Impact of partial oxidation in dense CO2 on the solid-state properties of (VO)2P2O7 catalyst
Andreas
Martin
*a,
Boris
Kerler
a,
Jean-Claude
Volta
b and
Jörg
Radnik
a
aInstitut für Angewandte Chemie Berlin-Adlershof e.V., Richard-Willstätter-Str. 12, D-12489 Berlin, Germany
bInstitut de Recherches sur la Catalyse—CNRS, 2 avenue A. Einstein, F-69626 Villeurbanne cedex, France
First published on 21st November 2002
Abstract
(VO)2P2O7 was used as a catalyst for the partial oxidation of propane to oxygenates (acrylic acid and acetic acid) in dense carbon dioxide (2–10 MPa) at 673 K using air as the oxidant. The catalytic studies showed a decreasing yield of acrylic acid with higher reaction pressure, with the opposite effect found for acetic acid. One of the reasons for such behaviour has recently been found to be a stronger interaction of the catalyst and the olefinic double bond via catalyst Lewis sites. Another reason could be changing vanadium oxidation state in the solid catalyst. Therefore, detailed XPS and 31P NMR spectroscopic studies using the spin echo mapping technique were carried out to reveal more information on the possible pressure dependency of the vanadium oxidation state. The investigations clearly revealed an increasing vanadium oxidation state in the direction of V5+ with increasing reaction pressure. This might be due to a higher oxygen concentration on the catalyst surface. The decrease in acrylic acid yield also seems to be due to the observed increase in the vanadium oxidation state leading to over-oxidation of a greater extent.
1 Introduction
Processing under supercritical reaction conditions seems to be a promising new direction in selective oxidation. The unusual physico-chemical properties of supercritical fluids1–4 include (i) ‘liquid-like’ density with high solubilities for organic compounds, (ii) ‘gas-like’ low dynamic viscosity and high diffusivity, (iii) high compressibility and thermal conductivity, and (iv) complete miscibility with permanent gases. Supercritical carbon dioxide (scCO2; pc = 7.37 MPa, Tc = 304 K), in particular, is an environmentally benign fluid; non-toxic, non-flammable and non-corrosive.In general, selective oxidation of hydrocarbons (especially alkanes) in the gas phase often suffers from low yields. Either the formed intermediates are subject to consecutive total oxidation, owing to their strong adsorption at the catalytic surface sites, or the reactant is directly converted in a parallel reaction to the undesired total oxidation products (CO, CO2). As an example, the heterogeneously catalysed direct oxidation of propane to acrolein and/or acrylic acid is one of the demanding and challenging objectives at present;5–8 the low oxygenate selectivities given in the literature are mostly the result of the limitations mentioned above. However, recent patents report acrylic acid yields of up to ca. 50 mol%.9,10
In our own preliminary studies on the selective oxidation of propane to oxygenates in dense CO2 on cobalt-containing oxide catalysts, we reported on a significant rise in the oxygenate selectivity at high pressures.11,12 Such increases were explained as due to the improved solvent power of the fluid as a result of the enhanced density, as has recently been proven by in situ FTIR spectroscopy for another selectivity-limited reaction, i.e. the partial oxidation of toluene to benzaldehyde.13
The main products of propane oxidation in dense CO2 on CoOx catalysts were acetic acid, methanol, acetone and acetaldehyde, apart from the formation of propylene. It was supposed that the CoOx-catalysed reaction proceeds via a radical mechanism initiated on the solid surface. The application of vanadium-containing redox catalysts (for example vanadium phosphates) may lead to a change in product distribution towards the desired products acrolein or acrylic acid due to the involvement of other mechanistic pathways.14,15 Recently, catalytic tests using vanadyl pyrophosphate [VPP; (VO)2P2O7] as catalyst revealed the formation of only acrylic and acetic acid, apart from total oxidation products.16,17
Vanadium phosphates (VPO) are well known solids used as catalysts for selective O- and N-insertion reactions of aliphatics and methyl aromatics to provide the corresponding aldehydes, carboxylic acids and nitriles.18–20 This class of catalysts has been intensively investigated in the last decade because of its immense significance in the partial oxidation of n-butane to maleic anhydride.18,21 VPP is the most heavily studied of the VPO solids,18 its synthesis by various routes and the preparation and transformation of the precursor [vanadyl hydrogen phosphate hemihydrate (VHP; VOHPO4·0.5 H2O)] are the objects of several publications per year. Interestingly, a very recent paper deals with the synthesis of VPP catalyst using supercritical CO2 as an antisolvent, which exclusively generates a microspheroidal X-ray amorphous solid.22
It was the aim of the present work to study the effect of a dense CO2 phase reaction medium on the partial oxidation of propane to oxygenates (p = 2–10 MPa, T = 673 K) on the solid-state properties of VPP. Beside the characterisation of fresh and spent samples by analyses of composition, vanadium oxidation state and BET surface area, further solid-state characterisation methods, such as X-ray photoelectron spectroscopy (XPS) and 31P nuclear magnetic resonance spectroscopy using the spin echo mapping technique (31P SEM-NMR)23,24 were used in order to obtain more detailed information concerning the vanadium oxidation state and the different vanadium species in the used catalysts.
2 Experimental
2.1 Catalyst synthesis
The parent VPP solid was obtained by a routine dehydration of a VHP precursor compound, synthesised by the aqueous route, described elsewhere.25 73 g vanadium(V) oxide (V2O5, 0.4 mol) were added to a hot mixture (353 K) of 102 g phosphoric acid [H3PO4 (85%), 1.04 mol] and 76 g oxalic acid [(COOH)2, 0.84 mol] in 160 ml distilled H2O with vigorous stirring for 18 h. This solution was cooled, filtered and the light-blue powder was dried for 14 h at 423 K. Precursor treatment under inert gas (6 h at 873 K) led to the desired VPP catalyst. The synthesised VPP powder was pressed into tablets (10 MPa, 0.5 min), crushed and classified by sieving into a fraction of 1.0–1.25 mm, which was used for the catalytic runs.2.2 Catalytic runs
The as-synthesised VPP was used as catalyst for the partial oxidation of propane to oxygenates in dense CO2 in a commercial high pressure set-up containing a 5 ml fixed-bed continuous flow reactor (BTRS Junior, Autoclave Engineers Inc.), described in more detail elsewhere.12 The catalytic tests were carried out at 673 K, at different reaction pressures up to ca. 10 MPa and with a residence time of 19 s (total reaction times 3–6 h). After reaction, the reactor was rapidly depressurised, cooled to room temperature under a constant N2 flow and the used VPP catalyst was examined by the various characterisation methods. Reaction products were analysed by on-line GC equipped with FID (hydrocarbons, oxygenates), as well as TCD for gases (O2, N2, CO); details of the GC analysis are described elsewhere.17 A GC calibration error of ±2% was observed, whereas the determination of FID correction factors led to an accuracy of ca. ±7%.All used catalyst samples are named as VPPn, with n representing the reaction pressure of the corresponding catalytic run.
2.3 Catalyst characterisation
The various VPPn samples after exposure to different reaction pressures were characterised and the data compared with those of the parent VPP.The catalyst surface area was determined by N2 adsorption at 77 K, according to the BET method (Micromeritics Gemini III 2375). The samples were pre-treated at 423–473 K for 1 h under vacuum. The composition of fresh and used catalyst samples was determined by ICP emission spectroscopy (Perkin-Elmer Optima 300 XL).
The vanadium oxidation state and vanadium content of fresh and used samples were measured by potentiometric titration using Ce4+ and Fe2+ solutions, according to a modified method described first by Niwa and Murakami.26
The surface analytical studies were performed with a Fisons ESCALAB 220iXL spectrometer, consisting of two vacuum chambers: the analyser and a fast entry air lock/preparation chamber. The powdered samples were fixed on a carbon tape (carbon conductive tape, Pelco International) at the top of the sample holder and transferred into the ultra-high vacuum. Monochromatic Al-Kα radiation (1486.6 eV) was used as the X-ray source, with an input power of 300 W. The emerging charge of the sample was equalised with the installed charge compensation. The final peak position was determined using the C 1s peak (shifted to 284.8 eV) corresponding to absorbed hydrocarbon species. The XPS measurements were performed at constant pass energy of 25 eV. The instrument was routinely calibrated with the appropriate XPS lines of Au, Ag and Cu.27 After background correction,28 the XPS spectra were described and the correct peak positions were determined from Gaussian–Lorentzian peaks.29 95% of the photoelectrons stem from a depth of 6 nm, estimated from the realistic mean free path of electrons in solid state. An instrumental standard deviation of ca. ±0.1 eV has to be taken into account.
The 31P SEM-NMR spectra were recorded at 161.9 MHz with a Bruker DSX400 spectrometer equipped with a standard 4 mm probe head. The spectra were obtained with a sweep width of 2 mHz, t = 20 ms and 90° pulse length of 1.5 ms. Reference VPO phases with vanadium oxidation states from +3 to +5 can be identified by their 31P SEM-NMR spectra.23 Indeed, the signal is shifted from ca. 0 ppm for VOPO4 materials (V5+) to 1600–2600 ppm for V4+ phases, and to approximately 4650 ppm for VPO4 (V3+) phases.24 The intermediate signals observed correspond to various densities around P and V atoms with different oxidation states and relative distributions [V4+–V5+ dimers at ca. 400–1000 ppm, isolated V5+ sites on (VO)2P2O7 at −25, −125 ppm].
3 Results and discussion
Fig. 1 depicts the catalytic performance of VPP at various reaction pressures during the partial oxidation of propane in dense CO2. The propane conversion increased with reaction pressure from ca. 70 mol% at 2.3 MPa up to 82 mol% at 9.7 MPa. The selectivities for oxygenates were rather small (only acetic acid and acrylic acid were observed), mainly due to the chosen reaction conditions: Although it is well known that both higher oxygen partial pressure as well as the admixture of water vapour to the reactants dramatically enhance the yield towards acrylic acid,14 we could not apply either of them to the reaction in dense CO2 for safety reasons and due to phase separation.16,17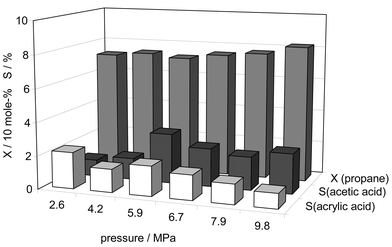 | ||
| Fig. 1 Partial oxidation of propane on (VO)2P2O7 used as catalyst at 673 K (CO2∶synthetic air∶propane molar ratio = 86∶13∶1, residence time = 19 s) at various reaction pressures: X = conversion of propane; S = selectivity towards acetic acid and acrylic acid. | ||
The selectivities of both the acids show distinct differences in their dependence on reaction pressure (see Fig. 1). Acetic acid was the favoured product at higher pressures; in contrast, the selectivity for acrylic acid declined with increasing pressure. This behaviour could be proven by using supercritical fluid chromatography (SFC) showing inhibited desorption of acrylic acid from the VPP surface in comparison to acetic acid.17 The reason for the differing trends in the selectivities is thought to be the stronger adsorption of acrylic acid at enhanced pressure due to additional interaction of the olefinic double bond with Lewis acid sites of the VPP catalyst. Therefore, hindered desorption may lead to further (over-)oxidation, resulting in diminishing acrylic acid yields with increasing pressure. However, other factors could also contribute to these observations, e.g. an increased vanadium oxidation state of the catalyst surface with higher reaction pressure.
Previous studies of the crystallographic and morphological properties of fresh and used VPP catalysts were carried out using powder X-ray diffractometry (XRD) and scanning electron microscopy (SEM). The XRD patterns of fresh and spent samples were found to be very similar, both showing good crystallinity.17 Slight differences were observed at 2θ = 22.9 (040) and 47.03° (020), pointing to changed orientation to the ‘010’ direction, correlating with the formation of the more plate-like habit observed by SEM.
Table 1 summarises the results of elemental analysis by ICP-OES and BET surface area measurements on the parent sample and two VPP specimen used in catalytic runs at different pressures. The composition remains virtually unchanged in the samples. However, the BET surface areas of used samples were markedly reduced in comparison to the fresh material, e.g. VPP catalyst used at 9.7 MPa suffered a reduction of 38% in surface area in comparison to the fresh sample.
| Catalyst | Elemental analysis (ICP-OES) | BET surface area/m2 g−1 | |
|---|---|---|---|
| V/wt% | P/wt% | ||
| Fresh | 32.5 | 20.1 | 6.4 |
| VPP26 | 32.2 | 20.0 | 5.3 |
| VPP97 | 32.3 | 19.9 | 4.0 |
Table 2 gives vanadium contents and oxidation numbers, both obtained by potentiometric titration. The potentiometrically determined vanadium contents agree well with those from the ICP-OES analysis (see Table 1). However, the data do not show significant differences in the bulk vanadium oxidation numbers of fresh and used VPP samples. Therefore, additional XPS and 31P SEM-NMR spectroscopic measurements were carried out to obtain a more detailed insight into possible pressure effects on the vanadium oxidation state on the surface of VPP as well as in the bulk.
| Catalyst | V4+/wt% | V5+/wt% | Vtotal/wt% | Oxidation number |
|---|---|---|---|---|
| Fresh | 32.7 | 0 | 32.7 | 4.000 |
| VPP23 | 31.4 | 0.13 | 31.5 | 4.004 |
| VPP78 | 32.8 | 0 | 32.8 | 4.000 |
| VPP95 | 32.0 | 0 | 32.0 | 4.000 |
The XP spectra were analysed and evaluated according to a method described by Richter et al. for VPO solids.30 They proposed measuring the distance, Δ, between the peak maxima of the V 2p3/2 and the O 1s peaks and found Δ = 14.7 eV for pure V4+ and 12.9 eV for pure V5+. This method was validated for different VPO materials.29 The advantage of this method is that it avoids errors/problems with the referencing of the XP spectra. Fig. 2 and Table 3 summarise the results obtained with VPP23 and VPP97 in comparison to the parent VPP. It can be seen that the peak distance Δ for VPP97 is smaller compared to both VPP23 and the fresh sample. The evaluation of the spectra shows the same surface vanadium oxidation number for VPP23 and the parent VPP of ca. 4.2–4.3, but an increased oxidation number for VPP97 of ca. 4.5–4.6. Thus, the XPS data clearly show a higher vanadium oxidation number at the surface of the sample exposed to the highest reaction pressure.
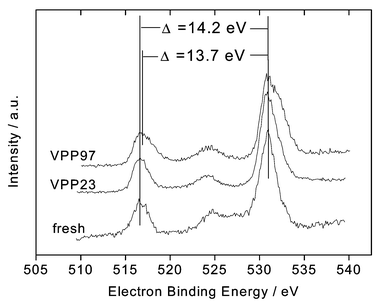 | ||
| Fig. 2 XP spectra of the parent VPP, VPP23 and VPP97 samples. The distances (Δ) between the maxima of the V 2p3/2 and the O 1s peaks are marked, including the corresponding binding energies referenced to the maxima of the O 1s peaks. | ||
| Catalyst | O 1s/eV | V 2p3/2/eV | Δ/eV | Oxidation number |
|---|---|---|---|---|
| Fresh | 530.8 | 516.6 | 14.2 | 4.2–4.3 |
| VPP23 | 531.3 | 517.1 | 14.2 | 4.2–4.3 |
| VPP97 | 530.5 | 516.8 | 13.7 | 4.5–4.6 |
Fig. 3 shows 31P SEM-NMR spectra of catalyst samples in the range ca. 4000 to −1000 ppm. Additionally, Fig. 4 presents details of the same spectra in the range ca. 100 to −200 ppm at higher resolution for better analysis of possible V5+ species. All the 31P SEM-NMR spectra are essentially characteristic for crystallised VPP solids with signals for V4+ located around PO4 tetrahedra appearing around 2550 ppm. Differences exist in the relative distribution of V4+–V5+ dimers (1700 to 700 ppm), V5+ micro-domains (90, 70, 50 ppm) in the presence (or absence) of δ-VOPO4 (−12 to −21 ppm) and in isolated V5+ sites (−25, −125 ppm).
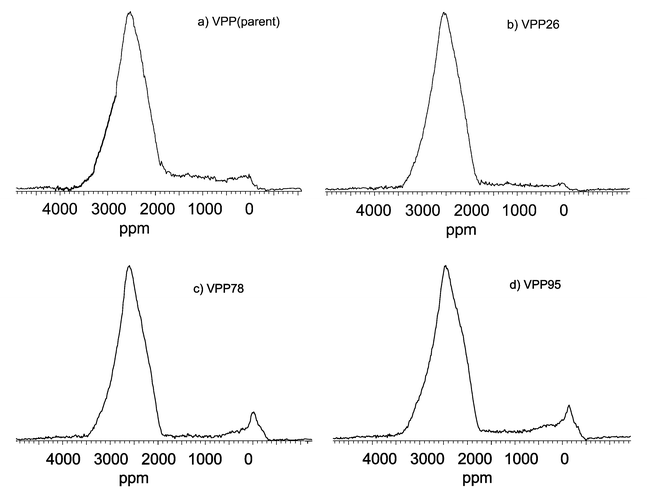 | ||
| Fig. 3 31P SEM-NMR spectra (4000 to −1000 ppm range) of parent VPP (a), VPP26 (b), VPP78 (c) and VPP95 (d). | ||
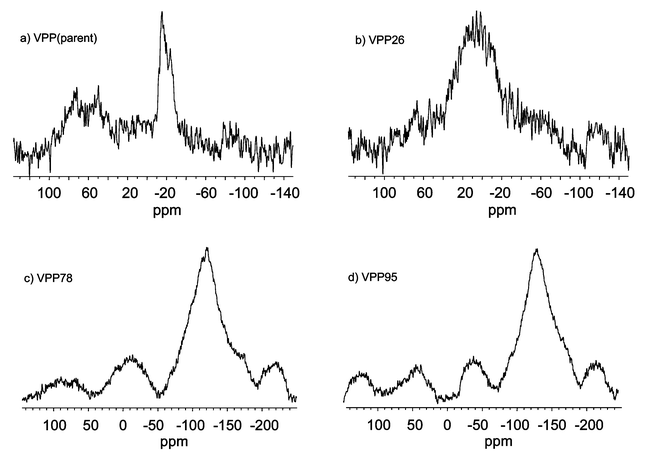 | ||
| Fig. 4 Higher resolution view of the 100 to −200 ppm range of the 31P SEM-NMR spectra of parent VPP (a), VPP26 (b), VPP78 (c) and VPP95 (d). | ||
Fig. 3(a) and 4(a) show the 31P SEM-NMR spectrum of the parent sample, which is typical for a crystalline VPP solid. The spectrum shows V4+–V5+ dimers, V5+ micro-domains, δ-VOPO4 and almost no isolated V5+ sites. After running the oxidation at 2.6 MPa, the spectrum of VPP26 [Fig. 3(b) and 4(b)] reveals a VPP solid that is less reduced in comparison to the parent sample (contribution of V3.69+ at 3200 ppm has diminished). Furthermore, less V4+–V5+ dimers, as well as a slightly increased proportion of V5+ micro-domains can be seen, and isolated V5+ sites begin to appear. It seems noteworthy to mention the appearance of a signal at ca. 0 ppm that points to an increase of “free phosphorus” (phosphate groups), which has lost its connection to vanadium in the previous δ-VOPO4 structure. The used catalyst obtained after increasing the reaction pressure to 7.8 MPa, VPP78, is a well-crystallised solid, very similar to VPP26 [Fig. 3(c) and 4(c)]. However, the amount of V4+–V5+ dimers in VPP78 giving rise to peaks in the 1000 ppm region diminishes [higher V4+∶V5+ ratio, Fig. 3(b)], but the V4+–V5+ contribution to the 400–500 ppm region increases [lower V4+∶V5+ ratio, Fig. 3(c)] compared to VPP26, i.e. this shift reflects an increased V5+ proportion due to these species. Free phosphate groups can still be found, but, interestingly, an increased proportion of isolated V5+ species is generated. They appear to be formed from V5+ micro-domains, and the isolation of V5+ stems from the free phosphate proportions. Fig. 3(d) and 4(d) show the spectrum of VPP95, which is similar to that of VPP78; there are still free phosphate and isolated V5+ sites. The 31P SEM-NMR investigations clearly show a higher number of various V5+ sites on the surface and in the catalyst bulk being generated with increased reaction pressure.
4 Conclusion
Our investigations indicate that the diminished yield of acrylic acid from the partial oxidation of propane in dense CO2 at high pressure may also be caused by increased oxidation of the vanadium species located near the solid surface, besides the recently reported effect of more hindered desorption. The characterisations reveal that the solid composition and the bulk oxidation state remained unchanged. However, XPS spectroscopy shows an increase in the surface vanadium oxidation state with rising reaction pressure. Similar findings were obtained from 31P SEM-NMR investigations. These results point to less reduced samples that contain an increasing number of isolated V5+ sites and a diminished V4+–V5+ ratio. Therefore, a further reason for the decreasing yield of acrylic acid seems to be a more oxidised catalyst, which also favours the over-oxidation of the desired product.Acknowledgements
The authors would like to thank Mrs M. Hartelt (ACA) for experimental assistance, Mrs M.-C. Durupty (IRC-CNRS) for the 31P SEM-NMR measurements, and Mr G.-U. Wolf (ACA) for potentiometric titration and discussion of various results.References
- P. E. Savage, S. Gopalan, T. I. Mizan, C. J. Martino and E. E. Brock, AIChE J., 1995, 41, 1723 Search PubMed.
- P. G. Jessop and W. Leitner, Chemical Synthesis Using Supercritical Fluids, Wiley-VCH, Weinheim, 1999 Search PubMed.
- A. Baiker, Chem. Rev., 1999, 99, 453 CrossRef CAS.
- R. Wandeler and A. Baiker, CatTech, 2000, 4, 34 Search PubMed.
- M. Baerns and O. V. Buyevskaya, Erdöl Erdgas Kohle, 2000, 1, 25 Search PubMed.
- M. M. Bettahar, G. Costentin, L. Savary and J. C. Lavalley, Appl. Catal., A, 1996, 48, 145.
- M. Baerns, O. V. Buyevskaya, M. Kubik, G. Maiti, O. Ovsitser and O. Seel, Catal. Today, 1997, 33, 85 CrossRef CAS.
- M. M. Lin, Appl. Catal., A, 2001, 207, 1 CrossRef CAS.
- T. Ushikubo, H. Nakamura, Y. Koyasu and S. Wajiki, Eur. Pat., 0 608 838 A2, 1994 Search PubMed.
- M. M. Lin and M. W. Linsen, US Pat., 6 180 825, 2001 Search PubMed.
- A. Martin and B. Kerler, Chem. Eng. Technol., 2001, 24, 41 CrossRef CAS.
- B. Kerler, A. Martin, A. Jans and M. Baerns, Appl. Catal., A, 2001, 222, 243 CrossRef.
- B. Müller, A. Martin and B. Lücke, J. Supercrit. Fluids, 2002, 23, 243 CrossRef CAS.
- T. Quandt, PhD Thesis, Ruhr-Universität Bochum, Germany, 1999.
- M. Ai, Catal. Today, 1998, 42, 297 CrossRef CAS.
- B. Kerler, A. Martin and M. Baerns, Chem.-Ing.-Tech., 2002, 74, 285 Search PubMed.
- B. Kerler, A. Martin, M.-M. Pohl and M. Baerns, Catal. Lett., 2002, 78, 259 Search PubMed.
- G. Centi, Catal. Today, 1993, 16, 1 CrossRef.
- F. K. Hannour, A. Martin, B. Kubias, B. Lücke, E. Bordes and P. Courtine, Catal. Today, 1998, 40, 263 CrossRef CAS.
- A. Martin and B. Lücke, Catal. Today, 2000, 57, 61 CrossRef CAS.
- E. Bordes, Catal. Today, 1987, 1, 499 CrossRef CAS.
- G. J. Hutchings, J. A. Lopez-Sanchez, J. K. Bartley, J. M. Webster, A. Burrows, C. J. Kiely, A. F. Carley, C. Rhodes, M. Hävecker, A. Knop-Gericke, R. W. Mayer, R. Schlögl, J. C. Volta and M. Poliakoff, J. Catal., 2002, 208, 197 Search PubMed.
- A. Tuel, L. Canesson and J. C. Volta, Colloids Surf., A, 1999, 158, 97 CrossRef CAS.
- M. T. Sananès, A. Tuel and J. C. Volta, J. Catal., 1994, 145, 251 CrossRef CAS.
- H. Berndt, K. Büker, A. Martin, M. Meisel, A. Brückner and B. Lücke, J. Chem. Soc., Faraday Trans., 1995, 91, 725 RSC.
- M. Niwa and Y. Murakami, J. Catal., 1982, 76, 9 CrossRef CAS.
- M. T. Anthony and M. P. Seah, Surf. Interface Anal., 1984, 6, 95 CAS.
- D. A. Shirley, Phys. Rev., 1972, B5, 4709 Search PubMed.
- R. O. Ansell, T. Dickinson, A. F. Povey and P. A. M. Sherwood, J. Electroanal. Chem., 1979, 98, 79 CrossRef CAS.
- F. Richter, H. Papp, G.-U. Wolf, Th. Götze and B. Kubias, Fresenius’ J. Anal. Chem., 1999, 365, 150 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |