Synthesis of well-dispersed nanoparticles within porous solid structures using surface-tethered surfactants in supercritical CO2†
Kai Man K.
Yu
,
Andrew M.
Steele
,
Jie
Zhu
,
Qijia
Fu
and
Shik Chi
Tsang
*
Surface and Catalysis Research Centre, Department of Chemistry, University of Reading, Whiteknights, Reading, UK RG6 6AD. E-mail: s.c.e.tsang@reading.ac.uk
First published on 25th November 2002
Abstract
We have developed a new method for the synthesis of Pd nanoparticles with controllable sizes within a silica matrix using solid-supported surfactants in supercritical CO2. XRD, HRTEM and CO chemisorption data show that unformly sized Pd nanoparticles are evenly distributed within the porous silica and are chemically tethered by surfactant molecules [poly(oxyethylene stearyl ether) and fluorinated poly(oxyethylene)]. It is postulated that tiny solid-supported surfactant assemblies act as nano-reactors for the template synthesis of nanoparticles or clusters from the soluble precursors therein.
Introduction
Recent literature reports have highlighted the advantages of using nanostructured materials in many areas. As a result, many governments/research councils have been advised to intensify their investment in ‘nanotechnology’ over the coming years. The motivation for this stems particularly from the unique electronic, optical, electro-optical, electrochemical and catalytic properties associated with colloidal metal particles of controlled size and composition. There are a number of ways to synthesise metal particles with diameters between 1 and 20 nm. These include precipitation,1 organometallic preparation and deposition,2 sonochemical methods,3 and via microemulsions created using organic stabilising agents.4,5 Other specific methods, such as laser ablation,6 electrochemical routes7 and cross-linking,8 for the preparation of colloid solutions of particles are also found in the literature. It is noted that most synthetic routes mentioned include nucleation, growth and stabilisation of the particles, which results in a range of particle sizes. For monodisperse size control, the use of organic stabilising agents, such as polymers, surfactants and bulky ligand appendages, as micelles or reverse micelles in liquid phases, creating ‘nano-vessels’ to tailor the particle size, is regarded as one of the most promising routes.4,5,8 Typically, size-defined metal nanoparticles can be synthesised from soluble metal ion reduction using surfactant assemblies in water. The hydrophobic segments of the surfactant molecule are directly adsorbed on the surface of the newly formed ‘hydrophobic’ solid metal, thus stabilising it, while the hydrophilic segments spread out into the water, preventing aggregation of metal particles during the reduction. There have also been some recent advances in the creation of micelles (fluorous surfactant assemblies) for the synthesis of well-defined nanoparticles in supercritical (sc) CO2.9,10 However, for many applications, such as catalysis and optics, the direct handling, processing and repeated use of nanoparticles is difficult if they are not stabilised in bulk solid matrices. Thus, the key challenge in this area (which is also the motivation behind this work) is to develop a general methodology that not only allows tailoring of size and composition of nanoparticles, but also facilitates handling (processing) of the ultra-fine particles through their ‘implantation’ into a range of porous bulk materials. A recently reported approach uses organic molecule-functionalised mesoporous silicates as templates for the synthesis of nanoparticles in these defined porous structures.11,12In this paper, we report the synthesis of surfactant molecules tethered to a silica surface and their use for the preparation of well-defined silica-supported metal nanoparticles. Poly(oxyethylene) and related fluorous poly(oxyethylene) surfactant molecules chemically tethered onto porous silica surfaces have been successfully synthesised. H2 treatment of these surfactant-modified silicas impregnated with a solution containing soluble metal ions (Pd salt) produces considerably smaller Pd nanoparticles with a narrower size distribution than those obtained using unmodified silica. The use of silica-tethered fluorous surfactant molecules and scCO2–ethanol produces the smallest Pd particles with the best size definition (11 nm) throughout the porous silica. The resulting bulk composite materials containing well-dispersed nanoparticles may find applications in catalysis, optics and electronics and as magnetic materials. Although, at this stage, the precise mechanism for the significantly smaller particles synthesised inside the surfactant-functionalised silica under the scCO2 conditions is not yet known, it is envisaged that the silica-tethered fluorous surfactant molecules may undergo self-assembly (the structure and morphology remain to be elucidated) somewhat akin to free micelles.13 These may act as templates for the synthesis and stabilisation of metal nanoparticles within the silica matrix.
Experimental
The primary aim was to compare the average size of Pd particles synthesised from an aqueous Pd precursor solution over unmodified silica and silica tethered with two forms of surfactant molecules under the same scCO2 treatment. The preparative procedures employed are as follows.Preparation of porous silica with tethered surfactant molecules
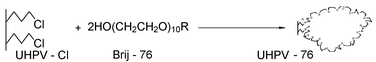 | ||
| Scheme 1 | ||
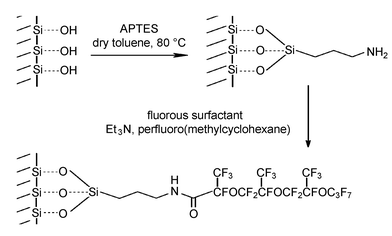 | ||
| Scheme 2 | ||
Preparation of Pd nanoparticles on unmodified and surfactant-modified silica
The Pd nanoparticles were formed by reduction of the silicas impregnated with Pd precursor solution in a 300 mL autoclave reactor using 5% H2–95% Ar at 2 bar and 25 °C for 5 h. This yielded three different products, denoted Pd@UHPV, Pd@UHPV-76 and Pd@UHPV-F.
Results
Silica with tethered surfactant molecules
The initial immobilisation of APTES and CPTES linker molecules onto the silica surface was achieved according to a modified method published by Sutra and Brunel.17 Evidence for the successful attachment of organic molecules onto the silica support comes from a range of analytical techniques. Typically, absorption peaks (2922.5, 860.0 and 698.6 cm−1) attributed to the C–H stretching and bending modes associated with organic molecules were clearly observed in the FT-IR spectra of UHPV-Cl and UHPV-NH2. Such peaks were not detected when the silica was immersed in either the linkers or surfactants at room temperature, filtered off and washed in a Soxhlet apparatus using 1∶1 dichloromethane–diethyl ether in an identical manner as described for UHPV-Cl and UHPV-NH2. This suggests that the linkers are chemically tethered to the silica surface in UHPV-Cl and UHPV-NH2 rather than being physically retained inside the porous structure. The TGA/DTA analyses of the samples (see Fig. S1–S3, ESI) clearly showed that all the modifications had successfully occurred. For pure UHPV silica, an endothermic weight loss (5%) at around 100 °C and a gradual weight loss (a further 2.3%) from 100 to 800 °C are attributed to the removal of physisorbed water and surface dehydroxylation, respectively. In the case of all the organically-modified silicas, two extra exothermic events were observed with associated additional weight loss from 100 to 800 °C when the samples were heated in air. These were assigned to the combustion of surface organic groups. A weight loss of 5% associated with the combustion of silica-anchored organic groups between 100 and 800 °C was found in the TG analysis of UHPV-Cl, while the analysis of UHPV-NH2 showed a weight loss of 3% in the same temperature range. There were 8 and >30% weight losses between 100 and 800 °C in the analyses of the silicas with tethered surfactants, UHPV-76 and UHPV-F, respectively (TGA/DTA for UHPV-F shown as a typical example in Fig. 1). These values are substantially higher than those found for the precursors with only the linker molecules attached, suggesting that the subsequent attachment of the surfactants to the linkers had been achieved in both cases. We estimate that most of the surface Si–OH groups (∼1 × 1019 m−2) were covered by surfactant molecules. The high weight loss in the case of UHPV-F is attributed to the bulkier fluorine-containing surfactant used in this case (EDX analysis shows the presence of the fluorine).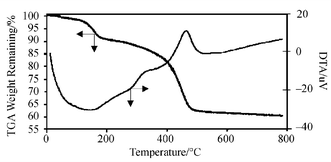
The BET surface area and the pore size analyses were also consistent with the chemical modification of the porous silica surface with bulky organic groups. Typically, the surface area of the UHPV-76 sample decreased from 304 to 223 m2 g−1, with a significant decrease in pore volume at 20 nm and the appearance of a shoulder at the smaller size of 16 nm, when compared with unmodified silica.
Characterisation of Pd particles on modified silicas
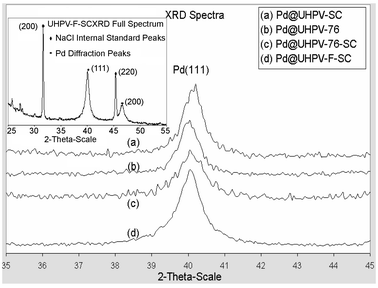 | ||
| Fig. 2 Selected Pd (111) angular broadenings from 35–45° 2θ for (a) Pd@UHPV-SC, (b) Pd@UHPV-76, (c) Pd@UHPV-76-SC and (d) Pd@UHPV-F-SC. Inset: typical XRD spectrum of Pd@UHPV-F-SC, showing reflection peaks due to Pd and internal standard. | ||
According to the calculated Pd particle sizes in Table 1, the Pd nanoparticles supported on surfactant-modified silica (Pd@UHPV-76) are significantly smaller than the Pd nanoparticles on the Pd@UHPV samples with and without supercritical CO2 pre-treatment (no significant difference was found between Pd@UHPV and Pd@UHPV-SC). When scCO2 pre-treatment was applied to Pd@UHPV-76 prior to reduction, there was a further reduction of about 2 nm in the average size of the Pd nanoparticles to 11.8 nm in Pd@UHPV-76-SC, this is also associated with significantly better dispersion compared to unmodified Pd@UHPV-SC, as shown in the TEM micrographs below (compare Fig. 6 to Fig. 4). It should be noted that the silica with tethered fluorinated surfactant after scCO2–ethanol pre-treatment (Pd@UHPV-F-SC) produced the smallest average Pd particle size of 10.1 nm with excellent dispersion (Fig. 8).
It is generally accepted that the sizes of supported noble metal particles evaluated by CO chemisorption and XRD are comparable, as is reflected in our results (Table 1), assuming the 1∶1 Pd∶CO stoichiometry is valid. Larger particle size can be derived if some surface metal sites are inaccessible or the CO chemisorption mode on the metal sites deviates from linear to bridging.20
We note that there is a significant difference in the particle sizes calculated for Pd@UHPV-SC using the two methods. It is believed that larger Pd particles of >23 nm with the dominant high coordinate metal sites on flat surfaces favour the bridging CO mode, with the result that the Pd particle size estimate is inflated. This result is consistent with the results of Sheu et al.,20 who showed that CO is preferentially bonded linear on low coordinate (corner or edge) Pd atoms of small particles and adopts the bridging mode on large particles. Despite this discrepancy, the fact that smaller Pd particles are formed on the surfactant-modified silicas compared with those on the unmodified material is obvious from both the XRD and CO chemisorption data.
 | ||
| Fig. 3 TEM image of Pd@UHPV. | ||
 | ||
| Fig. 4 TEM image of Pd@UHPV-SC. | ||
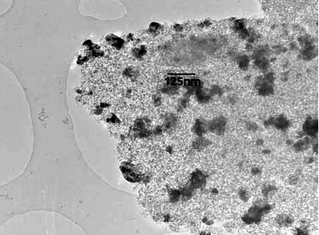 | ||
| Fig. 5 TEM image of Pd@UHPV-76. | ||
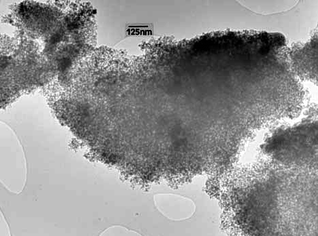 | ||
| Fig. 6 TEM image of Pd@UHPV-76-SC. | ||
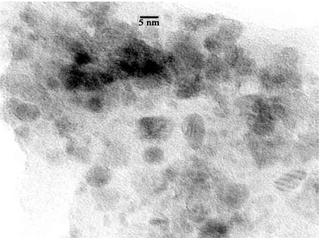 | ||
| Fig. 7 HRTEM image of Pd@UHPV-F. | ||
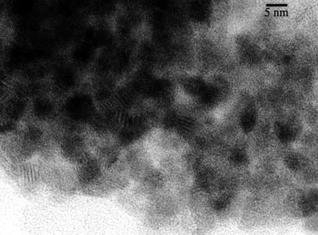 | ||
| Fig. 8 HRTEM image of Pd@UHPV-F-SC. | ||
Discussion
Solid-supported metal particles are conventionally prepared by wet impregnation via mixing of solutions containing metal precursors with high surface area porous solids, followed by drying and H2 reduction. Generally, the sizes of particles prepared by this method span a broad range of sizes (3–30 nm21,22), with the average particle size depending nucleation, growth and stabilisation of the particle precursors, which in turn depends on metal–support interactions, the solvent used and the drying and reduction conditions, etc. There are considerable practical advantages to synthesising more uniformly sized metal nanoparticles which are better dispersed within the solid support matrix than those prepared by the conventional method.The use of free surfactant assemblies as micelles or reverse micelles in solvents including supercritical CO2 (using fluorous surfactant molecules) is well documented. Here, we have emplyed silica-supported surfactants in order to determine whether the surface-tethered surfactant molecules influence the size of particles synthesised thereupon. It is apparent from the TEM, XRD and CO chemisorption studies that a much better particle dispersion and smaller and more uniformly sized particles are obtained on these surfactant-modified silicas than with the unmodified solid.
Thus, this work clearly demonstrates that surfactant molecules tethered to the surface of silica allow the synthesis of smaller and more unformly sized particles than can be obtained using conventional impregnation. We believe that this method is different to that recently reported in work on the template synthesis of nanoparticles using mesoporous silicate (i.e. MCM-41) funtionalised with organic molecules, where the particle nucleation is initated by the surface functional groups and size restriction is imposed by the channels.11,12 In our case, it should be noted that uniformly dispersed nanoparticles can equally be synthesised within silica that possesses no intrinic uniform pore structure. Presently, the mechanism for this interesting process is unknown although some possibilities present themselves. One suggestion is that the steric effects arising from the different lengths of the surfactants used may assist in modulating the ‘effective pore size’ for nanoparticle growth. Other possibilities include the influence of the chelating ability of the surfactant moieties to the Pd ions in the reductive nucleation leads to nanoparticles of defined size. It should be noted, however, that the number of Pd atoms used far exceeds the number of surfactant molecules on the surface of the support. On the other hand, with regard to the length of the tethered surfactant molecules (containing both hydrophobic and hydrophilic moieties), we postulate that these tethered molecules may be capable of assembling themselves into tethered micelles within the internal porous solid matrix. These assemblies may not necessarily be identical to the free micelle form, however, the hydrophobic–hydrophilic interactions of the surface molecules at high coverage, with solvent, may also provide a similar driving force to enable these anchored molecules to organise themselves into localised micelles on the internal solid structure, thereby providing the necessary ‘solvent droplets’ of controlled size for the accommodation of the soluble metal ion precursor. A tentative model is presented in Scheme 3, showing surface-tethered surfactant molecules creating micelles within the pores of the solid matrix and acting as templates for nanoparticle formation. It is emphasised that, although this surface micelle model is consistent with our other work on biphasic catalysis over silica-tethered surfactants,14 direct evidence on surface micelle formation is required before more concrete conclusions can be drawn.
 | ||
| Scheme 3 | ||
The drying stage is very important as regards the sizes of the particles formed from the soluble precursor. Unevenly distributed puddles on the internal solid structure will create local deposition of non-uniform particles, and these may be large if large puddles are involved. It is known that scCO2 has many interesting properties, such as efficient mass/heat transport coefficients, the ability to dissolve and carry organic molecules in its fluid phase, unique solubility and particularly strong interactions with fluorinated species.16,23 Thus, the scCO2–ethanol mixture clearly assists the dispersion of the ethanol-soluble Pd precursor throughout the modified silicas, however, in the absence of surfactant, no such effect is observed. This suggests that the scCO2–ethanol fluid interacts with the tethered surfactant molecules (hydrophobic–hydrophilic interactions), providing solvent penetration, re-dispersion and controlled drying of the precursors to give finer particle control. Accordingly, the materials with tethered fluorous surfactant molecules (with the moieties containing carbon–fluorine groups aligned towards the scCO2 solvent and the oxygen species aligned towards the ethanol solution) afford the best dispersion and provide particles with the smallest average sizes. This observation is consistent with claims in the literature for the synthesis of small nanoparticles in scCO2 using free fluorous surfactant assemblies.9,10
Conclusion
It is shown in this work that surfactant-modified silica supports provide ‘nano-reactors’ inside the internal silica structure for the preparation of uniformly dispersed Pd nanoparticles on the supports. Thus, evenly distributed Pd nanoparticles of a uniform size are obtained via addition of chloropalladate H2O–ethanol solution to surfactant-modified silica, followed by scCO2–ethanol pre-treatment to promote extensive species distribution and, finally, H2 reduction. The best performance in terms of particle size and dispersion is obtained using fluorinated surfactant-modified silica along with the scCO2–ethanol pre-treatment.References
- P. Claus, A. Brückner, C. Mohr and H. Hofmeister, J. Am. Chem. Soc., 2000, 122, 11430 CrossRef CAS.
- U. A. Paulus, U. Endruschat, G. J. Feldmeyer, T. J. Schmidt, H. Bonnemann and R. J. Behm, J. Catal., 2000, 195, 383 CrossRef CAS.
- R. A. Salkar, P. Jeevanandam, S. T. Aruna, Y. Koltypin and A. Gedanken, J. Mater. Chem., 1999, 9, 1333 RSC.
- A. Martino, S. A. Yamanaka, J. S. Kawola and D. Loy, Chem. Mater., 1997, 9, 423 CrossRef CAS.
- T. Li, J. Moon, A. A. Morrone, J. J. Mecholsky, D. R. Talham and J. H. Adair, Langmuir, 1999, 15, 4328 CrossRef CAS.
- C. B. Hwang, Y. S. Fu, Y. L. Lu, S. W. Jang, P. T. Chou, C. R. C. Wang and S. J. Yu, J. Catal., 2000, 195, 336 CrossRef CAS.
- Y. Y. Yu, S. S. Chang, C. L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661 CrossRef CAS.
- K. V. Sarathy, G. U. Kulkarni and C. N. R. Rao, Chem. Commun., 1997, 537 RSC.
- H. Ohde, J. M. Rodriguez, X. R. Ye and C. M. Wai, Chem. Commun., 2000, 2353 RSC.
- H. Ohde, F. Hunt and C. M. Wai, Chem. Mater., 2001, 13, 4130 CrossRef CAS.
- J. P. M. Niederer, Top. Catal., 2002, 18, 265 Search PubMed.
- P. Mukherjee, C. R. Patra, R. Kumar and M. Sastry, PhysChemComm, 2001, 4, 24 Search PubMed.
- K. P. Johnston, K. L. Harrison, M. J. Clarke, S. M. Howdle, M. P. Heitz, F. V. Bright, C. Carlier and T. W. Randolph, Science, 1996, 271, 624 CrossRef CAS.
- S. C. Tsang, N. Zhang, L. Fellas and A. M. Steele, Catal. Today, 2000, 61, 29 CrossRef CAS.
- P. M. Jenkins and S. C. Tsang, unpublished results.
- L. T. Taylor, Supercritical Fluid Extraction, John Wiley & Sons, Inc., New York, 1996, p. 21 Search PubMed.
- P. Sutra and D. Brunel, Chem. Commun., 1996, 21, 2485 RSC.
- G. Neri, M. G. Musolino, C. Milone, D. Pietropaolo and S. Galvagno, Appl. Catal. A, 2001, 208, 307 CrossRef CAS.
- N. Mahata, K. V. Raghavan, V. Vishwanathan, C. Park and M. A. Keane, Phys. Chem. Chem. Phys., 2001, 3, 2712 RSC.
- L. L. Sheu, Z. Karpinksi and W. M. H. Sachtler, J. Phys. Chem., 1989, 93, 4890 CrossRef CAS.
- F. Pinna, M. Selva, M. Signoretto, G. Strukul, F. Boccuzzi, A. Benedetti, P. Canton and G. Fagherazzi, J. Catal., 1994, 150, 356 CrossRef CAS.
- G. Fagherazzi, A. Benedetti, G. Deganello, D. Duca, A. Martorana and G. Spoto, J. Catal., 1994, 150, 117 CrossRef CAS.
- A. Baiker, Chem. Rev., 1999, 99, 453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA and DTA curves for UHPV-Cl, UHPV-76 and UHPV-F-SC (Fig. S1–S3); HRTEM image of UHPV-76-SC (Fig. S4). See http://www.rsc.org/suppdata/jm/b2/b207064b/ |
| This journal is © The Royal Society of Chemistry 2003 |