasing temperature (see Fig. 4 later) accompanied by an overall increase in unit cell volume of 7.6%. The triclinic α and γ angles also showed small increases with temperature, however, the β angle behaved differently, decreasing from 68.7° at 100 K to 66.6° at 296 K. In order to investigate this phenomenon fully a series of X-ray crystal structure determinations were undertaken in the accessible temperature range. This resulted in a clear picture of the variation in unit cell dimensions and atomic coordinates with temperature (Figs. 2 and 4). The X-ray data were supplemented by the collection of single crystal neutron data at 20 K and 250 K in order to accurately locate the water and nitric acid protons.
 |
| | Fig. 2 Change in triclinic unit cell angles α, β and γ as a function of temperature from 20 K to 296 K. | |
The 20 K neutron structure (Fig. 3a) proved to be particularly precise and confirmed the neutral formulation of the complex with the acid O2NO–H bond length 1.063(2)
Å and a hydrogen bonded distance H2O⋯HNO3 1.433(2)
Å, Table 1. The nitric acid OH bond length is significantly elongated compared to the water OH units, while the OH⋯O hydrogen bond angle is near linear at 165.87(18)°. The combined neutron and X-ray data on hydrogen bond length variation with temperature are shown in Fig. 4. While the three crystallographically independent hydrogen bonding interactions do not behave in an identical manner, all show the expected increase in length with temperature, arising from the increased thermal motion of the acidic hydrogen atoms. Indeed on going to 250 K the covalent O2NO–H bond length apparently gets shorter as the hydrogen bond lengthens in response to increased thermal motion (although covalent bond lengths are uncorrected for thermal motion). The dynamic behaviour of H(41), H(42) and H(51) is clearly demonstrated by the 250 K neutron data, Fig. 3b.
 |
| | Fig. 3 Thermal ellipsoid plots (70% probability) of the single crystal neutron structure of 1
(a) at 20 K and (b) at 250 K. (CH hydrogen atoms omitted for clarity.) | |
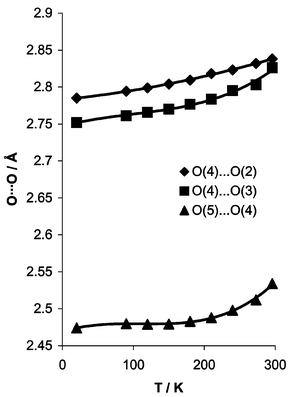 |
| | Fig. 4 Variation in O⋯O hydrogen bond lengths as a function of temperature. | |
Importantly the crown ether does not adopt the usual D3d symmetry18 but distorts, such that the diameter approximately parallel to the H2O⋯HNO3 vector is markedly longer than that perpendicular to it. This implies that hydrogen bonding to the water ‘pinches in’ the pair of oxygen atoms involved. As the temperature increases the OH⋯Ocrown interactions become longer and weaker and their ability to distort the crown ether becomes less pronounced. As a result the pinching decreases and the macrocycle becomes more symmetrical. This is equivalent to a decrease in the distance between O(1) and O(1)′, which are not involved in hydrogen bonding, from 6.16 to 6.01 Å between 20 and 296 K, Scheme 1. The O(1)⋯O(1)′ vector is aligned along {101}. Thus shortening O(1)⋯O(1)′ has the immediate consequence of decreasing the crystallographic β angle, Fig. 5. Thus there is an intimate and readily appreciated link between the gross structural features of the system and its solid-state dynamic behaviour. Despite the fact that the 18-crown-6·2 H2O·2 HNO3 unit is centrosymmetric the directionality of the hydrogen bonding interactions results in a distinctive shape change which is intimately correlated to the bulk physical properties of the crystal. Establishing such a chain of cause and effect from atomic features to macroscopic properties necessarily represents the first step in the rational design of materials with controllable physical properties.
 |
| | Scheme 1 Change in shape of 18-crown-6 in 1 from 20–296 K. | |
 |
| | Fig. 5 Crystal packing in 1 viewed along the b axis (CH hydrogen atoms omitted for clarity). (a) Single molecule showing orientation with respect to the unit cell axes, (b) packing diagram showing the alignment of the O(1)⋯O(1)′ vectors. Increases in the three independent hydrogen bond distances increase the width of the 18-crown-6 molecule in the direction of the hydrogen bonding interactions with concomitant decrease in the perpendicular direction as measured by the O(1)⋯O(1)′
(non-hydrogen bonded) separation which decreases from 6.16 Å at 20 K to 6.01 Å at 296 K. The distance between oxygen atoms hydrogen bonding to the water molecule O(2)⋯O(3)
increases from 4.61 Å to 4.68 Å over the same temperature range. The shrinking O(1)⋯O(1)′ vector is shown as a thin solid line. | |
Conclusion
This study shows that ‘soft’ non-covalent interactions in organic materials have significant, anisotropic effects on bulk material properties of the substance. Such highly directional interactions are subject to design and manipulation in a way that is the topic of intense activity in crystal engineering. Thus, the possibility of soft materials with designer thermal properties is raised. A great variety of materials are known with strongly directional hydrogen bonding interactions. The next step in this work will be to establish an empirical correlation of hydrogen bonding properties and thermal expansion in a variety of such compounds as a necessary prelude to prediction and, ultimately, control.
Experimental
Crystallography
Crystal data for 1
(X-ray, 100 K): C12H30N2O14, M 426.38 g mol−1, P, a=7.4650(15)
Å, b=7.8137(16)
Å, c=9.6921(19)
Å, α=73.160(4)°, β=68.710(3)°, γ=87.818(4)°, U=502.73(17)
Å3, Z=1, unique data 2274 (2θ⩽55°), parameters 140, R1
[F2>2σ(F2)] 0.0367, wR2
(all data) 0.0840. Details of X-ray crystallographic procedures at King's College have been described previously.14 Crystal data (neutron, 20 K). C12H30N2O14, M 426.38 g mol−1, P, a=7.4256(9)
Å, b=7.7483(9)
Å, c=9.6413(11)
Å, α=73.019(3)°, β=69.031(5)°, γ=87.728(5)°, U=494.09(10)
Å3, Z=1, unique data 1530 (2θ⩽59.80°, λ=0.8390(3)
Å), parameters 263, R1
[F2>2σ(F2)] 0.0195, wR2
(all data) 0.0439. Neutron 250 K: C12H30N2O14, M 426.38 g mol−1, P, a=7.5600(15)
Å, b=7.9619(15)
Å, c=9.7801(17)
Å, α=73.314(6)°, β=67.323(7)°, γ=87.935(8)°, U=518.44(17)
Å3, Z=1, unique data 1154 (2θ⩽59.80°, λ=0.8390(3)
Å), parameters 263, R1
[F2>2σ(F2)] 0.0352, wR2
(all data) 0.0772. For neutron experiments a colourless plate shaped crystal (4.0×2.6×1.7 mm) was mounted using glass wool in quartz capillaries and cooled to 20 K using an Air Products 201 helium Displex19 on the D9 instrument at ILL. Two standard reflections showed no change in intensity throughout the data collection. Integration and geometric corrections were carried out using the ‘advance’ software.20 A face-indexed absorption correction was applied. Neutron coherent scattering lengths were taken from ref. 21. The X-ray data (vide supra) were taken as a starting point for structure refinement using conventional alternating cycles of least squares refinement and difference Fourier synthesis (SHELXL-97).22 The initial difference Fourier map showed clearly the positions of all acidic hydrogen atoms and these were incorporated into the model with full anisotropic refinement. Over the course of cooling from 298 K to 20 K a strong reflection was monitored and showed a smooth change in peak position, see discussion regarding temperature dependent change in unit cell dimensions.
CCDC reference number 196886 and 196887. See http://www.rsc.org/suppdata/nj/b2/b207292k/ for crystallographic data in CIF or other electronic format.
Preparation of 1.
18-Crown-6 (0.26 g, 1.0 mmol) was dissolved in aqueous nitric acid (50%, 3 cm3) and the resulting colourless solution left to stand at room temperature. After nine days colourless needle crystals of 1 were obtained. Continued standing with gradual evaporation of water over a period of 12 months resulted in the growth of very large single crystals suitable for the neutron study in essentially quantitative yield based on 18-crown-6.
Acknowledgements
We thank the Institut Laue Langevin, Grenoble, France and King's College London for financial support.
References
- M. Staikova and D. J. Donaldson, Phys. Chem. Chem. Phys., 2001, 3, 1999 RSC.
- A. Katrusiak and M. Szafranski, Chem. Phys. Lett., 2001, 340, 302 CrossRef CAS.
- A. M. Pivovar, M. D. Ward, T. Yildirim and D. A. Neumann, J. Chem. Phys., 2001, 115, 1909 CrossRef CAS.
- J. Wasicki, M. Grottel, A. Kozak and Z. Pajak, J. Phys. Condens. Matter, 1994, 6, 2491 CrossRef CAS.
- M. Szafranski, Phys. Status Solidi B Basic Res., 1997, 201, 343 Search PubMed.
- L. A. Villaescusa, P. Lightfoot, S. J. Teat and R. E. Morris, J. Am. Chem. Soc., 2001, 123, 5453 CrossRef CAS.
- T. G. Amos and A. W. Sleight, J. Solid State Chem., 2001, 160, 230 CrossRef CAS.
- K. Fukuda, J. Ceram. Soc. Jpn., 2001, 109, 846 Search PubMed.
-
C. De Meyer, I. Van Driessche and S. Hoste, in Euro Ceramics VII, Pt. 1–3, eds. C. Kermel, V. Lardot, D. Libert and I. Urbain, Trans Tech Publications, Zurich, 2002, vol. 206-2, p. 11 Search PubMed.
- C. Lind, A. P. Wilkinson, C. J. Rawn and E. A. Payzant, J. Mater. Chem., 2001, 11, 3354 RSC.
- P. C. Junk, B. J. McCool, B. Moubaraki, K. S. Murray, L. Spiccia, J. D. Cashion and J. W. Steed, J. Chem. Soc., Dalton Trans., 2002, 1024 RSC.
- J. W. Steed and P. C. Junk, J. Chem. Soc., Dalton Trans., 1999, 2141 RSC.
- P. C. Junk, B. J. McCool, B. Moubaraki, K. S. Murray and L. Spiccia, Angew. Chem., Int. Ed., 1999, 38, 2224 CrossRef CAS.
- M. Calleja, K. Johnson, W. J. Belcher and J. W. Steed, Inorg. Chem., 2001, 40, 4978 CrossRef CAS.
- M. Calleja, S. A. Mason, P. D. Prince, J. W. Steed and C. Wilkinson, New J. Chem., 2001, 25, 1475 RSC.
- J. L. Atwood and P. C. Junk, J. Chem. Soc., Dalton Trans., 1997, 4393 RSC.
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, OUP, Oxford, 1997.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171 CrossRef CAS.
- J. Archer and M. S. Lehmann, J. Appl. Crystallogr., 1986, 21, 471.
- C. Wilkinson, H. W. Khamis, R. F. D. Stansfield and G. J. McIntyre, J. Appl. Crystallogr., 1988, 21, 471 CrossRef.
- V. F. Sears, Neutron News, 1992, 3, 26 Search PubMed.
-
G. M. Sheldrick, SHELXL-97, University of Göttingen, 1997.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |
Click here to see how this site uses Cookies. View our privacy policy here.