Cyclopropanation and Diels–Alder reactions catalyzed by the first heterobimetallic complexes with bridging phosphinooxazoline ligands†
Pierre
Braunstein
,
Guislaine
Clerc
and
Xavier
Morise
Laboratoire de Chimie de Coordination, UMR CNRS 7513, Université Louis Pasteur, 4 rue Blaise Pascal, F-67070, Strasbourg Cedex, France. E-mail: braunst@chimie.u-strasbg.fr
First published on 25th October 2002
Abstract
The bimetallic complex trans-[(OC)3Fe![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) (μ-LP,N)2Cu
(μ-LP,N)2Cu![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) ]BF4
(2), which contains two bridging phosphinooxazoline ligands, is the first metal–metal bonded six-membered ring system with P,N donors and its crystal structure shows a unique bimetallic cradle conformation. This complex is an efficient catalyst for the cyclopropanation of styrene by ethyl diazoacetate and for the Diels–Alder reaction between cyclopentadiene and methacrolein, these reactions being catalyzed for the first time by heterometallic complexes.
]BF4
(2), which contains two bridging phosphinooxazoline ligands, is the first metal–metal bonded six-membered ring system with P,N donors and its crystal structure shows a unique bimetallic cradle conformation. This complex is an efficient catalyst for the cyclopropanation of styrene by ethyl diazoacetate and for the Diels–Alder reaction between cyclopentadiene and methacrolein, these reactions being catalyzed for the first time by heterometallic complexes.
Introduction
Heterometallic complexes are extensively studied in the search for cooperative effects between metal centres of different natures, for metallosite selective reactions and/or chemical transformations occurring at both metal centres in sequence or in a concomitant manner.1 Special interest arises of course when their reactivity is different from that of their homometallic or mononuclear anologues. Such differences may be related to the presence of a new chemical bond (direct metal–metal interaction), to changes in the coordination sphere of a metal centre resulting from the stereoelectronic properties of an adjacent metal, or to their combination.Bi- or polyfunctional ligands, in particular functional phosphines, are often used as assembling ligands to stabilise bimetallic structures and have mostly led to the formation of five-membered metallocycles.2 Metal–metal bonded systems with larger bimetallic cycles have been less studied3 and in the case of (P,N) ligands there appears to be no six-membered ring system known and only one seven-membered macrocycle has been very recently reported.4
Although phosphinooxazoline ligands are being much investigated as P-monodentate or P,N-chelating ligands,5 there is no reported example, to the best of our knowledge, of bridging behaviour for such ligands. We have now investigated their potential as assembling ligands to form metal–metal bonded six-membered ring systems.
Results and discussion
Using the ligand (2-oxazoline-2-ylmethyl)diphenylphosphine (LP,N),6 we have prepared trans-[Fe(CO)3(LP,N)2] 1 and used it to form the Fe–Cu complex trans-[(OC)3Fe(μ-LP,N)2Cu]BF4
(2), with which the catalytic properties of heterometallic complexes have been examined for the first time in olefin cyclopropanation and Diels–Alder reactions. Complex 1, whose structure has been determined by X-ray diffraction (Fig. 1), was reacted with a stoichiometric amount of [Cu(NCMe)4][BF4]
(Scheme 1) to afford the bimetallic complex 2 in 91% yield.
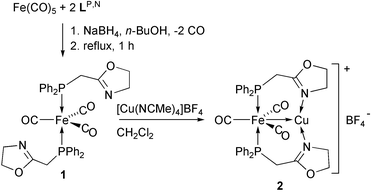
![View of the crystal structure of complex trans-[Fe(CO)3(LP,N)2]
(1). Ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (°): Fe–C(1) 1.763(9), Fe–C(2) 1.78(1), Fe–C(3) 1.79(1), Fe–P(1) 2.197(3), Fe–P(2) 2.213(3), C(5)–N(1) 1.28(1), C(21)–N(2) 1.28(1), N(1)–C(6) 1.48(1), N(2)–C(22) 1.48(1), P(1)–C(4) 1.848(9), P(2)–C(20) 1.87(1), C(5)–O(4) 1.34(1), C(21)–O(5) 1.34(1); C(1)–Fe–C(2) 118.7(4), C(1)–Fe–C(3) 120.4(5), C(2)–Fe–C(3) 120.9(5), P(1)–Fe–P(2) 176.6(1), P(1)–C(4)–C(5) 116.1(6), P(2)–C(20)–C(21) 115.2(7).](/image/article/2003/NJ/b206410n/b206410n-f1.gif) | ||
| Fig. 1 View of the crystal structure of complex trans-[Fe(CO)3(LP,N)2] (1). Ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (°): Fe–C(1) 1.763(9), Fe–C(2) 1.78(1), Fe–C(3) 1.79(1), Fe–P(1) 2.197(3), Fe–P(2) 2.213(3), C(5)–N(1) 1.28(1), C(21)–N(2) 1.28(1), N(1)–C(6) 1.48(1), N(2)–C(22) 1.48(1), P(1)–C(4) 1.848(9), P(2)–C(20) 1.87(1), C(5)–O(4) 1.34(1), C(21)–O(5) 1.34(1); C(1)–Fe–C(2) 118.7(4), C(1)–Fe–C(3) 120.4(5), C(2)–Fe–C(3) 120.9(5), P(1)–Fe–P(2) 176.6(1), P(1)–C(4)–C(5) 116.1(6), P(2)–C(20)–C(21) 115.2(7). | ||
 | ||
| Scheme 1 | ||
Despite the “anti-type” spatial arrangement of the oxazoline groups in 1, which minimizes steric repulsion, the LP,N ligands can function as bridges and lead to two fused six-membered rings in 2. The ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) vibration at 1643 cm−1 is consistent with the N-coordination of the oxazoline to Cu(I) and a singlet is observed in the 31P{1H} NMR spectrum at δ 71.71. No signal characteristic of MeCN was observed, indicating complete displacement from the precursor. The PCH2 carbon atoms give rise in 13C{1H} NMR to an AA′XX′ multiplet from which a 2JPP coupling constant of 34.8 Hz was extracted (gNMRV3.6 simulation). This value is similar to those reported in the literature for related trans-[Fe(CO)3LL′] complexes (L
N) vibration at 1643 cm−1 is consistent with the N-coordination of the oxazoline to Cu(I) and a singlet is observed in the 31P{1H} NMR spectrum at δ 71.71. No signal characteristic of MeCN was observed, indicating complete displacement from the precursor. The PCH2 carbon atoms give rise in 13C{1H} NMR to an AA′XX′ multiplet from which a 2JPP coupling constant of 34.8 Hz was extracted (gNMRV3.6 simulation). This value is similar to those reported in the literature for related trans-[Fe(CO)3LL′] complexes (L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =tertiary phosphines).7 The ν(CO) vibrations (CH2Cl2) at 1986(w), 1915(s) and 1882(vs) cm−1 are consistent with a meridional arrangement of the carbonyls around the Fe centre. The structure of 2·1.5CH2Cl2 was established by X-ray diffraction (Fig. 2). The molecule contains a mirror plane which includes the metals and the CO ligands. The coordination geometry about the Fe atom may be described as distorted octahedral with the P atoms trans to one another and the CO ligands in a mer arrangement. Alternatively, if one ignores the Fe–Cu bond, it may be viewed as trigonal bipyramidal, as in 1, a geometry consistent with a formally zero-valent Fe centre. The Cu(I) atom exhibits a trigonal coordination involving the Fe atom and the two nitrogen atoms from the LP,N ligands. The Fe–Cu distance of 2.5441(7)
Å is longer than in a four-membered cycle with a bridging aminosilyl ligand [2.530(2)
Å],8 in five-membered ring complexes with bridging 2-(diphenylphosphino)pyridine (Ph2PPy)
[2.501(2)
Å, 2.512(2)
Å],2d or bridging dppm [2.497(2),9a 2.540(2)
Å9b] or in a bis-seven-membered macrocyclic compound [2.4572(7)
Å],4 but is similar to that in a complex with bridging N-(diphenylphosphinomethyl)morpholine [2.550(1)
Å].10 The Fe–Cu distance in 2 is equal to the sum of the atomic radii of iron and copper (2.54 Å). Owing to the presence of a mirror plane in the molecule, the P, CH2 and oxazoline groups of one ligand are respectively eclipsed with those of the other ligand. This creates a sort of molecular cradle cavity (Fig. 3), which results from the ring size and the ligand conformation and contrasts with the generally rather flat structures exhibited by dinuclear complexes containing other types of assembling ligands, such as Ph2PPy or dppm. The C(1)–Fe–C(2) angle of 131.0(2)° has opened up compared to 1 owing to the presence of the Cu centre and this leads to a C(1)–Cu separation of 2.339(4)
Å and a Fe–C(1)–O(1) angle of 174.32(1)°.
=tertiary phosphines).7 The ν(CO) vibrations (CH2Cl2) at 1986(w), 1915(s) and 1882(vs) cm−1 are consistent with a meridional arrangement of the carbonyls around the Fe centre. The structure of 2·1.5CH2Cl2 was established by X-ray diffraction (Fig. 2). The molecule contains a mirror plane which includes the metals and the CO ligands. The coordination geometry about the Fe atom may be described as distorted octahedral with the P atoms trans to one another and the CO ligands in a mer arrangement. Alternatively, if one ignores the Fe–Cu bond, it may be viewed as trigonal bipyramidal, as in 1, a geometry consistent with a formally zero-valent Fe centre. The Cu(I) atom exhibits a trigonal coordination involving the Fe atom and the two nitrogen atoms from the LP,N ligands. The Fe–Cu distance of 2.5441(7)
Å is longer than in a four-membered cycle with a bridging aminosilyl ligand [2.530(2)
Å],8 in five-membered ring complexes with bridging 2-(diphenylphosphino)pyridine (Ph2PPy)
[2.501(2)
Å, 2.512(2)
Å],2d or bridging dppm [2.497(2),9a 2.540(2)
Å9b] or in a bis-seven-membered macrocyclic compound [2.4572(7)
Å],4 but is similar to that in a complex with bridging N-(diphenylphosphinomethyl)morpholine [2.550(1)
Å].10 The Fe–Cu distance in 2 is equal to the sum of the atomic radii of iron and copper (2.54 Å). Owing to the presence of a mirror plane in the molecule, the P, CH2 and oxazoline groups of one ligand are respectively eclipsed with those of the other ligand. This creates a sort of molecular cradle cavity (Fig. 3), which results from the ring size and the ligand conformation and contrasts with the generally rather flat structures exhibited by dinuclear complexes containing other types of assembling ligands, such as Ph2PPy or dppm. The C(1)–Fe–C(2) angle of 131.0(2)° has opened up compared to 1 owing to the presence of the Cu centre and this leads to a C(1)–Cu separation of 2.339(4)
Å and a Fe–C(1)–O(1) angle of 174.32(1)°.
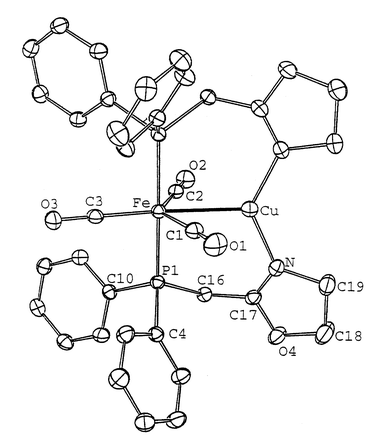 | ||
| Fig. 2 View of the crystal structure of complex 2. Ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (°): Fe–Cu 2.5441(7), Fe–C(1) 1.795(4), Fe–C(2) 1.777(4), Fe–C(3) 1.786(4), Fe–P 2.230(1), Cu–C(1) 2.339(4), Cu–C(2) 2.514(4), Cu–N 1.939(2), C(17)–N 1.270(3); C(1)–Fe–C(2) 131.0(2), C(1)–Fe–C(3) 108.1(2), C(2)–Fe–C(3) 121.0(2), P(1)–Fe–P(1′) 173.75(4), Cu–Fe–C(1) 62.4(1), Cu–Fe–C(2) 68.5(1), Cu–Fe–C(3) 170.5(1), Cu–Fe–P 90.16(2), Fe–Cu–N 113.33(7), N–Cu–N′ 132.7(1), Fe–C(1)–O(1) 174.32(1), Fe–C(2)–O(2) 178.32(1), Fe–C(3)–O(3) 178.96(1). | ||
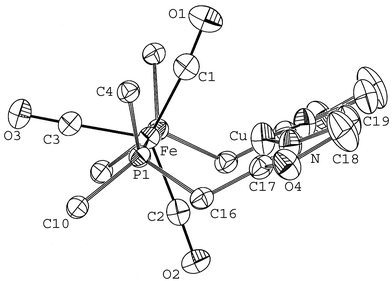 | ||
| Fig. 3 Perspective view of the structure of 2 along the P–Fe–P axis showing the curvature generated by the two, mutually eclipsed assembling phosphinooxazoline ligands. The Fe–Cu bond has been omitted for clarity and only the ipso carbons of the phenyl groups are shown. | ||
Complex 2 is an efficient catalyst for the cyclopropanation of styrene with ethyl diazoacetate [eqn. (1)]. The reaction was carried out in 1,2-dichloroethane at room temperature (1 mol% of 2 based on ethyl diazoacetate), with an olefin/ethyl diazoacetate ratio of 2∶1. The ethyl diazoacetate was added very slowly over a period of 35 h.11Trans- and cis-ethyl 2-phenyl-1-cyclopropanecarboxylates were obtained, in 91% isolated yield in a 70∶30 ratio, and complex 2 could still be detected after complete consumption of the ethyl diazoacetate. These results compare favourably with literature data for mononuclear copper catalysts.12 However, we found that with [Cu(NCMe)4][BF4] as a catalyst under similar conditions, trans- and cis-ethyl 2-phenyl-1-cyclopropanecarboxylates were obtained in 97% isolated yield in a 64∶36 ratio.
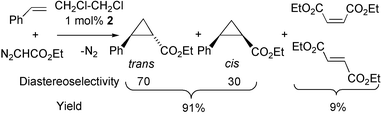 | (1) |
=bis(2-oxazoline-2-ylmethyl)phenylphosphine)],13 in which the coordination sphere of the Cu(I) ion contains two oxazoline ligands disposed similarly to those in 2
(in which additionally the Fe centre behaves like a metalloligand). With this complex as a catalyst, trans- and cis-ethyl 2-phenyl-1-cyclopropanecarboxylates were obtained in 91% isolated yield in a 68∶32 ratio.However, to the best of our knowledge, 2 is the first metal–metal bonded heterometallic catalyst to be used in olefin cyclopropanation.
Furthermore, 2 was found to catalyze the Diels–Alder reaction of methacrolein with cyclopentadiene [eqn. (2)]. The reaction reached 76% convertion after stirring for 162 h at −20°C.14
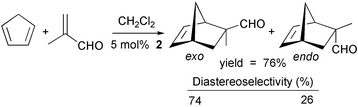 | (2) |
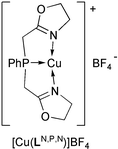
°C and the exo∶endo ratio was 85/15 (determined by 1H NMR). In contrast, (CuLN,P,N)BF4 did not catalyze this reaction, although the utilization of phosphinooxazoline ligands and Cu(I) salts has been reported for the catalytic enantioselective aza-Diels–Alder reaction of imines.16
Conclusion
The complex trans-[Fe(CO)3(LP,N)2] 1 is a readily available precursor for the construction of new bimetallic complexes with phosphinooxazoline assembling ligands, as exemplified with the formation of trans-[(OC)3Fe(μ-LP,N)2Cu]BF4
(2), a formally Fe(0)→Cu(I) complex. The latter appears to be the first metal–metal bonded heterometallic complex successfully tested in olefin cyclopropanation and Diels–Alder reactions. Although our preliminary catalytic results do not demonstrate a clear advantage of the bimetallic system over a well-chosen mononuclear Cu(I) catalyst, we had to resort to different Cu(I) precursors to observe catalysis in each reaction. We expect further developments in the context of bimetallic effects in catalysis1 and of enhanced selectivity properties associated with the unique “bimetallic cavity-like” shape of complex 2. Current extensions include the use of chiral systems and we have already prepared the Fe–Cu complex 2*
(see Experimental) containing the enantiopure ligand LP,NPh.17 Its evaluation in catalysis will be the subject of further studies.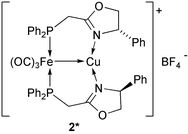
Experimental
All reactions were performed under purified nitrogen. Solvents were purified and dried under nitrogen by conventional methods. The 1H and 31P{1H} NMR spectra were recorded at 300.13 and 121.5 MHz, respectively, on a FT Bruker AC300 instrument, 13C NMR spectra at 75.477 or 100.628 MHz on FT Bruker AC300 or AC400 instruments, respectively, and the IR spectra in the 4000–400 cm−1 range on a Bruker IFS66 FT spectrometer. The ligands (LP,N),6 (LN,P,N)13 and (LP,NPh)17 were prepared according to the literature.Synthesis of the complexes
×10 ml) to remove the borate salts. The solid product was dried under vacuum. The IR and 31P{1H} NMR spectra showed the isolated product to be exclusively trans-[Fe(CO)3(LP,N)2]
(4.82 g, 90%). IR (CH2Cl2): ν(CO) 1974 (w), 1886 (vs), ν(CN) 1661(m) cm−1; (KBr): ν(CO) 1973 (vw), 1931 (sh), 1876 (vs), ν(CN) 1658 (ms) cm−1. 1H NMR (CDCl3, 298 K): δ 7.78–7.42 (m, 20H, aromatics), 4.06 (t, 4H, 3JHH=9.0 Hz, OCH2), 3.75 (t, 4H, 3JHH=9.0 Hz, NCH2), 3.60 (m, 4H, JPH=7.9 Hz, PCH2). 13C{1H} NMR (CD2Cl2): δ 213.2 (t, 3CO, 2JPC=22 Hz), 162.7 (s, CN), 137.0 (d, 1JPC=22 Hz, ipso-aryl), 132.8 (s, m-aryl), 130.6 (s, p-aryl), 128.6 (s, o-aryl), 67.9 (s, OCH2), 55.2 (s, NCH2), 34.1 (m, PCH2). 31P{1H} NMR (CDCl3, 298 K): δ 78.4 (s) Anal. Calcd. for C35H32FeN2O5P2: C, 61.94; H, 4.76; N, 4.13. Found: C, 61.81; H, 5.01; N, 3.89%.
°C. The mixture was stirred vigorously for 20 min, the yellow solution became deeper almost immediately. The mixture was concentrated and slow diffusion of pentane afforded yellow crystals (1.11 g, 91%). IR (CH2Cl2): ν(CO) 1986 (mw), 1915 (s) 1882 (vs), ν(CN) 1643 (m) cm−1. 1H NMR (CDCl3, 298 K): δ 7.58–7.52 (m, 20H, aromatics), 4.46 (t, 4H, 3JHH=9.7 Hz, OCH2), 4.17 (t, 4H, 3JHH=9.7 Hz, NCH2), 3.56 (appearance of “filled-in d” analyzed as an AA′XX′ spin system (X=P), 4H, 2+4JPH=9.1, 2JPP=34.8 Hz, PCH2). 13C{1H} NMR (CD2Cl2): δ 210.0 (t, 3CO 2JPC=25 Hz), 168.7 (s, CN), 133.9–128.9 (aromatics), 68.6 (s, OCH2), 55.3 (s, NCH2), 31.6 (m, AA′XX′ spin system (X=P), 2+3JPC=19.6, JPP=34.8 Hz, PCH2). 31P{1H} NMR (CDCl3, 298 K): δ 71.7 (s). Anal. Calcd. for C35H32BCuF4FeN2O5P2·1.5CH2Cl2·0.5MeCN: C, 46.12; H, 3.77; N, 3.59. Found: C, 46.15; H, 3.57; N, 3.87%.
°C. The mixture was stirred vigorously for 20 min and a white precipitate formed after hexane was added (1.04 g, 98%). IR (KBr): ν(CN) 1644 (vs) cm−1. 1H NMR (CD2Cl2): δ 1.97 (s, 3H, CH3CN), 3.17 (m, 2 H, part A of an ABX system (A=B=H, X=P), PCHAHB), 3.45 (m, 2 H, part B of an ABX system, PCHAHB), 3.74 (m, 2 H, part A of an ABFG system (A=B=F=G=H), NCHAHB), 3.89 (m, 2 H, part G of an ABFG system, NCHAHB), 4.48 (m, 2 H, part F of an ABFG system, OCHFHG), 4.55 (m, 2H, part B of an ABFG system, OCHFHG), 7.58–7.89 (m, 5 H, aromatics), 31P{1H} NMR (DMSO-d6): δ
−22.3 (s). Anal. Calcd. for C14H17BCuF4N2O2P·CH3CN: C, 41.11; H, 4.32; N, 8.99. Found: C, 40.57; H, 4.30; N, 9.00%. At this stage, we cannot state whether the molecule of acetonitrile found in solution and in the solid (elemental analysis) corresponds to a molecule of solvation or to a Cu(I)-bound ligand.
×10 ml) to remove the borate salts. The solid product was dried under vacuum. Both IR and 31P NMR spectra showed the isolated product to be exclusively trans-[Fe(CO)3(LP,NPh)2]
(0.382 g, 9.70%). IR (CH2Cl2): ν(CO) 1970 (w), 1882 (vs), ν(CN) 1657 (m) cm−1. 1H NMR (CDCl3, 298 K): δ 8.10–6.90 (m, 30H, aromatics), 5.09 (dd, 2H, A part of an AB spin system 2J(HAHB)=9.8 Hz, 3J(HAH)=8.6 Hz, OCHAHB), 4.45 (dd, 2H, B part of an AB spin system 2J(HAHB)=9.8 Hz, 3J(HBH)=8.6 Hz, OCHAHB), 3.89 (t, 2H, 3J(HAH)≈3J(HBH)=8.6 Hz, NCH), 3.75 (m, 2H, A part of an AB spin system, PCHAHB), 3.64 (m, 2H, B part of an AB spin system, PCHAHB). 31P{1H} NMR (CDCl3, 298 K): δ 77.7 (s). Anal. Calcd. for C47H40FeN2O5P2: C, 67.94; H, 4.86; N, 3.37. Found: C, 67.99; H, 4.86; N, 3.39%.
![[upper bond 1 start]](https://www.rsc.org/images/entities/b_char_e010.gif) (μ-LP,NPh)2Cu
(μ-LP,NPh)2Cu![[upper bond 1 end]](https://www.rsc.org/images/entities/b_char_e011.gif) ]BF4
(2*).
Solid [Cu(NCMe)4]BF4
(0.145 g, 0.46 mmol) was added to a solution of trans-[Fe(CO)3(LP,NPh)2]
(1*)
(0.382 g, 0.46 mmol) in CH2Cl2 at 25°C. The yellow solution became deeper almost immediately and the mixture was stirred vigorously for 20 min. Concentration and slow diffusion of pentane afforded a yellow powder (0.411 g, 91%). IR (CHCl3): ν(CO) 1988 (mw), 1919 (s) 1885 (vs), ν(CN) 1640 (m) cm−1. 1H NMR (CDCl3, 298 K): δ 8.00–6.70 (m, 30H, aromatics), 4.68 (br, 4H, OCH2), 4.11 (br, 2H, NCH), 3.45 (m, 2H, A part of an AB spin system, PCHAHB), 3.61 (m, 2H, B part of an AB spin system, PCHAHB). 31P{1H} NMR (CDCl3, 298 K): δ 69.65 (s). Anal. Calcd. for C47H40BCuF4FeN2O5P2·CH3CN: C, 57.58; H, 4.24; N, 4.11. Found: C, 58.00; H, 4.07; N, 4.03%.
]BF4
(2*).
Solid [Cu(NCMe)4]BF4
(0.145 g, 0.46 mmol) was added to a solution of trans-[Fe(CO)3(LP,NPh)2]
(1*)
(0.382 g, 0.46 mmol) in CH2Cl2 at 25°C. The yellow solution became deeper almost immediately and the mixture was stirred vigorously for 20 min. Concentration and slow diffusion of pentane afforded a yellow powder (0.411 g, 91%). IR (CHCl3): ν(CO) 1988 (mw), 1919 (s) 1885 (vs), ν(CN) 1640 (m) cm−1. 1H NMR (CDCl3, 298 K): δ 8.00–6.70 (m, 30H, aromatics), 4.68 (br, 4H, OCH2), 4.11 (br, 2H, NCH), 3.45 (m, 2H, A part of an AB spin system, PCHAHB), 3.61 (m, 2H, B part of an AB spin system, PCHAHB). 31P{1H} NMR (CDCl3, 298 K): δ 69.65 (s). Anal. Calcd. for C47H40BCuF4FeN2O5P2·CH3CN: C, 57.58; H, 4.24; N, 4.11. Found: C, 58.00; H, 4.07; N, 4.03%.
Catalytic reactions
°C for 162 h and then filtered through silica gel with Et2O elution to give after concentration (exo)-2-methylbicyclo[2.2.1]hept-5-ene-2-carboxaldehyde and (endo)-2-methyl bicyclo[2.2.1]hept-5-ene-2-carboxaldehyde as a clear colourless oil.
Crystallographic analysis of 1 and 2·1.5CH2Cl2
Single crystals were selected and mounted on a Kappa CCD diffractometer. Data were collected using phi-scans and the structure was solved using direct methods and refined against F2 using SHELX 97 software.18,19 No absorption correction was used. All non-hydrogen atoms were refined anisotropically with H atoms introduced as fixed contributors (dC–H=0.95 Å, U11=0.04).
Crystal data for 1: C35H32FeN2O5P2, M=678.45, orthorhombic, a=41.966(1)
Å, b=34.575(1)
Å, c=9.741(1)
Å, V=14133(2)
Å3, T=173 K, space group: Fdd2, Z=16, μ(Mo-Kα)=0.559 mm−1
(λ=0.71073 Å), 17857 reflections measured, 2614 reflections with I>3σ(I), R=0.046 and Rw=0.079.
For 2·1.5CH2Cl2: C35H32BCuF4FeN2O5P2·1.5CH2Cl2, M=956.2, monoclinic, a=24.4623(7)
Å, b=14.3872(6)
Å, c=12.4708(4)
Å, β=111.598(4),°
V=4080.9(5)
Å3, T=173 K, space group C2/m, Z=2, μ(Mo-Kα)=1.213 mm−1
(λ=0.71073 Å), 13114 reflections measured, 4571 reflections with I>3σ(I), R=0.046 and Rw=0.057.
CCDC reference numbers 177707 and 177708.
See http://www.rsc.org/suppdata/nj/b2/b206410n/ for crystallographic data in CIF or other electronic format.
Acknowledgements
This work was supported by the CNRS and the Ministère de la Recherche and we are also grateful to the COST D-17 programme of the European Commission (DG-XII). We are grateful to Ms. N. Gruber and Dr. A. DeCian for the X-ray structure determinations.References
- (a) T. Nakajima, I. Shimizu, K. Kobayashi, H. Koshino and Y. Wakatsuki, Inorg. Chem., 1997, 36, 6440 CrossRef CAS; (b) P. Braunstein and X. Morise, Organometallics, 1998, 17, 540 CrossRef CAS; (c) P. Braunstein and J. Rosé, Heterometallic Clusters in Catalysis, in Metal Clusters in Chemistry, eds. P. Braunstein, L. A. Oro and P. R. Raithby, Wiley-VCH, Weinheim, 1999, vol. 2, p. 616 Search PubMed; (d) N. Wheatley and P. Kalck, Chem. Rev., 1999, 99, 3379 CrossRef CAS; (e) J. R. Torkelson, O. Oke, J. Muritu, R. McDonald and M. Cowie, Organometallics, 2000, 19, 854 CrossRef CAS; (f) P. Braunstein, J. Durand, M. Knorr and C. Strohmann, Chem. Commun., 2001, 211 RSC; (g) P. Braunstein, M. Chetcuti and R. Welter, Chem. Commun., 2001, 2508 RSC; (h) P. Braunstein, G. Clerc and X. Morise, Organometallics, 2001, 20, 5036 CrossRef CAS.
- (a) G. R. Newcome, Chem. Rev., 1993, 93, 2067 CrossRef CAS; (b) J. T. Mague, J. Cluster Sci., 1995, 6, 217 CAS; (c) Z.-Z. Zhang and H. Cheng, Coord. Chem. Rev., 1996, 147, 1 CrossRef; (d) S. L. Li, T. C. W. Mak and Z.-Z. Zhang, J. Chem. Soc., Dalton Trans., 1996, 3475 RSC; (e) P. Braunstein, M. Knorr and C. Stern, Coord. Chem. Rev., 1998, 178–180, 903 CrossRef CAS; (f) P. Espinet and K. Soulantica, Coord. Chem. Rev., 1999, 193–195, 499 CrossRef CAS.
- P. Braunstein, C. Charles, A. Tiripicchio and F. Ugozzoli, J. Chem. Soc., Dalton Trans., 1996, 4365 RSC.
- D.-J. Cui, Q.-S. Li, F.-B. Xu, X.-B. Leng and Z.-Z. Zhang, Organometallics, 2001, 20, 4126 CrossRef CAS.
- (a) M. Gomez, G. Muller and M. Rocamora, Coord. Chem. Rev., 1999, 193–195, 769 CrossRef CAS; (b) G. Helmchen and A. Pfalz, Acc. Chem. Res., 2000, 33, 336 CrossRef CAS; (c) P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680 CrossRef CAS.
- P. Braunstein, M. D. Fryzuk, M. Le Dall, F. Naud, S. J. Rettig and F. Speiser, J. Chem. Soc., Dalton Trans., 2000, 1067 RSC.
- (a) G. Bellachioma, G. Cardaci, A. Macchioni and G. Reichenbach, J. Organomet. Chem., 1990, 391, 367 CrossRef CAS; (b) R. L. Keiter, J. W. Benson, E. A. Keiter, T. A. Harris, M. W. Hayner, L. L. Mosimann, E. E. Karch, C. A. Boecker, D. M. Olson, J. van der Veen, D. E. Brandt, A. L. Rheingold and G. P. A. Yap, Organometallics, 1997, 16, 2246 CrossRef CAS.
- P. Braunstein, C. Stern, C. Strohmann and N. Tong, Chem. Commun., 1996, 2237 RSC.
- (a) P. Braunstein, M. Knorr, B. E. Villarroya and J. Fischer, New J. Chem., 1990, 14, 583 Search PubMed; (b) P. Braunstein, M. Knorr, U. Schubert, M. Lanfranchi and A. Tiripicchio, J. Chem. Soc., Dalton Trans., 1991, 1507 RSC.
- H.-B. Song, Q.-M. Wang, Z.-Z. Zhang and T. C. W. Mak, J. Organomet. Chem., 2000, 605, 15 CrossRef CAS.
- H. Fritschi, U. Leutenegger and A. Pfaltz, Helv. Chim. Acta, 1988, 71, 1553 CrossRef CAS.
- (a) M. P. Doyle, R. L. Dorow, W. E. Buhro, J. H. Griffin, W. H. Tamblyn and M. L. Trudell, Organometallics, 1984, 3, 44 CrossRef CAS; (b) D. A. Evans, K. A. Woerpel and M. J. Scott, Angew. Chem., Int. Ed. Engl., 1992, 31, 430 CrossRef; (c) A. I. Meyers and A. Price, J. Org. Chem., 1998, 63, 412 CrossRef CAS; (d) D.-J. Cho, S.-J. Jeon, H.-S. Kim and T.-J. Kim, Synlett, 1998, 617 CAS; (e) Y. Imai, W. Zhang, T. Kida, Y. Nakatsuji and I. Ikeda, J. Org. Chem., 2000, 65, 3326 CrossRef CAS; (f) M. J. Alcon, E. Gutierrez-Puebla, M. Iglesias, M. A. Monge and F. Sánchez, Inorg. Chim. Acta, 2000, 306, 116 CrossRef CAS; (g) T. Portada, M. Roje, Z. Raza, V. Caplar, M. Zinic and V. Sunjic, Chem. Commun., 2000, 1993 RSC; (h) H. L. Wong, Y. Tian and K. S. Chan, Tetrahedron Lett., 2000, 41, 7723 CrossRef CAS; (i) R. Annunziata, M. Benaglia, M. Cinquini and F. Cozzi, Eur. J. Org. Chem., 2001, 1045 CrossRef CAS; (j) M. M. Díaz-Requejo and P. J. Pérez, J. Organomet. Chem., 2001, 617–618, 110 CrossRef; (k) M. Bühl, F. Terstegen, F. Löffler, B. Meynhardt, S. Kierse, M. Müller, C. Näther and U. Lüning, Eur. J. Org. Chem., 2001, 2151 CrossRef CAS and references cited.
- P. Braunstein, M. D. Fryzuk, F. Naud and S. J. Rettig, J. Chem. Soc., Dalton Trans., 1999, 589 RSC.
- D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. von Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 7582 CrossRef CAS and references cited therein.
- A. A. H. van der Zeijden, J. Organomet. Chem., 1996, 518, 147 CrossRef CAS.
- S. Yao, S. Saaby, R. G. Hazell and K. A. Jørgensen, Chem. Eur. J., 2000, 6, 2435 CrossRef CAS.
- F. Speiser, Thèse de Doctorat, Université Louis Pasteur, Strasbourg, France, April 2002.
- Kappa CCD Operation Manual, Nonius B. V., Delft, The Netherlands, 1997.
- G. M. Sheldrick, SHELXL97, Program for the refinement of crystal structures, University of Gottingen, Germany, 1997.
Footnote |
| † Dedicated to Prof. R. Ugo on the occasion of his 65th birthday, with our congratulations and best wishes. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2003 |