Replacing proline at the apex of heptapeptide-based chloride ion transporters alters their properties and their ionophoretic efficacy
Paul H.
Schlesinger
*a,
Riccardo
Ferdani
b,
Jolanta
Pajewska
b,
Robert
Pajewski
b and
George W.
Gokel
*b
aCell Biology and Physiology, Washington University School of Medicine, 660 S. Euclid Ave., St. Louis, MO 63110, USA
bProgram in Bioorganic Chemistry, Division of Bioorganic Chemistry, Departments of Molecular Biology & Pharmacology, Washington University School of Medicine, 660 S. Euclid Ave., St. Louis, MO 63110, USA. E-mail: ggokel@molecool.wustl.edu; Fax: 314/362-9298 or 7058; Tel: 314/362-9297
First published on 12th December 2002
Abstract
A membrane anchored heptapeptide, (C18H37)2NHCOCH2OCH2CO–NH–Gly–Gly–Gly–Pro–Gly–Gly–Gly–OCH2Ph, has proved to be a selective chloride anion transporter that functions in phospholipid bilayer membranes. When Pro was replaced by the natural amino acid Leu, the activity decreased dramatically. In the present study, Pro is replaced by pipecolic acid (homoproline, Pip); the resulting anchored heptapeptide is a membrane active, chloride selective transporter.
Chloride membrane transport is a ubiquitous process in living systems. An array of chloride channels has been studied for some time and understanding their function is clearly an important goal.1,2 The only solid state structures of chloride transporters were reported very recently.3 The X-ray structures of the ClC Cl− channels from the prokaryotes Salmonella enterica serovar typhimurium and Escherichia coli were reported at 3.0 Å and 3.5 Å resolutions, respectively. These structures reveal that the chloride channels are quite complicated and distinct from those of voltage gated cation channels. Both ClC channels incorporate two subunits of 18 α-helices that function as independent channel pathways. Their association is coordinated through a 2300 Å2 contact surface. To date, four major families of chloride channels have been described.4,5
The complexity of the chloride channel structures results from many regulatory influences and a long evolutionary history as a fundamental cellular function. In early organisms, chloride transport essential to volume and pH control must have been achieved by significantly simpler structures. Certainly, chloride permeability in contemporary cells is essential for volume, pH, and membrane potential regulation and these same features must have been required, at least to some extent, in early organisms.6,7 The work reported here results from an effort to develop a simple, abiotic compound that would function as a chloride transporter in phospholipid bilayers.
An effort by Tomich and coworkers8–10 has been reported to develop a chloride channel by using modified proteins. Specifically, C-K4-M2GlyR is a peptide prepared by modifying either the N- or C-terminus of the M2 transmembrane sequence of the brain glycine receptor by the addition of lysine residues. The resulting peptide, C-K4-M2GlyR, was found to act as a chloride channel when it was inserted into the apical membrane of cultured renal epithelial monolayers. Our effort to develop a chloride transporter differed. We used what is currently known about chloride channel proteins as a guide and inspiration for the design of a synthetic, chloride selective transporter. As a result, we recently reported the preparation and limited characterization of the first example of this new family of abiotic chloride channels.11,12 We now report related compounds that have quite different activities depending on what appear to be very subtle differences.
Results and discussion
Design and synthesis of SCMTR
Our design schematic for a chloride ion transporter (![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) ynthetic
ynthetic ![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif) hloride
hloride ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) embrane
embrane ![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif)
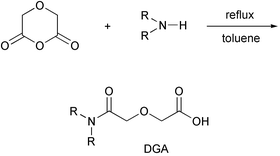
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ansporter, SCMTR or “scimitar”) involved the development of three molecular sectors. These included a hydrocarbon anchor, a peptide fragment expected to serve as the channel portal and filter, and the connector. Diglycolic acid, HOCOCH2OCH2COOH, was selected to be the link between the peptide and the hydrocarbon anchors. The latter were constructed from a dialkylamine, giving an overall anchor of R2NCOCH2OCH2COOH. The peptide was joined to the anchor's C-terminus to give, schematically, R2NCOCH2OCH2CO-peptide.
ansporter, SCMTR or “scimitar”) involved the development of three molecular sectors. These included a hydrocarbon anchor, a peptide fragment expected to serve as the channel portal and filter, and the connector. Diglycolic acid, HOCOCH2OCH2COOH, was selected to be the link between the peptide and the hydrocarbon anchors. The latter were constructed from a dialkylamine, giving an overall anchor of R2NCOCH2OCH2COOH. The peptide was joined to the anchor's C-terminus to give, schematically, R2NCOCH2OCH2CO-peptide.
The anchor unit was designed to correspond approximately to the size and shape of a phospholipid membrane monomer. The twin diamine chains mimicked the two fatty acid chains of a natural phospholipid. Phospholipids contain a glycerol unit, to which the fatty acids are attached by acyl links, resulting in a region of intermediate polarity. The midpolar regime was emulated by diglycolic acid, which contains carbonyl groups and an ether oxygen. Diglycolic acid is especially attractive for this application because its readily formed anhydride reacts cleanly with diamines as shown in Scheme 1.
 | ||
| Scheme 1 | ||
The peptide's design was guided by structural patterns found in natural chloride transporters. It was hoped that these would lead to chloride selectivity in our abiotic compound. Based upon the following lines of evidence, we have identified a critical role for the peptide in the ion selectivity of naturally occurring chloride transporters.6 (1) Proline is present in the conserved motif GKxGPxxH in the anion pathway of all ClC chloride protein channels.13,14 (2) The cation selectivity of nicotinic acetylcholine receptor is altered to anion selectivity when proline is inserted into the native channel's selectivity filter.15,16 (3) The presence of a so-called “hinge-bend” regime in channel-forming peptides is attributed to the proline in the sequence GxxP.17 (4) Proline is reported to induce a surface “kink” in various proteins.18 (5) Studies with proinsulin's C-peptide show that the presence of a helix–loop–helix motif at the apex of the peptide's “loop” conformation is required for ion channel activity.19 The combination of structural observations and functional implications suggested to us the importance of incorporating this particular element into our ion portal design.
We settled on a heptapeptide sequence having proline as its central point for the reasons described above. The simplest heptapeptide containing a “central” proline is Gly–Gly–Gly–Pro–Gly–Gly–Gly (GGGPGGG). Analyses using molecular models and simple computational methods suggested that GGGPGGG would have a v-shaped conformation. Further, distance measurements across a computer-generated model of the “v” show that the C- and N-termini have a maximum center-to-center separation of ∼8 Å and the apex of the “v” makes an angle of about 75°. Space-filling (CPK) molecular models show that the available space within the “v” is 6–8 Å, depending on how compressed are the two GGG chains. A computational model of CH3CH2CONH–GGGPGGG–OCH2Ph is shown in Fig. 1. Measurement of the model shows that the two remotest carbonyl groups are 8.8 Å apart in the illustrated conformation.
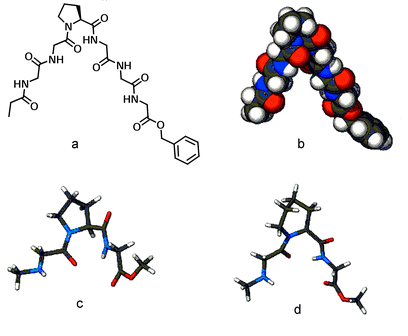 | ||
| Fig. 1 Ac–GGGPGGG–Bzl shown in a structural figure (a) and rendered in the CPK representation (b). Calculated structures of CH3NH–Gly–Pro–Gly–OCH3 (c) and CH3NH–Gly–Pip–Gly–OCH3 (d). | ||
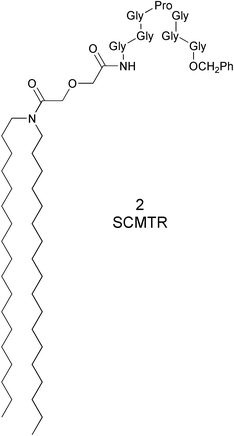
In 2, [CH3(CH2)17]2NCOCH2OCH2CO–GGGPGGG–OCH2Ph, the peptide sequence places proline at the pinnacle of an “arch” flanked with glycine residues. We propose that having the heptapeptide reside at the top of the mid-polar regime of the membrane bilayer is critical. In this position it can form an uncharged, chloride-selective portal. The resulting formation of an aqueous pathway for ions to the interior of the bilayer was anticipated. If this pathway was maintained long enough for ion diffusion through the bilayer to occur, channel activity should result. We anticipated that by adding a hydrophobic membrane anchor of appropriate length, the peptide would be positioned to generate a functional “hinge-bend” portal at the membrane surface.19
The membrane anchor
As noted above, the membrane anchor was designed to mimic a phospholipid monomer in size, polarity, and functional group position. The anchor is a dialkylamino derivative of diglycolic acid: R2NCOCH2OCH2COOH.20 A related anchor system was reported several years ago21,22 but no experimental details were included and no yield or characterization data appear in the Beilstein database.23 We were able to construct R2NCOCH2OCH2COOH conveniently by the reaction of one equivalent each of diglycolic anhydride and dialkylamine (e.g., dioctadecylamine) in refluxing toluene. After heating for 48 hours, the solvent was removed and the nearly pure R2NCOCH2OCH2COOH was crystallized from CHCl3 to afford the analytically pure product in nearly 90% yield. It is interesting to note that the reaction of bis(2-methoxyethyl)amine with diglycolic anhydride is reported to be much faster than the rates we observed for the anhydride with simple dialkylamines.21 We confirmed this rate difference, although the experimental procedure in the literature does not work as reported for the methoxyethylamine.We surmised that an anchored peptide would insert deeply into the outer leaflet of a phospholipid bilayer. This, we felt, would lead to a controlled membrane disruption of the second leaflet resulting in a transmembrane pore. The advantage of this approach was thought to be that the kinetics of insertion into the outer leaflet of the bilayer would be more favorable than any system that required transverse relaxation (flip-flop). Further, by limiting the length of our anchor to the thickness of a single bilayer, we anticipated that the detergent activity of this compound would be minimized, limiting the formation of membrane disks at high concentrations.24
Synthesis of the compounds studied
Four target compounds were prepared for the studies reported here. Each procedure began with the reaction of either didecylamine or dioctadecylamine with diglycolic anhydride. The product was (C10H21)2NCOCH2OCH2COOH or (C18H37)2NCOCH2OCH2COOH, respectively. We use shorthand to designate these diglycolic amide (DGA) derivatives: 102DGA–OH and 182DGA–OH. The C-terminal tripeptide was prepared as a benzyl ester. It was coupled (1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride) with TsOH·H2N–GGG–OCH2Ph to give R2DGA–Gly–Gly–Gly–OCH2Ph. The C-terminal benzyl group was removed by hydrogenolysis in EtOH.The tetrapeptide required to complete the heptapeptide sequence was prepared by coupling TsOH·H2N–GGG–OCH2Ph with N–Boc–Pro–OH to give Boc–Pro–Gly–Gly–Gly–OCH2Ph (Z–PGGG–Bzl). The Boc group was removed (HCl, dioxane) and the final coupling between R2DGA–Gly–Gly–Gly–OH and H2N–PGGG–OCH2Ph gave either 102DGA–GGGPGGG–Bzl (1) or 182DGA–GGGPGGG–Bzl (2). Compounds 3 and 4 were prepared analogously except that pipecolic acid (homoproline) replaced proline in the heptapeptide. The sequence is shown below in Scheme 2 for the preparation of either 1 or 2 and details for the synthesis of compounds 1–4 are recorded in the Experimental Section.
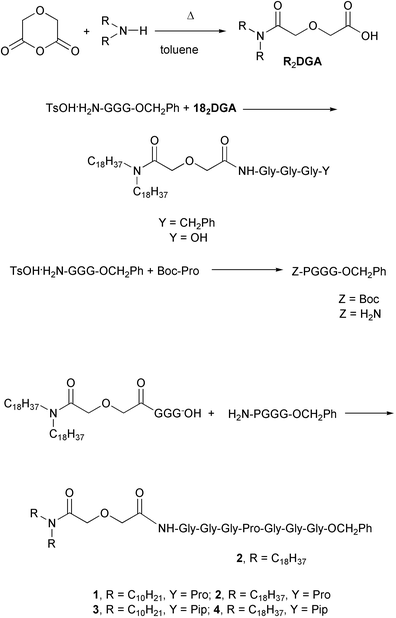 | ||
| Scheme 2 | ||
Carboxyfluorescein dequenching and pore characterization
Carboxyfluorescein (CF) is a fluorescent dye that may readily be incorporated into liposomes. When concentrated within the vesicle, CF self-quenches. Under appropriate conditions, its exit from vesicles may readily be detected. We prepared phospholipid liposomes in the presence of 20 mM carboxyfluorescein (CF). Dequenching of intravesicular CF was followed spectrofluorometrically in a thermostated cuvette (see Experimental Section). Data obtained for 2 over a range of concentrations are shown in Fig. 2. On the timescale shown, the data for 2 at 190 μM and 253 μM are nearly superimposable.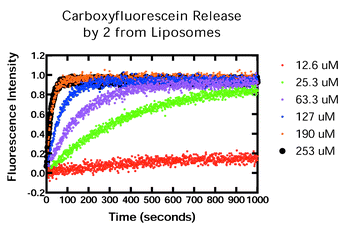 | ||
| Fig. 2 Carboxyfluorescein release by 2 from liposomes. Concentrations of 2: (bottom to top): 12.6 μM, 25.3 μM, 63.3 μM, 127 μM, 190 μM, and 253 μM. The two highest concentrations are nearly superimposed in this graph. | ||
The CF transport data show that 2 forms well-behaved pores that release the dye in a controlled fashion. The critical test, however, of a putative chloride transporter is whether it fosters chloride release from liposomes. We used unilamellar liposomes (150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ±16 nm) prepared from 30%
(w/w) 1,2-dioleoyl-sn-glycero-3-phosphate and 70% 1,2-dioleoyl-sn-glycero-3-phosphocholine.
±16 nm) prepared from 30%
(w/w) 1,2-dioleoyl-sn-glycero-3-phosphate and 70% 1,2-dioleoyl-sn-glycero-3-phosphocholine.
Compound 2 mediated chloride release that was rapid, complete, and concentration dependent.11 Both 1 and 2 release carboxyfluorescein, a process related to chloride transport. We have recently noted that CF release by 1 (10Pro), which has shorter anchor chains, is about 10-fold greater25 than for 2 (18Pro).11 Thus CF release was complete in less than 100 s when either 1 or 2 was present at a concentration of ∼15 μM or ∼250 μM respectively. We infer from this that the as yet untested chloride transport rates will show a comparable relationship.
Replacement of proline by leucine in 2
(182DGA–GGGPGGG–Bzl→182DGA–GGGLGGG–Bzl, 5) led to reduced CF release. Thus, equal CF release rates could be achieved when [5]=154 μM and [2]=24.1 μM.11 This is at least a 6-fold reduction in activity, apparently resulting from the loss of the rigid “hinge-bend” enforced by proline's cyclic structure.
Pore conductance and rectification in planar bilayers
Planar lipid bilayers were prepared from asolectin and studied under voltage clamp conditions (see Experimental Section and ref. 26). The addition and mixing of 89.7 μM 182DGA–GGGPGGG–Bzl (2, 18Pro, SCMTR) on the trans side of the planar bilayer resulted in a current that was mildly rectifying at negative potentials. The I–V relationship (−60 mV to +60 mV) had a limiting slope of 349 pS. The 102DGA–GGGPGGG–Bzl (1, 10Pro, SCMTR) at 5.2 μM was also mildly rectifying and had a limiting slope of 573 pS. We have previously noted that the 18Pro is selective for chloride over potassium by >10∶1. The larger conductance of 10Pro with an identical pore size as determined in Fig. 1 indicates that this pore is relatively non-selective for chloride versus potassium.Substitution of proline in the heptapeptide sequence by leucine or by pipecolic acid
The known peptide sequence of Cl−-conducting C-peptide27 contains proline at the loop apex in humans and in rats.28 In contrast, the pig sequence has leucine in this position and this peptide is not a chloride ion transporter.27 As noted above, we found in previous studies that a Pro→Leu replacement led to diminished (∼6-fold) activity. A more subtle variation is the change from proline to pipecolic acid.29 This alteration formally involves a 1-carbon ring expansion of the essential amino acid. The structures of the bis(octadecyl) derivatives are shown below as 2 and 4.
These two amino acids differ in several respects (Fig. 3).29 The difference in ring size changes the N–C–CO angles from ∼112.5° to ∼110.5°. The presence of the additional carbon atom in the ring also increases hydrophobicity but, as with the bond angles, the change is expected to be small. The molecular weights of these two compounds are 1168 Da and 1182 Da, respectively. This is a difference of only ∼1%. We also consider the differences in bond angles and hydrophobicity to be modest.
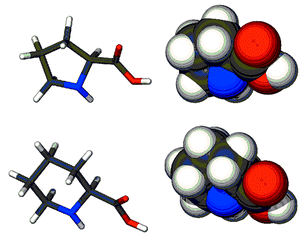 | ||
| Fig. 3 Models of proline (top) and pipecolic acid in tube and space filling representations. | ||
We have assessed the efficacy of the proline (1, 2) and pipecolic acid (3, 4) derivatives by CF dequenching (see above). Selected data are presented in Fig. 4. The graph shows four traces corresponding to various concentrations of 1–4. The two nearly superimposed traces seen in the middle of the graph are for 102DGA–GGGPGGGOBzl (1) and 182DGA–GGGPGGG–OBzl (2). The CF release is essentially identical but the concentration difference is dramatic: their activities differ by nearly 400-fold ([1]=0.167 μM, [2]=63.3 μM).
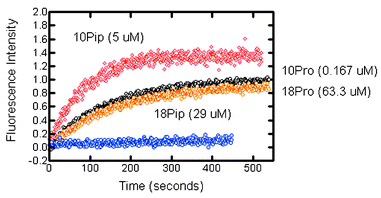 | ||
| Fig. 4 Carboxyfluorescein release from phospholipid liposomes mediated by compounds 1–4. The top (red, 10Pip, 3, 5 μM) and bottom (blue, 18Pip, 4, 29 μM) traces are for 3 (10Pip) pipecholic acid derivatives. The overlapping central traces (black above, 1, 10Pro, 0.167 μM; orange, 2, 18Pro, 63.3 μM) are for the proline-containing heptapeptide. | ||
We may compare the activity of 102DGA–GGGPGGG–OBzl (10Pro, 1) with 102DGA–GGGPipGGG–OBzl (10Pip, 3). If the traces were superimposed, the ratio of activity would be 5 μM/0.167 μM or 29.9. The didecyl-pipecolic (10Pip) acid derivative (3) is somewhat more active at 5 μM than is the proline analog at 0.167 μM, so we estimate that 10Pip (3) is about 20-fold less active than the proline counterpart (10Pro, 1). This is a significant reactivity difference that may result from variations in bond angles, hydrophobicity, and/or other factors. The more striking observation is that at a concentration of 29 μM, 18Pip (4) showed essentially no transport activity. A direct comparison of 2 with 4 at identical concentrations would have been more satisfactory. Unfortunately, when [18Pip]>29 μM, precipitation of the transporter occurred and the experiment was compromised. We therefore undertook the fluorescence dequenching experiment with 18Pro (2) at a concentration of 25.3 μM. Within the error of these experiments, this is an identical concentration. The results are shown in Fig. 4. At identical concentrations, the activity of 4 is unmeasurable compared to that of 2.
A comparison of the data presented in the graphs of Figs. 2 and 4 raises several interesting questions. First, do we expect any activity from a compound in which pipecolic acid replaces proline? As noted above, previous work showed that the anchored heptapeptide lost nearly all of its activity when Leu replaced Pro.11 This result correlated to known C-peptide activity (see above).27 Pipecolic acid is far closer to proline29 than is leucine, although Pro and Leu have the same number of carbon atoms. Proline and pipecolic acids are similar in size and shape and the latter should function to form a conformational kink in the heptapeptide chain as does proline. Indeed, in the C10-anchored system, pore formation for 10Pro (1) was ∼20-fold greater than for 10Pip (3). Significant activity for 10Pip was observed only at or above 5 μM concentrations, compared to 167 nM for 10Pro.
A second question is why no pore formation is observed for 4 when 2 shows reasonable kinetics at an identical concentration. Note that 18Pro (2) is significantly less active than is 10Pro (1). A more compelling question, given the design and conceptualization of these compounds, is why more effective transport is observed generally with shorter anchors (1, 3 are more active than 2 or 4). We have experimentally confirmed this relationship in separate work but the explanation remains obscure.
Pore size for 18Pro, 2, vs. 10Pip, 3
Planar bilayer conductance studies permit us to assess the effective size of the pore that is formed. This is typically given in terms of conductance rather than diameter because ion slectivity as well as pore size can determine the current. As shown in the graph of Fig. 5, “C18” represents 182DGA–GGGPGGG–OBzl, 2, and “C10” corresponds to 102DGA–GGGPipGGG–OBzl, 3. The size of the resulting pores was determined to be the same when using CF (Fig. 2) so that the increase in conductance by 10Pro probably results from lowered ion selectivity. This pore passes both chloride and potassium; since they are traveling in opposite directions in response to the applied voltage, the resulting current is summed. The graph shows identical chloride selectivity (x-intercept), which indicates that the striking difference in slope results from a change in pore size. Admittedly, these compounds display large stability and activity differences in pore activation. However, these are single pore experiments in which thermodynamic and kinetic characteristics are only indirectly reflected. We have previously observed that pore sizes for 1 and 2 (C10Pro vs. C18Pro chains, proline derivative) do not differ significantly.25 Thus, we did not expect ring size per se to lead to a difference in pore size. The data shown in Fig. 5 show that the pore sizes for 2 and 3 differ by nearly 4-fold. This is a remarkable difference considering that the peptide chains of 2 and 3 differ in such a subtle way.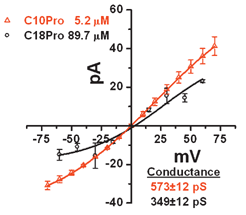 | ||
| Fig. 5 Planar lipid bilayers were generated on a 0.25 mm orifice in a plastic cuvette. When a bilayer was present, C18Pro or C10Pip was added to the cis chamber at 40 or 9 μM respectively in a 450∶150 mM KCl gradient (cis to trans). Currents were then determined as the voltage across the bilayer was varied. The slope of the current–voltage plot is the conductance as determined from the inverse of Ohm's law (1/R=I/E). The x intercept is the reversal potential that is calculated by the Nernst equation to be ∼29 mV for a 450∶150 KCl gradient with a highly selective chloride channel. The slopes for the lines are shown in picoSiemens (pS) and millivolts (mV). | ||
Conclusion
Our study of these anchored heptapeptides was based upon the premise that a synthetic, chemically simple, chloride transporter would be instructive for the study of anion transport across bilayer lipid membranes. We hoped that defined changes in transporter structure would alter activity and provide insight into important factors in anion transport. The results reported here support this hypothesis. The “SCMTR” family of molecules has solubility behavior with dramatic structural sensitivity and aggregation properties that are complex and structurally dependent. The pore activation step clearly depends upon the anchor chain length; pore size is altered dramatically by the subtle change from proline to pipecolate. We now anticipate that this novel family of compounds will be useful to explore in detail the chemistry and biophysics of pore formation and anion transport in lipid bilayers.Experimental
General
All reaction solvents were freshly distilled and reactions were conducted under N2 unless otherwise specified. Et3N was distilled from KOH and stored over KOH. CH2Cl2 was distilled from CaH2. Column chromatography was performed on silica gel 60 (230–400 mesh). Thin layer chromatography was performed with silica gel 60 F254 plates with visualization by UV light (254 nm) and/or by phosphomolybdic acid (PMA) spray. Starting materials were purchased from Aldrich Chemical Co., and used as received unless otherwise indicated. 1H-NMR spectra were recorded at 300 MHz and are reported in the following manner: chemical shifts are reported in ppm downfield from internal tetramethylsilane (integrated intensity, multiplicity (b=broad; s=singlet; d=doublet; t=triplet; m=multiplet, bs=broad singlet, etc.), coupling constants in Hertz, assignment). 13C-NMR spectra were obtained at 75 MHz and referenced to CDCl3
(δ 77.0). Infrared spectra were recorded in KBr unless otherwise noted and were calibrated against the 1601 cm−1 band of polystyrene. Melting points were determined on a Thomas Hoover apparatus in open capillaries and were uncorrected. Optical rotations were recorded on a Perkin-Elmer Model 214 polarimeter.
N,N-Didecyl-3-oxaglutaramide (102DGA)
A solution of didecylamine (5.0 g, 16.8 mmol) and diglycolic anhydride (2.2 g, 18.5 mmol) in THF (40 mL) was refluxed for 48 h. The solvent was evaporated and the crude product was dissolved in Et2O (150 mL) and washed with 10% aqueous HCl (2×20 mL), dried (MgSO4) and crystallized from Et2O to give a white solid (5.61 g, 88%), mp 51–52°C. 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (28H, pseudo-s, CH3(CH2)7CH2CH2N), 1.54 (4H, bs, CH3(CH2)7CH2CH2N), 3.07 (2H, t, J=7.8 Hz, CH3(CH2)7CH2CH2N), 3.34 (2H, t, J=7.8 Hz, CH3(CH2)7CH2CH2N), 4.21 (2H, s, COCH2O), 4.38 (2H, s, COCH2O). 13C-NMR: 14.1, 22.7, 26.8, 26.9, 27.4, 28.6, 29.2, 29.3, 29.4, 29.5, 31.8, 46.8, 71.3, 73.2, 170.6, 171.7. IR (CHCl3): 3401, 3269, 2957, 2923, 2853, 1713, 1627, 1467, 1431, 1377, 1255, 1150, 1137, 1062, 923 cm−1.
TsOH·GGG–OCH2Ph
H2N–GGG–OH (3.0 g, 15.9 mmol) and p-toluenesulfonic acid (monohydrate, 3.6 g, 18.9 mmol) were added to a mixture of benzyl alcohol (20 mL) and toluene (30 mL). The mixture was heated to reflux and water was removed by using a Dean–Stark trap. When no more water appeared in the distillate, heating was stopped. The mixture was cooled to room temperature, diluted with ether (50 mL) and cooled in an ice water bath for 2 h. Crystalline TsOH·H2N–GGG–Bzl was collected on a filter, washed with ether (50 mL) and dried. After recrystallization from methanol–ether (1∶1), the salt (5.5 g, 77%) melted at 176–177°C. 1H-NMR CDCl3: 2.34 (3H, s, CH3Ph), 3.74 (2H, s, Gly NCH2), 3.97 (4H, s, Gly NCH2), 5.14 (2H, s, PHCH2O), 7.21 (2H, d, J=8.4 Hz, tosyl HAr), 7.30–7.35 (5H, m, Ph HAr), 7.69 (2H, d, J=8.4 Hz, tosyl HAr). 13C-NMR: 21.4, 41.7, 42.1, 43.2, 68.1, 127.2, 129.5, 129.6, 129.9, 130.2, 137.5, 142.1, 143.7, 168.4, 171.4, 172.2. IR (KBr): 3331, 3083, 1747, 1670, 1545, 1456, 1406, 1362, 1202, 1125, 1035, 1011, 913, 817, 736, 685 cm−1.
102DGA–GGG–OCH2Ph
To 102DGA (2 g, 4.8 mmol) dissolved in CH2Cl2 (30 mL), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.0 g, 5.3 mmol) was added and the mixture was stirred at room temperature. After 0.5 h, TsOH·GGG–OCH2Ph (2.2 g, 4.8 mmol) and Et3N (2.0 mL) were added and reaction was stirred at ambient temperature for two days. The reaction mixture was evaporated in vacuo, the residue successively washed with 5% citric acid (20 mL), water (20 mL), 5% NaHCO3 (20 mL), and brine (20 mL), dried (MgSO4), and evaporated. The crude, oily product was purified by chromatography (SiO2, 2% MeOH–CH2Cl2) and afforded a colorless oil (2.42 g, 76%). 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (28H, pseudo-s, CH3(CH2)7CH2CH2N), 1.50 (4H, bs, CH3(CH2)8CH2CH2N), 3.05 (2H, t, J=7.5 Hz, CH3(CH2)7CH2CH2N), 3.25 (2H, t, J=7.5 Hz, CH3(CH2)7CH2CH2N), 3.95–4.05 (6H, m, Gly NCH2), 4.10 (2H, s, COCH2O), 4.30 (2H, s, COCH2O), 5.13 (2H, s, PHCH2O), 7.30–7.35 (5H, m, HAr), 7.96 (1H, t, J=5.7 Hz, Gly CONH), 8.27 (1H, t, J=5.7 Hz, Gly CONH). 13C-NMR: 13.9, 22.5, 26.7, 26.9, 27.4, 28.6, 29.2, 29.3, 31.8, 41.0, 42.9, 46.3, 46.7, 66.9, 69.6, 71.7, 128.2, 128.4, 128.6, 135.3, 168.6, 169.7, 169.8, 170.0, 171.5. IR (film): 3292, 2915, 2848, 1750, 1650, 1538, 1465, 1195, 1130, 1031, 721, 697 cm−1.
102DGA–GGG–OH
102DGA–GGG–OCH2Ph (2.3 g, 3.4 mmol) was dissolved in absolute ethanol (40 mL) and 10% Pd/C (0.15 g) was added and this mixture was shaken under 60 psi H2 for 3 h. The reaction mixture was heated to reflux and filtered through a celite pad. The solvent was evaporated under reduced pressure to afford a white solid (1.92 g, 96%), mp 120–122°C. 1H-NMR CD3OD: 0.90 (6H, t, J=6.9 Hz, –CH2CH3), 1.29 (32H, pseudo-s, CH3(CH2)8CH2N), 1.57 (4H, bs, CH3(CH2)7CH2CH2N), 3.21 (2H, t, J=7.8 Hz, CH3(CH2)8CH2N), 3.35 (2H, t, J=7.8 Hz, CH3(CH2)7CH2N), 3.93 (2H, s, Gly NCH2), 3.94 (2H, s, Gly NCH2), 3.97 (2H, s, Gly NCH2), 4.12 (2H, s, COCH2O), 4.40 (2H, s, COCH2O).13C-NMR: IR (KBr): 3319, 3269, 3084, 2922, 2852, 1727, 1651, 1604, 1557, 1467, 1428, 1413, 1377, 1283, 1248, 1210, 1129, 1031, 955, 905, 722, 684 cm−1.
Boc–PGGG–OCH2Ph
Boc-L-Proline (1.43 g, 6.7 mmol), TsOH·GGG–O–CH2Ph (3.0 g, 6.7 mmol), and Et3N (2.80 mL) were dissolved in CH2Cl2 (40 mL) and cooled to 5°C. EDCI (1.34 g, 7 mmol) was added and reaction was stirred at ambient temperature for 3 days. Solvent was evaporated and residue was dissolved in EtOAc (50 mL) and washed with saturated aqueous NH4Cl (25 mL) and brine (25 mL), dried over MgSO4 and evaporated. The crude, oily product was purified by chromatography (SiO2, 5% MeOH–CH2Cl2) and afforded colorless crystals (2.25 g, 71%, mp 54–55°C). 1H-NMR CDCl3: 1.42 (9H, s, C(CH3)3), 1.80–2.20 (4H, m, Pro NCH2CH2CH2), 3.35–3.55 (2H, m, Pro NCH2CH2CH2), 3.85–4.20 (7H, m, Gly NCH2, Pro NCH), 5.15 (2H, s, PHCH2O), 7.05 (2H, bs, Gly CONH), 7.30–7.35 (5H, m, HAr), 7.80 (1H, bs, Gly CONH). 13C-NMR: 24.6, 28.3, 29.4, 41.1, 43.0, 43.3, 47.2, 60.7, 66.9, 80.9, 128.4, 128.5, 128.7, 135.4, 155.8, 169.6, 170.0, 173.9. IR (KBr): 3310, 3066, 2976, 2933, 1753, 1667, 1540, 1455, 1408, 1366, 1245, 1174, 1129, 1031, 974, 912, 773, 739, 698. Anal. Calcd for C23H32N4O7: C, 57.97; H, 6.77; N, 11.76%. Found: C, 57.87; H, 6.76; N, 11.39%.
HCl·H2N–PGGG–OCH2Ph
Boc–PGGG–OCH2Ph (0.2 g, 0.42 mmol) was dissolved in 4 M HCl in dioxane (10 mL) at 5°C and the reaction mixture was stirred for 1 h. The solvent was evaporated in vacuo and the residue was crystallized from 1∶1 MeOH/Et2O (0.18 g, 100%) to give the title compound as a colorless solid (mp 145–146°C). 1H-NMR (CD3OD): 2.00–2.25 (4H, m, Pro NCH2CH2CH2), 3.35–3.45 (2H, m, Pro NCH2CH2CH2), 3.90–4.05 (6H, m, Gly NCH2), 4.30–4.40 (1H, m, Pro NCH), 5.18 (2H, s, PHCH2O), 7.30–7.40 (5H, m, HAr). 13C-NMR: 25.2, 30.9, 42.1, 43.3, 43.7, 47.6, 61.4, 68.1, 129.5, 129.6, 129.9, 137.5, 170.9, 171.4, 171.8, 172.4.
102DGA–GGGPGGG–OCH2Ph, 1
To 102DGA–GGG–OH (0.18 g, 0.31 mmol) suspended in CH2Cl2 (20 mL), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.07 g, 0.34 mmol) was added and the reaction was stirred for 0.5 h. Then HCl·PGGG–OCH2Ph (0.13 g, 0.31 mmol) in CH2Cl2 (10 mL) containing Et3N (0.10 mL) was added and reaction mixture was stirred for 48 h at room temperature. Then solvent was evaporated and the crude, oily product was purified by chromatography (SiO2, 10% MeOH–CH2Cl2) and afforded 1 as colorless crystals (0.17 g, 60%), mp 127–128°C. [α]20D
−11.4°
(c 1.265, CHCl3). 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (28H, pseudo-s, CH3(CH2)7 CH2CH2N), 1.47 (4H, bs, CH3(CH2)7CH2CH2N), 1.80–2.20 (4H, m, Pro NCH2CH2CH2), 3.03 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.23 (2H, t, J=7.5 Hz, CH3(CH2)8CH2N), 3.40–3.45 (1H, m, Pro NCH2CH2CH2), 3.50–3.55 (1H, m, Pro NCH2CH2CH2), 3.90–4.05 (12H, m, Gly NCH2), 4.12 (2H, s, COCH2O), 4.26 (2H, s, COCH2O), 4.35 (1H, bs, Pro NCH), 5.12 (2H, s, PHCH2O), 7.33 (5H, s, HAr), 7.66 (1H, bs, Gly CONH), 7.76 (1H, br, Gly CONH), 7.85 (1H, br, Gly CONH), 8.06 (1H, bs, Gly CONH), 8.31 (1H, bs, Gly CONH), 8.47 (1H, bs, Gly CONH). 13C-NMR: 13.9, 22.5, 24.9, 26.8, 26.9, 27.5, 28.6, 29.2, 29.5, 29.6, 31.8, 41.1, 41.8, 42.5, 42.6, 42.8, 43.1, 46.1, 46.7, 61.1, 67.0, 69.1, 70.9, 128.2, 128.4, 128.6, 135.4, 168.5, 169.0, 170.3, 170.5, 170.6, 170.9, 171.2, 173.7. IR (KBr): 3300, 2921, 2852, 1741, 1652, 1541, 1457, 1241, 1131, 1028, 720 cm−1. Anal. Calcd for C48H78N8O11: C, 61.12; H, 8.34; N, 11.88%. Found: C, 60.87; H, 8.26; N, 11.65%.
N,N-Dioctadecyl 3-oxaglutaramide (182DGA)
A solution of dioctadecylamine (2.0 g, 3.8 mmol) and diglycolic anhydride (0.44 g, 3.8 mmol) in toluene (50 mL) was refluxed for 48 h. The solvent was evaporated and the crude product crystallized from CHCl3 to give a white solid (2.12 g, 87%), mp 80–81°C. 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (60H, pseudo-s, CH3(CH2)15CH2CH2N), 1.55 (4H, bs, CH3(CH2)15CH2CH2N), 3.07 (2H, t, J=7.8 Hz, CH3(CH2)16CH2N), 3.34 (2H, t, J=7.8 Hz, CH3(CH2)16CH2N), 4.21 (2H, s, COCH2O), 4.38 (2H, s, COCH2O). 13C-NMR: 14.2, 22.8, 26.9, 27.0, 27.5, 28.7, 29.4, 29.5, 29.6, 29.7, 29.8, 32.0, 47.0, 71.4, 73.2, 171.0, 172.2. IR (KBr): 2918, 2850, 1748, 1602, 1488, 1472, 1463, 1431, 1356, 1224, 1159, 1135, 1045, 1013, 990, 920, 885, 729, 720, 689, 643 cm−1.
182DGA–GGG–OCH2Ph
To 182DGA–OH (1g, 1.5 mmol) dissolved in CH2Cl2 (30 mL), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.31 g, 1.6 mmol) was added and the mixture was stirred at room temperature. After 0.5 h, TsOH·H2N–GGG–OCH2Ph (0.66 g, 1.5 mmol) and Et3N (0.6 mL) were added and the mixture was stirred at ambient temperature overnight. The reaction mixture was successively washed with water (20 mL), 0.5 M HCl (20 mL), water (20 mL), 10% Na2CO3 (20 mL), and brine (20 mL), dried (MgSO4), evaporated and the residue crystallized from MeOH to afford a white solid (1.26 g, 89%), mp 41–42°C. 1H-NMR CDCl3: 0.86 (6H, t, J=6.9 Hz, –CH2CH3), 1.24 (60H, pseudo-s, CH3(CH2)15CH2CH2N), 1.49 (4H, bs, CH3(CH2)15CH2CH2N), 1.61 (1H, H2O), 3.04 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.24 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.95–4.05 (6H, m, Gly NCH2), 4.09 (2H, s, COCH2O), 4.29 (2H, s, COCH2O), 5.12 (2H, s, PHCH2O), 7.23 (1H, t, J=6.0 Hz, Gly CONH), 7.30–7.35 (5H, m, HAr), 7.93 (1H, t, J=5.7 Hz, Gly CONH), 8.27 (1H, t, J=5.7 Hz, Gly CONH). 13C-NMR: 13.9, 22.5, 26.7, 26.9, 27.4, 28.6, 29.2, 29.3, 29.6, 31.8, 41.0, 42.9, 46.3, 46.7, 66.9, 69.6, 71.7, 128.2, 128.4, 128.6, 135.3, 168.6, 169.7, 169.8, 170.0, 171.5. IR (KBr): 3293, 2916, 2849, 1749, 1651, 1544, 1467, 1196, 1128, 1031, 721, 697 cm−1. Anal. Calcd for C53H94N4O7+0.5 H2O: C, 70.11; H, 10.54; N, 6.17%. Found: C, 70.18; H, 10.55; N, 6.18%.
182DGA–GGG–OH
182DGA–GGG–OCH2Ph (1.0 g, 1.1 mmol) was dissolved in absolute ethanol (100 mL) and 10% Pd/C (0.2 g) was added and this mixture was shaken under 60 psi H2 for 3 h. The reaction mixture was heated to reflux and filtered hot through a celite pad. The solvent was evaporated under reduced pressure to afford a white solid (0.86 g, 96%), mp 163–164°C. 1H-NMR CD3OD: 0.90 (6H, t, J=6.9 Hz, –CH2CH3), 1.29 (60H, pseudo-s, CH3(CH2)15CH2CH2N), 1.57 (4H, bs, CH3(CH2)15CH2CH2N), 3.21 (2H, t, J=7.8 Hz, CH3(CH2)16CH2N), 3.35 (2H, t, J=7.8 Hz, CH3(CH2)16CH2N), 3.93 (2H, s, Gly NCH2), 3.94 (2H, s, Gly NCH2), 3.97 (2H, s, Gly NCH2), 4.12 (2H, s, COCH2O), 4.40 (2H, s, COCH2O). 13C-NMR: IR (KBr): 3285, 3084, 2925, 2852, 1740, 1650, 1551, 1467, 1420, 1378, 1219, 1128, 1033, 1011, 721, 681 cm−1. Anal. Calcd for C46H88N4O7: C, 68.28; H, 10.96; N, 6.92%. Found: C, 67.97; H, 10.92; N, 6.81%.
182DGA–GGGPGGG–OCH2Ph, 2
182DGA–GGG–OH (0.31 g, 0.39 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.08 g, 0.42 mmol) were stirred for 0.5 h in CH2Cl2 (40 mL) and HCl·H2N–PGGG–OCH2Ph (0.16 g, 0.39 mmol) in CH2Cl2 (10 mL) containing Et3N (0.15 mL) was added. The mixture was stirred for 48 h at room temperature, solvent was evaporated, and the residue was crystallized from MeOH to give 2 as a white solid (0.37 g, 82%), mp 116–118°C. [α]20D
−8.9 (c 1.055, CHCl3). 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (60H, pseudo-s, CH3(CH2)15CH2CH2N), 1.47 (4H, bs, CH3(CH2)15CH2CH2N), 1.80–2.20 (4H, m, Pro NCH2CH2CH2), 3.03 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.23 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.40–3.45 (1H, m, Pro NCH2CH2CH2), 3.50–3.55 (1H, m, Pro NCH2CH2CH2), 3.90–4.05 (12H, m, Gly NCH2), 4.12 (2H, s, COCH2O), 4.26 (2H, s, COCH2O), 4.35 (1H, bs, Pro NCH), 5.12 (2H, s, PHCH2O), 7.33 (5H, s, HAr), 7.66 (1H, bs, Gly CONH), 7.76 (1H, br, Gly CONH), 7.85 (1H, br, Gly CONH), 8.06 (1H, bs, Gly CONH), 8.31 (1H, bs, Gly CONH), 8.47 (1H, bs, Gly CONH). 13C-NMR: 13.9, 22.5, 24.9, 26.8, 26.9, 27.5, 28.6, 29.2, 29.5, 29.6, 31.8, 41.1, 41.8, 42.5, 42.6, 42.8, 43.1, 46.1, 46.7, 61.1, 67.0, 69.1, 70.9, 128.2, 128.4, 128.6, 135.4, 168.5, 169.0, 170.3, 170.5, 170.6, 170.9, 171.2, 173.7. IR (KBr): 3301, 2922, 2853, 1740, 1653, 1540, 1457, 1242, 1131, 1029, 720. MS (ESI): m/z
[M+Na]+ calculated for C64H110N8O11Na 1189, observed 1190. Anal. Cald for C64H110N8O11: C, 65.83; H, 9.50; N, 9.60%. Found: C, 65.38; H, 9.68; N, 9.16%.
Boc–PipGGG–Bzl
Boc-L-Pipecolic acid (0.50 g, 2.2 mmol), TsOH·GGG–O–CH2Ph (0.98 g, 2.2 mmol), and Et3N (1.50 mL) were dissolved in CH2Cl2 (20 mL) and cooled to 5°C. EDCI (0.46 g, 2.4 mmol) was added and the reaction was stirred at ambient temperature for 3 days. Solvent was evaporated and the residue was dissolved in EtOAc (50 mL), washed with citric acid (5%, 25 mL), Na2CO3
(25 mL), brine (25 mL), dried over MgSO4, and evaporated. The crude, oily product was purified by chromatography (SiO2, 5% MeOH–CH2Cl2) to afford colorless crystals (0.97 g, 91%), mp 56–58°C. [α]20D
−36.1°
(c 1.045, CHCl3). 1H-NMR CDCl3: 1.45 (9H, s, C(CH3)3), 1.59 (4H, bs, Pip CH2), 2.15 (2H, bs, Pip CH2), 3.00 (1H, bs, Pip CH2), 3.85–4.15 (7H, overlapping signals due to Gly CH2 and Pip CH2), 4.69 (1H, s, Pip CH), 5.15 (2H, s, CH2Ph), 6.96 (1H, bs, NH), 7.10 (1H, bs, NH), 7.30–7.50 (5H, m, HAr). 13C-NMR: 20.4, 24.7, 25.8, 28.3, 41.3, 42.6, 42.9, 43.2, 55.1, 67.2, 80.8, 128.3, 128.5, 128.6, 135.2, 156.1, 169.1, 169.6, 172.6. IR (KBr): 3325, 2938, 1752, 1664, 1530, 1457, 1409, 1366, 1253, 1189, 1164, 1032, 989, 870, 751, 699 cm−1.
H2N–PipGGG–OCH2Ph·HCl
Boc–PipGGG–OCH2Ph (0.2 g, 0.25 mmol) was dissolved in 4 M HCl in dioxane (10 mL) at 5°C and the reaction mixture was stirred for 1 h. The solvent was evaporated in vacuo and the residue was used in the next reaction without further purification.
102DGA–GGGPipGGG–OCH2Ph, 3
To 102DGA–GGG–OH (0.36 g, 0.61 mmol) suspended in CH2Cl2 (20 mL), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.12 g, 0.64 mmol) and HOBt (0.09 g, 0.64 mmol) were added and the reaction mixture was stirred for 0.5 h. Then HCl·PipGGG–OCH2Ph (0.29 g, 0.61 mmol) in CH2Cl2 (20 mL) containing Et3N (0.09 mL, 0.64 mmol) was added and the reaction mixture was stirred for 48 h at room temperature. The reaction mixture was evaporated in vacuo, the residue successively washed with 5% citric acid (20 mL), water (20 mL), 5% NaHCO3 (20 mL), and brine (20 mL), dried (MgSO4), and evaporated. The crude, oily product was purified by chromatography (SiO2, 5% MeOH–CH2Cl2) and afforded 3 as a white solid (0.49 g, 84%), mp 78–80°C. [α]20D
−22.5 (c 1.065, CHCl3). 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (28H, pseudo-s, CH3(CH2)7CH2CH2N), 1.50 (8H, bs, overlapping signals due to CH3(CH2)7CH2CH2N and Pip CH2), 2.13 (2H, bs, Pip CH2), 2.85 (1H, bs, Pip CH2) 3.05 (2H, t, J=7.5 Hz, CH3(CH2)7CH2CH2N), 3.25 (2H, t, J=7.5 Hz, CH3(CH2)7CH2CH2N), 3.55–3.65 (1H, m, Pip CH2), 3.80–4.15 (12H, m, Gly NCH2), 4.10 (2H, s, COCH2O), 4.27 (2H, s, COCH2O), 5.06 (1H, bs, Pip CH), 5.14 (2H, s, PHCH2O), 7.34 (5H, pseudo s, HAr), 7.49 (1H, bs, Gly CONH), 7.55 (2H, bs, Gly CONH), 7.85 (1H, bs, Gly CONH), 7.98 (1H, bs, Gly CONH), 8.29 (1H, bs, Gly CONH). 13C-NMR: 14.0, 20.2, 22.6, 24.9, 25.7, 26.8, 27.0, 27.6, 28.8, 29.2, 29.3, 29.4, 29.5, 31.8, 41.2, 41.4, 42.7, 43.0, 43.3, 46.2, 46.8, 53.4, 67.0, 69.2, 71.2, 128.1, 128.3, 128.5, 135.3, 168.3, 169.1, 170.0, 170.1, 170.2 170.4, 170.8, 171.9. IR (CHCl3): 3309, 3069, 2926, 2854, 1748, 1651, 1540, 1457, 1259, 1193, 1129, 1029 cm−1. Anal. Calcd for C49H80N8O11: C, 61.48; H, 8.42; N, 11.71%. Found: C, 61.12; H, 8.35; N, 11.43%.
182DGA–GGGPipGGG–OCH2Ph, 4
182DGA–18–GGG–OH (0.20 g, 0.25 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.05 g, 0.28 mmol) and HOBt (0.04 g, 0.28 mmol) were suspended in CH2Cl2 (20 mL) and stirred for 0.5 h. HCl·H2N–PipGGG–OCH2Ph (0.12 g, 0.25 mmol) in CH2Cl2 (5 mL) containing N-methylmorpholine (0.05 mL, 0.46 mmol) was added and the mixture was stirred for 48 h at room temperature. The solvent was evaporated off and the residue was crystallized from MeOH to give 4 as a white solid (0.24 g, 82%), mp 164–165°C. [α]20D
−15.1°
(c 1.10, CHCl3). 1H-NMR CDCl3: 0.87 (6H, t, J=6.9 Hz, –CH2CH3), 1.25 (60H, pseudo-s, CH3(CH2)15CH2CH2N), 1.50 (8H, bs, overlapping signals due to CH3(CH2)15CH2CH2N and Pip CH2), 2.13 (2H, bs, Pip CH2), 2.85 (1H, bs, Pip CH2) 3.05 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.25 (2H, t, J=7.5 Hz, CH3(CH2)16CH2N), 3.55–3.65 (1H, m, Pip CH2), 3.80–4.15 (12H, m, Gly NCH2), 4.10 (2H, s, COCH2O), 4.27 (2H, s, COCH2O), 5.04 (1H, bs, Pip CH), 5.14 (2H, s, PHCH2O), 7.34 (5H, pseudo s, HAr), 7.49 (1H, bs, Gly CONH), 7.55 (2H, bs, Gly CONH), 7.85 (1H, bs, Gly CONH), 7.98 (1H, bs, Gly CONH), 8.29 (1H, bs, Gly CONH). 13C-NMR: 14.1, 20.1, 22.7, 24.8, 25.8, 26.9, 27.1, 27.6, 29.3, 29.4, 29.6, 29.7, 31.9, 41.2, 41.4, 42.8, 43.0, 43.3, 46.3, 46.9, 50.7, 53.6, 67.0, 69.4, 71.3, 128.2, 128.4, 128.6, 135.4, 168.4, 169.4, 169.9, 170.0, 170.1, 170.3, 170.4, 170.9, 172.0. IR (KBr): 3315, 2921, 2851, 2359, 1749, 1656, 1541, 1468, 1262, 1130, 1029, 722, 698 cm−1. Anal. Calcd for C65H112N8O11: C, 66.07; H, 9.55; N, 9.48%. Found: C, 66.34; H, 9.63; N, 9.32%.
Acknowledgements
We gratefully acknowledge support of this work by the NIH (GM-36262) and by the Kilo Foundation.References
- M. Maduke, C. Miller and J. A. Mindell, Annu. Rev. Biomol. Struct., 2000, 29, 411–438 Search PubMed.
- C. Miller, Curr. Opin. Chem. Biol., 2000, 4, 148–151 CrossRef CAS.
- R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS.
- N. S. Heiss and A. Poustka, Genomics, 1997, 45, 224–228 CrossRef CAS.
- T. J. Jentsch and W. Gunther, Bioessays, 1997, 117–26 CAS.
- D. R. Halm and R. A. Frizzell, Intestinal Chloride Secretion, Raven, New York, 1990 Search PubMed.
- K. Steinmeyer, C. Ortland and T. J. Jentsch, Nature, 1991, 354, 301–304 CrossRef CAS.
- J. M. Tomich, D. Wallace, K. Henderson, K. E. Mitchell, G. Radke, R. Brandt, C. A. Ambler, A. J. Scott, J. Grantham, L. Sullivan and T. Iwamoto, Biophys. J., 1998, 74, 256–267 Search PubMed.
- D. P. Wallace, J. M. Tomich, J. W. Eppler, T. Iwamoto, J. J. Grantham and L. P. Sullivan, Biochim. Biophys. Acta, 2000, 1464, 69–82 CrossRef CAS.
- J. R. Broughman, K. E. Mitchell, R. L. Sedlacek, T. Iwamoto, J. M. Tomich and B. D. Schultz, Am. J. Physiol., 2001, 280, C451–C458 Search PubMed.
- P. H. Schlesinger, R. Ferdani, J. Liu, J. Pajewska, R. Pajewski, M. Saito, H. Shabany and G. W. Gokel, J. Am. Chem. Soc., 2002, 124, 1848–1849 CrossRef CAS.
- P. H. Schlesinger, R. Ferdani, R. Pajewski, J. Pajewska and G. W. Gokel, Chem. Commun., 2002, 840–841 RSC.
- C. Fahlke, H. T. Yu, C. L. Beck, T. H. Rhodes and A. L. George, Nature, 1997, 390, 529–532 CrossRef CAS.
- C. Fahlke, R. R. Desai, N. Gillani and A. L. George, J. Biol. Chem., 2001, 276, 1759–1765 CAS.
- P. J. Corringer, S. Bertrand, J. Galzi, A. Devillers-Thiery, J.-P. Changeux and D. Bertrandt, Neuron, 1999, 22, 831–843 CrossRef CAS.
- J. L. Galzi, A. Devillers-Thièry, N. Hussy, S. Bertrand, J. P. Changeux and D. Bertrand, Nature, 1992, 359, 500–505 CrossRef CAS.
- N. Gibbs, R. B. Sessions, P. B. Williams and C. E. Dempsey, Biophys. J., 1997, 72, 2490–2495 Search PubMed.
- C. J. Brandl and C. M. Deber, Proc. Natl. Acad. Sci. USA, 1986, 83, 917–921 CAS.
- D. P. Tieleman, M. S. P. Sansom and H. J. C. Berendsen, Biophys. J., 1999, 76, 40–49 Search PubMed.
- H. Ihara, Y. Hashiguchi and T. Kunitake, Chem. Lett., 1983, 733–736 CAS.
- B. D. Gildea, S. Casey, J. MacNeill, H. Perry-O'Keefe, D. Soerensen and J. M. Coull, Tetrahedron Lett., 1998, 7255–7258 CrossRef CAS.
- R. McCrindle and A. J. McAlees, J. Chem. Soc., Perkin Trans. 1, 1981, 741–745 RSC.
- H. Ebato, J. N. Herron, W. Müller, Y. Okahata, H. Ringsdorf and P. Suci, Angew. Chem., Int. Ed. Engl., 1992, 31, 1087–1090 CrossRef.
- J. Dufourcq, J. F. Faucon, G. Fourche, J. L. Dasseux, M. Le Maire and T. Gulik-Krzywicki, Biochim. Biophys. Acta, 1986, 859, 33–48 CAS.
- P. H. Schlesinger, N. K. Djedovič, R. Ferdani, J. Pajewska, R. Pajewski and G. W. Gokel, J. Am. Chem. Soc., 2002 , submitted.
- P. Schlesinger, A. Gross, X. Xin, K. Yamamoto, M. Saito, G. Waksman and S. J. Korsmeyer, Proc. Natl. Acad. Sci. USA, 1997, 94, 11357–11362 CrossRef CAS.
- P. H. Schlesinger, Y. Ido and J. R. Williamson, Diabetes, 1998, 47, A29 Search PubMed.
- Y. Ido, A. Vindigni, K. Chang, L. Stramm, R. Chance, W. F. Heath, R. D. DiMarchi, E. DiCera and J. R. Williamson, Science, 1997, 277, 563–566 CrossRef CAS.
- (a) W.-J. Wu and D. P. Raleigh, J. Org. Chem., 1998, 63, 6689–6698 CrossRef CAS; (b) Y. Takeuchi and F. R. Marshall, J. Am. Chem. Soc., 1998, 120, 5363–5372 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |