Synthesis and characterization of fluorinated β-ketoiminate and imino-alcoholate Pd complexes: precursors for palladium chemical vapor deposition
Yan-Hsiu
Liu
a,
Yi-Ching
Cheng
a,
Yung-Liang
Tung
a,
Yun
Chi
*a,
Yao-Lun
Chen
a,
Chao-Shiuan
Liu
*a,
Shie-Ming
Peng
b and
Gene-Hsiang
Lee
b
aDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: ychi@mx.nthu.edu.tw
bDepartment of Chemistry and Instrumentation Center, National Taiwan University, Taipei 10764, Taiwan
First published on 18th November 2002
Abstract
The design and synthesis of Pd complexes with two β-ketoiminate or with two imino-alcoholate chelate ligands is reported. In order to establish their structures in the solid-state, methoxyethyl substituted complexes 1c and 2c were characterized by single crystal X-ray diffraction, showing a square-planar local coordination for the Pd atom, but the β-ketoiminate ligands of 1c gave a bent basal plane involving two chelating hexagons, which was in sharp contrast to the boat configuration of the imino-alcoholate ligands observed in the second complex 2c. Chemical vapor deposition (CVD) experiments were conducted at deposition temperatures of 250–350 °C. Scanning electron micrographs (SEM) were taken to reveal the surface morphologies and grain sizes of the Pd metal thin films. The resulting thin films were found to contain a low level of carbon and oxygen impurities using O2 as the carrier gas, as measured by X-ray photoelectron spectroscopy (XPS).
Palladium has been used extensively in depositing metal-containing thin films in microelectronics device processing1 as well as for the preparation of finely dispersed metal particles on the catalytic surface of porous supports.2 Among various deposition methods that have been investigated, chemical vapor deposition (CVD) has becoming increasingly attractive,3 as CVD allows direct deposition of the Pd-containing material on the substrate and greatly simplifies the deposition processes. However, studies of palladium CVD have been restricted by the lack of suitable precursors, as most Pd complexes are either non-volatile under typical CVD conditions, or suffer poor stability in moist air, which makes their preparation and handling a tedious manipulation, the latter includes complexes such as PdMe2(TMEDA), PdMe2(PMe3)2 and Pd(η3–MeC3H4)2.4 This problem was partly solved by Puddephatt and coworkers, who synthesized a series of allyl–Pd complexes containing one β-diketonate ligand, which gave the required volatility and thermal stability for CVD applications.5
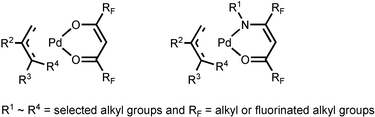
Recently, our group has initiated a program focused on Pd CVD, and a series of allyl complexes with a β-ketoiminate ligand was then synthesized.6 Similarly to Puddephatt's complexes, our compounds also showed a substantial improvement in thermal stability as well as a lowering of the melting points, making these allyl–ketoiminate complexes equally suitable for Pd metal deposition. In order to extend this research endeavor, we have now prepared two new series of Pd complexes (1 and 2), for which the central Pd atom is coordinated by either two β-ketoiminate ligands or two imino-alcoholate ligands (Scheme 1). Despite the obvious distinction in the design of the ligand framework, they can easily form stable complexes through chelation involving both oxygen and nitrogen donor atoms. Moreover, as they possess two electron withdrawing CF3 substituents, the ligands as well as the resulting metal complexes are expected to be more volatile and somewhat more robust with respect to their non-fluorinated counterparts.7 The as-deposited thin films also contain minimal amounts of impurities, due to the effective removal of intact ligands that are less susceptible to exhaustive oxidation, especially during Pd deposition using O2 as the carrier gas.
 | ||
| Scheme 1 | ||
Experimental
General information and materials
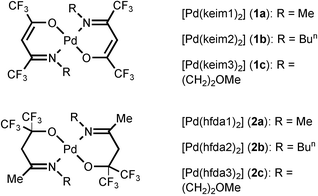
1H, 13C and 19F NMR spectra were recorded on a Bruker AMX-300 instrument or a Varian Unity INOVA 500 instrument. Mass spectra were obtained on a JEOL SX-102A instrument operating in electron impact (EI) mode. The thermogravimetric analyses (TGA) were recorded on a Seiko TG/DTA 300 instrument under an atmospheric pressure of N2 with a flow rate of 100 sccm and with a heating rate of 10 °C min−1. All reactions were performed in air using anhydrous solvents or solvents treated with an appropriate drying reagent. A THF solution of Li2[PdCl4] was prepared by mixing of PdCl2 and approximate 2 equiv of LiCl at room temperature.8 The fluorinated β-ketoimine ligands were prepared from the reaction of hexafluoroacetylacetone with amines using montmorillonite K10 as catalyst.9 These β-ketoimine molecules include: (keim1)H = CF3C(O)CH2C(CF3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMe, (keim2)H = CF3C(O)CH2C(CF3)NBun and (keim3)H = CF3C(O)CH2C(CF3)N(CH2)2OMe. The second fluorinated imino chelate ligands were similarly prepared from direct condensation of amines with a fluorinated keto-alcohol HOC(CF3)2CH2C(Me)O, the latter was prepared according to the literature.10 These imino-alcohol ligands are listed as the following: (hfda1)H = HOC(CF3)2CH2C(Me)NMe, (hfda2)H = HOC(CF3)2CH2C(Me)NBun and (hfda3)H = HOC(CF3)2CH2C(Me)N(CH2)2OMe.
NMe, (keim2)H = CF3C(O)CH2C(CF3)NBun and (keim3)H = CF3C(O)CH2C(CF3)N(CH2)2OMe. The second fluorinated imino chelate ligands were similarly prepared from direct condensation of amines with a fluorinated keto-alcohol HOC(CF3)2CH2C(Me)O, the latter was prepared according to the literature.10 These imino-alcohol ligands are listed as the following: (hfda1)H = HOC(CF3)2CH2C(Me)NMe, (hfda2)H = HOC(CF3)2CH2C(Me)NBun and (hfda3)H = HOC(CF3)2CH2C(Me)N(CH2)2OMe.
Identification of the as-deposited Pd thin films was carried out using X-ray diffraction (XRD) with Cu Kα radiation. Scanning electron microscopy (SEM) was recorded on a Hitachi S-4000 system. The electrical resistivity onf the films was measured by a four-point probe method at room temperature, for which the instrument is assembled using Keithley 2182 nanovoltmeter and a Keithley 2400 constant current source. The elemental composition was determined by X-ray photoelectron spectroscopy (XPS) utilizing a Physical Electronics PHI 1600 system with an Al/Mg dual anode X-ray source, and the XPS spectra were collected after 1–2 min sputtering with argon at 4 keV until a constant composition was obtained.
Synthesis of 1a
A solution of Li2[PdCl4] was first prepared by mixing PdCl2 (0.37 g, 2.1 mmol) and LiCl (0.18 g, 4.2 mmol) in 40 mL of THF at room temperature. To this was added dropwise a second solution of K(keim1), which was in situ prepared from (keim1)H (1.0 g, 4.5 mmol) and KOH (0.30 g, 5.3 mmol) in 30 mL of THF. The mixture was stirred at 0 °C for 60 min until the color of the solution changed from wine-red to red–orange. The THF solvent was then removed under vacuum, the residue was dissolved in minimum amount of CH2Cl2, and the resulting solution was washed with de-ionized water three-times (50 mL × 3) to remove any unreacted ketoiminate reactant and water soluble co-products. The solvent was then removed under vacuum and the resulting oily residue purified by vacuum sublimation (500 mTorr, 55 °C), giving 0.63 g of orange–red complex Pd(keim1)2 (1a, 1.2 mmol, 55%). Single crystals suitable for XRD study were grown from a mixed solution of CH2Cl2 and heptane at room temperature.Spectral data of 1a: MS (FAB, 107Pd): m/z 546 (M+). 1H NMR (300 MHz, CDCl3, 298 K): δ 5.88 (s, CH, 2H), 3.28 (q, NCH3, JHF = 1.8 Hz, 6H). 13C NMR (125 MHz, CDCl3, 298 K): δ 162.8 (q, CN, 2JCF = 34 Hz, 2C), 154.6 (q, CO, 2JCF = 29 Hz, 2C), 117.3 (q, CF3, 1JCF = 281 Hz, 2C), 116.9 (q, CF3, 1JCF = 281 Hz, 2C), 90.8 (s, CH, 2C), 40.3 (s, NCH3, 2C). 19F NMR (470.3 MHz, CDCl3, 298 K): δ −64.4(s, CF3, 6F), −73.1(s, CF3, 6F). Anal. Calcd for C12H10F12N2O2Pd: C, 30.28; H, 2.54; N, 4.41. Found: C, 30.26; H, 2.54; N, 4.40.
Synthesis of 1b
In a similar fashion to 1a, 0.34 g of PdCl2 (1.9 mmol), 0.17 g of LiCl (4.0 mmol), 1.06 g of ketoiminate ligand (keim2)H (4.0 mmol) and 0.25 g of KOH (4.5 mmol) were used. After carrying out the routine workup procedures, vacuum sublimation (470 mTorr, 60 °C) gave an orange–red solid Pd(keim2)2 (1b, 0.77 g, 1.2 mmol) in 64% yield.Spectral data of 1b: MS (FAB, 107Pd): m/z 630 (M+). 1H NMR (400 MHz, CDCl3, 298 K): δ 5.58 (s, CH, 2H), 3.54 (t, NCH2, 3JHH = 7.6 Hz, 4H), 1.74 (m, NCH2CH2, 3JHH = 7.6 Hz, 4H), 1.33 (m, CH2CH3, 3JHH = 7.6 Hz, 4H), 0.92 (t, CH3, 3JHH = 7.6 Hz, 6H). 13C NMR (125 MHz, CDCl3, 298 K): δ 162.7 (q, CN, 2JCF = 34 Hz, 2C), 153.4 (q, CO, 2JCF = 29 Hz, 2C), 117.3 (q, CF3, 1JCF = 286 Hz, 2C), 116.9 (q, CF3, 1JCF = 282 Hz, 2C), 91.0 (s, CH, 2C), 51.8 (s, NCH2, 2C), 35.6 (s, NCH2CH2, 2C), 20.0 (s, CH2CH3, 2C), 13.7 (s, CH2CH3, 2C). 19F NMR (470.3MHz, CDCl3, 298 K): δ −63.1(s, CF3, 6F), −72.9(s, CF3, 6F). Anal. Calcd for C18H20F12N2O2Pd: C, 34.27; H, 3.20; N, 4.44. Found: C, 34.38; H, 3.31; N, 4.76.
Synthesis of 1c
Procedures identical to that of 1a were followed, using 0.35 g of PdCl2 (2.0 mmol), 0.18 g of LiCl (4.2 mmol), 1.10 g of ketoiminate ligand (keim3)H (4.1 mmol) and 0.26 g of KOH (4.6 mmol). After carrying out the routine workup procedures, vacuum sublimation (480 mTorr, 62 °C) gave an orange–red solid Pd(keim3)2 (1c, 0.88 g, 1.4 mmol) in 70% yield. Single crystals suitable for XRD study were grown from a mixed solution of CH2Cl2 and heptane at room temperature.Spectral data of 1c: MS (FAB, 107Pd): m/z 634 (M+). 1H NMR (300 MHz, CDCl3, 298 K): δ 5.88 (s, CH, 2H), 3.79 (t, CH2OCH3, 3JHH = 5.1 Hz, 4H), 3.73 (t, NCH2, 3JHH = 5.1 Hz, 4H), 3.38 (s, OCH3, 6H). 13C NMR (125 MHz, CDCl3, 298 K): δ 163.2 (q, CN, 2JCF = 34 Hz, 2C), 155.0 (q, CO, 2JCF = 29 Hz, 2C), 117.1 (q, CF3, 1JCF = 286 Hz, 2C), 116.8 (q, CF3, 1JCF = 281 Hz, 2C), 91.5 (s, CH, 2C), 73.2 (s, CH2OCH3, 2C), 59.1 (s, NCH2, 2C), 51.1 (s, OCH3, 2C). 19F NMR (470.3 MHz, CDCl3, 298 K): δ −62.5(s, CF3, 6F), −73.0(s, CF3, 6F). Anal. Calcd for C16H16F12N2O4Pd: C, 26.37% H, 1.48% N, 5.12. Found: C, 26.36; H, 1.51; N, 5.14.
Synthesis of 2a
A solution of Li2[PdCl4] was first prepared by mixing PdCl2 (0.32 g, 1.8 mmol) and LiCl (0.17 g, 4.0 mmol) in 40 mL of THF at room temperature. To this was added dropwise a second solution of K(hfda1), which was in situ obtained by mixing (hfda1)H (0.9 g, 3.8 mmol) and KOH (0.23 g, 4 mmol) in 30 mL of THF. The reaction mixture was stirred at 0 °C for 60 min until the color of the solution changed from wine-red to turbid yellow. The THF solvent was then removed under vacuum, the residue dissolved in the minimum amount of CH2Cl2, and the resulting solution washed with de-ionized water three-times (50 mL × 3) to remove any excess unreacted iminoalcohol and water-soluble co-products. The solvent was removed under vacuum and the resulting solid material purified by vacuum sublimation (490 mTorr, 72 °C), giving 0.57 g of yellow complex Pd(hfda1)2 (2a, 1.0 mmol, 55%).Spectral data of 2a: MS (FAB, 107Pd): m/z 578 (M+). 1H NMR (300 MHz, CDCl3, 298 K): δ 3.24 (s, CH2, 4H), 3.20 (s, NCH3, 6H), 2.16 (s, CH3, 6H). 13C NMR (75 MHz, CDCl3, 298 K): δ 174.8 (s, CN, 2C), 123.7 (q, CF3, 1JCF = 290 Hz, 4C), 76.1 (m, CO, 2JCF = 28 Hz, 2C), 46.7 (s, CH2, 2C), 40.1 (s, NCH3, 2C), 22.1 (s, CH3, 2C). 19F NMR (470.3 MHz, CDCl3, 298 K): δ −76.5 (s, CF3, 12F). Anal. Calcd for C14H16F12N2O2Pd: C, 29.06; H, 2.79; N, 4.84. Found: C, 28.95; H, 2.82; N, 4.91.
Synthesis of 2b
Procedures identical to that of 2a were followed, using 0.38 g of PdCl2 (2.0 mmol), 0.19 g of LiCl (4.5 mmol), 1.19 g of the hfda ligand (hfda2)H (4.3 mmol) and 0.25 g of KOH (4.5 mmol). After removal of the solvent, vacuum sublimation (510 mTorr, 70 °C) gave a yellow solid Pd(hfda2)2 (2b, 1.0 g, 1.5 mmol) in 74% yield.Spectral data of 2b: MS (FAB, 107Pd): m/z 661 (M+). 1H NMR (500 MHz, CDCl3, 298 K): δ 3.46 (t, NCH2, 3JHH = 7.8 Hz, 4H), 3.19 (s, CH2, 4H), 2.16 (s, CH3, 6H), 1.76 (m, NCH2CH2, 3JHH = 7.8 Hz, 4H), 1.42 (m, CH2CH3, 3JHH = 7.8 Hz, 4H), 0.94 (t, CH2CH3, 3JHH = 7.8 Hz, 6H). 13C NMR (125 MHz, CDCl3, 298 K): δ 173.3 (s, CN, 2C), 123.7 (q, CF3, 1JCF = 291 Hz, 2C), 51.5 (s, NCH2, 2C), 46.9 (s, CH2, 2C), 31.3 (s, NCH2CH2, 2C), 21.9 (s, CH2CH3, 2C), 20.2(s, CH3, 2C), 14.0 (s, CH2CH3, 2C). 19F NMR (470.3 MHz, CDCl3, 298 K): δ −76.2 (s, CF3, 12F). Anal. Calcd for C20H28F12N2O2Pd: C, 36.24; H, 4.26; N, 4.23. Found: C, 36.18; H, 4.26; N, 4.57.
Synthesis of 2c
Procedures identical to that of 2a were followed, using 0.34 g of PdCl2 (1.9 mmol), 0.19 g of LiCl (4.5 mmol), 1.15 g of the hfda ligand (hfda3)H (4.1 mmol) and 0.26 g of KOH (4.6 mmol). After evaporation of the solvent, vacuum sublimation (520 mTorr, 76 °C) gave a yellow solid Pd(hfda3)2 (2c, 1.18 g, 1.8 mmol) in 92% yield. Single crystals suitable for XRD study were grown from a solution of CH2Cl2 and heptane at room temperature.Spectral data of 2c: MS (FAB, 107Pd): m/z 666 (M+). 1H NMR (300 MHz, CDCl3, 298 K): δ 3.87 (t, CH2OCH3, 3JHH = 4.8 Hz, 4H), 3.73 (t, NCH2, 3JHH = 4.8 Hz, 4H), 3.30 (s, OCH3, 6H), 3.20 (s, CH2, 4H), 2.34 (s, CH3, 6H). 13C NMR (75 MHz, CDCl3, 298 K): δ 177.0 (s, CN, 2C), 124.9 (q, CF3, 1JCF = 290 Hz, 4C), 78.2 (m, CO, 2JCF = 28 Hz, 2C), 72.2 (s, CH2OCH3, 2JCF = 28 Hz, 2C), 58.9 (s, NCH2, 2C), 51.2 (s, OCH3, 2C), 47.4 (s, CH2, 2C), 22.9(s, CH3, 2C). 19F NMR (470.3 MHz, CDCl3, 298 K): δ −76.4 (s, CF3, 12F). Anal. Calcd for C18H24F12N2O4Pd: C, 32.42; H, 3.63; N, 4.20. Found: C, 32.43; H, 3.63; N, 4.27.
X-Ray crystallography
XRD was carried out on a Nonius Kappa CCD diffractometer at room temperature. Intensities of the diffraction signals were corrected for Lorentz, polarization and absorption effects (ψ scans). The structure was solved using the NRCC-SDP-VAX package. All the non-hydrogen atoms had anisotropic temperature factors, while all hydrogen atoms were placed at the calculated positions with UH = UC + 0.1. The crystallographic refinement parameters of complexes 1c and 2c are summarized in Table 1.| Compound | 1c | 2c |
|---|---|---|
| Formula | C16H16F12N2O4Pd | C18H24F12N2O4Pd |
| M W | 634.71 | 666.79 |
| Temperature/K | 150(1) | 295(2) |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 8.7507(2) | 5.8906(1) |
| b/Å | 9.4424(2) | 10.2740(2) |
| c/Å | 13.7058(2) | 10.9422(1) |
| α/° | 74.0139(9) | 77.307(1) |
| β/° | 86.8875(10) | 89.906(1) |
| γ/° | 81.3120(10) | 75.714(1) |
| Volume/Å3; Z | 1076.09(4); 2 | 625.09(2); 1 |
| D c/g cm−3 | 1.959 | 1.771 |
| μ(Mo-Kα)/mm−1 | 0.993 | 0.859 |
| Reflections collected | 26095 | 8392 |
| Independent reflections | 4952 (Rint = 0.0498) | 2874 (Rint = 0.0278) |
| Data/restraints/parameters | 4952/0/344 | 2874/0/170 |
| Goodness-of-fit on F2 | 1.087 | 1.061 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0323, wR2 = 0.0792 | R 1 = 0.0262, wR2 = 0.0630 |
| R indices (all data) | R 1 = 0.0428, wR2 = 0.0859 | R 1 = 0.0273, wR2 = 0.0636 |
CCDC reference numbers 192599 and 192600.
See http://www.rsc.org/suppdata/jm/b2/b208535f/ for crystallographic data in CIF or other electronic format.
CVD procedures
The thermal CVD reactions were carried using a horizontal hot-wall Pyrex reactor, consisting of a Pyrex tube of 25 mm internal diameter, placed within a tube furnace, as described in Scheme 2. The precursor was loaded in the glass container. The carrier gas was introduced through the sidearm of the container and became saturated with vapors of palladium precursors before entering the hot zone, which is approximately 25 cm in length. The Si substrates were cleaned using the RCA cleaning method, while SiO2 substrates were placed in dilute HF solution for 12 h, then washed with de-ionized water, and dried at 120 °C for 10 min. The flow rate of O2 carrier gas was adjusted to 30 sccm and the deposition time was kept at approximately 3 h. The deposition time was obtained by visually observing the time for complete consumption of the source compound.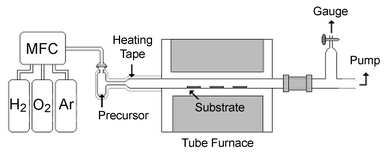 | ||
| Scheme 2 | ||
Results and discussion
Synthesis and characterization of Pd complexes
The synthesis of β-ketoimine ligands involved direct condensation of hexafluoroacetylacetone with a primary amine H2NR (R = Me, Bun and methoxyethyl), conducted in a CHCl3 solution using montmorillonite K10 as solid catalyst. The CF3 substituted β-ketoimine ligands were obtained in 60–85% yield after reduced pressure distillation or vacuum sublimation. Formation of ammonium salt [hfac][NH3R],11 a kinetic product obtained upon addition of amine into the solution of hexafluoroacetylacetone, was not detected using this modified approach. The corresponding imino-alcohols (hfda)H were similarly prepared from treatment of fluorinated keto-alcohols with amines H2NR in the presence of montmorillonite K10. These chemical transformations are best indicated in Scheme 3. | ||
| Scheme 3 | ||
For preparation of the Pd complexes, either the β-ketoimine ligand or the imino-alcohol ligand was first converted to the potassium saltine complex by treatment with excess of KOH in THF. Mixing this solution with a second solution of Li2[PdCl4] would easily afford the anticipated Pd complexes Pd(keim)2 or Pd(hfda)2. These reactions were typically conducted at 0 °C for approximately 60 min. After the standard workup routines, vacuum sublimation gave a series of the expected Pd metal complexes 1a–1c and 2a–2c in good yields (Scheme 1).
Single crystal XRD of complexes 1c and 2c were then carried out to establish their solid-state geometries. The molecular structure of 1c is shown in Fig. 1 and the bond distances and angles are given in Table 2. The Pd atom adopts a square-planar geometry, on which each of the ketoiminate chelates forms a distorted hexagonal arrangement and is essentially planar. This may well be accounted for by an extended conjugation of the π-orbitals along the ketoiminate backbone atoms. The bond distances along the backbone indicated that the O–C bonds and N–C bonds are in the range 1.282–1.286(3)
Å and 1.297–1.300(3)
Å, while the C–C bonds adjacent to the O–C vectors (cf. C(2)–C(3)
= 1.357(4)
Å and C(7)–C(8)
= 1.358(4)
Å) are shortened (∼0.068 Å) with respect to the second C–C bonds that resided next to the imino N–C fragments (cf. C(3)–C(4)
= 1.425(3)
Å and C(8)–C(9)
= 1.425(4)
Å). These observations are in good agreement with formation of a non-delocalized π-interaction on the ketoiminate backbone, for which the C–C double bond is located adjacent to the oxygen atom, and the second C–C linkage, which is next to the imino CN fragment, adopts the single-bond interaction. For comparison, a similar non-delocalized β-ketoiminate bonding interaction was observed in two other Pd complexes, such as [Pd{N(C6H4Br-o)C(Me)CHC(Me)O}2]·C6H612 and [Pd(π-methallyl)(phenylethylimino-3-pentene-4-olate)].13
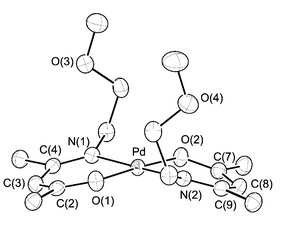 | ||
| Fig. 1 ORTEP drawing of complex 1c with thermal ellipsoids shown at 50% probability level, fluorine atoms of the CF3 substituents are removed for clarity. | ||
| Pd–O(1) | 1.974(2) | Pd–O(2) | 1.986(2) |
| Pd–N(1) | 2.041(2) | Pd–N(2) | 2.047(2) |
| N(1)–C(4) | 1.300(3) | O(1)–C(2) | 1.282(3) |
| C(2)–C(3) | 1.357(4) | C(3)–C(4) | 1.425(3) |
| N(2)–C(9) | 1.297(3) | O(2)–C(7) | 1.286(3) |
| C(7)–C(8) | 1.358(4) | C(8)–C(9) | 1.425(4) |
| ∠O(1)–Pd–N(1) | 92.35(8) | ∠O(2)–Pd–N(2) | 92.02(8) |
Moreover, it is notable that both of the chelate skeletons are tilting in the same direction with the intersecting angle between the Pd–O(1)–N(1)–C(2)–C(3)–C(4) and the Pd–O(2)–N(2)–C(7)–C(8)–C(9) least-square hexagons being 158°, giving a virtually curved molecular structure that has no precedence for the related Pd chelate complexes. Fig. 2 shows the projections of the three-dimensional packing arrangement along the b axis. It appears that the bending of the basal skeleton is related to the long alkyl chain attached to the imino functional group, as the alkyl chains are found to point in the same direction and are away from the basal plane. As a result, each individual molecule is packed in an alternated layer structure, composed of a parallel arrangement of alkyl chains and perpendicularly aligned double basal planes. Moreover, the observed curvature is a natural consequence of the face-to-face and back-to-back stacking of the molecules, for which the CF3 substituents of one ketoiminate ligand are inserted into the concavity near the Pd metal atom of the adjacent complex, which then gave the observed inter-locking arrangement as well as the inward distortion involving the basal plane. The hexagonal chelate skeleton of adjacent molecules are well separated from each other by ∼3.62 Å, which is larger than the interlayer distance of graphite (3.35 Å), indicating a weaker π–π interaction. For a direct comparison, no such distortion induced by crystal packing was observed in the β-diketonate complexes such as Pd(tmhd)2, tmhd = 2,2,6,6-tetramethylheptane-3,5-dionate, for which the basal plane shows an essentially planar arrangement.14
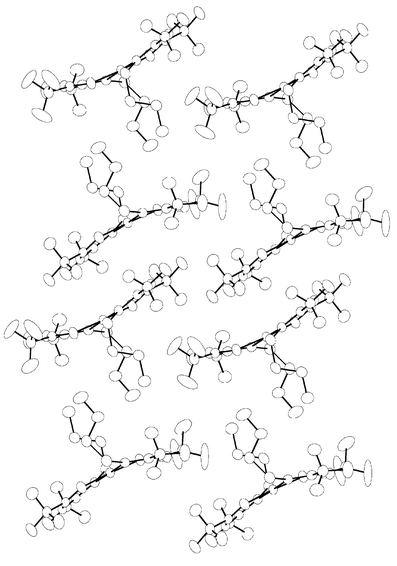 | ||
| Fig. 2 Crystal packing for complex 1c along the b axis of the unit cell. | ||
The crystal structure of 2c is shown in Fig. 3 and the corresponding bond distances and angles are presented in Table 3. In contrast to that of 1c, this molecule consists of a square-planar geometry in the usual all trans-PdN2O2 fashion, and the Pd atom is lying on a crystallographic center of inversion. The imino-alcoholate ligand adopts a boat configuration, with the methylene groups C(5) and C(5A) being located at the opposite sides of the PdN2O2 plane. Moreover, the Pd–O and Pd–N distances are longer than those observed in 1c with a margin of ∼0.01 Å, showing the existence of the reduced ligand-to-metal bonding interactions. The imino N(1)–C(4) bond of the chelate is found to be 1.285(3)
Å, showing an intermediate value with respect to those of 1c
(1.297–1.300(3)
Å) and the NC double bonds detected in the imino Cu complexes (1.264–1.274 Å).15
 | ||
| Fig. 3 ORTEP drawing of complex 2c with thermal ellipsoids shown at 30% probability level, fluorine atoms of the CF3 substituents are removed for clarity. | ||
| Pd–N(1) | 2.053(2) | Pd–O(2) | 2.008(1) |
| N(1)–C(3) | 1.481(3) | N(1)–C(4) | 1.285(3) |
| O(2)–C(6) | 1.375(3) | C(4)–C(5) | 1.504(3) |
| C(5)–C(6) | 1.559(3) | ||
| ∠N(1)–Pd–O(2) | 90.16(6) | ∠O(2)–Pd–O(2A) | 180 |
| ∠N(1)–Pd–O(2A) | 89.83(6) |
The volatility and thermal stability of complexes 1a, 1b, 2a and 2c were then investigated by TGA under an atmospheric pressure of N2 (Fig. 4). The ketoiminate Pd complexes 1a and 1c are more volatile than the corresponding imino-alcoholate complexes. This is revealed by a rapid loss of weight, which started at 100 °C and ended at about 200 °C for 1a and 1c; while the on-set temperatures for the imino-alcoholate complexes 2a and 2c initiates at least 50 °C higher. This notable difference is probably due to the more even distribution of the CF3 substituents on the ketoiminate backbone, which can effectively reduce the intermolecular van der Waals interactions and hence increase the volatility with respect to the imino-alcoholate complexes 2a and 2c, for which both the CF3 groups are gathered in an adjacent area to the alkoxide groups, showing a situation similar to that observed in the related Sr and Ba fluoroalkoxide complexes.16 Moreover, all of these Pd complexes gave residue weights ≤3 wt.% upon raising the temperature to 450 °C. This is in good agreement with the effective conversion from solid to gaseous phase. Other physical properties relevant to CVD studies, such as melting points, deposition temperature and T1/2, which is the temperature at which 50 wt.% of sample was lost during TGA runs, are all listed in Table 4. These systematic trends allow modification of the physical properties by simply changing the alkyl substituent of the imino functional group. In particular, we can systematically lower the melting point of the samples, so that the resulting Pd source complexes would be utilized as liquid precursors, taking advantage of their more reproducible and steady rates of evaporation during actual CVD runs.17
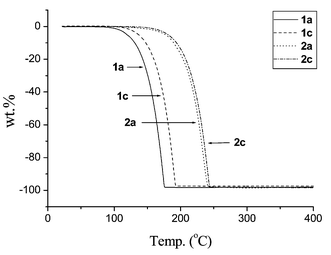 | ||
| Fig. 4 TGA data; all experiments were carried out at atmospheric pressure with N2 as carrier (100 sccm) and a heating rate of 10 °C min−1. | ||
| Entry | Compound | mp/°C | Dec./°C | T 1/2/°Ca | Residueb (%) |
|---|---|---|---|---|---|
| a The temperature at which 50 wt.% of the sample has been lost during TGA analysis (heating rate = 10 °C min−1 and N2 flow rate = 100 cm3 min−1). b Total wt.% of the sample observed at 400 °C during TGA analysis. c This complex exhibits a decomposition over a wide temperature range starting from 280 °C. | |||||
| 1a | Pd(keim1)2 | 110–112 | 190 | 177 | 1.9 |
| 1b | Pd(keim2)2 | 56–57 | 190 | 180 | 0.3 |
| 1c | Pd(keim3)2 | 101–103 | 160 | 160 | 2.7 |
| 2a | Pd(hfda1)2 | 267–269 | c | 224 | 1.9 |
| 2b | Pd(hfda2)2 | 132–133 | 175 | 207 | 3.4 |
| 2c | Pd(hfda3)2 | 129–130 | 204 | 227 | 1.6 |
CVD of Pd thin films
The CVD experiments were conducted in a horizontal hot wall reactor using Pd complexes 1c and 2c at approximately 300 mTorr. Other complexes were not used for the CVD experiments as we expected that their behaviors would be more or less identical. All experiments were carried out at temperatures of 250–350 °C, and precursor 1c was kept at 80 °C while the less volatile 2c was kept at 100 °C to ensure an equal evaporation rate. For the deposition experiments carried out under vacuum or using argon as the carrier gas, no thin film deposition was observed at the substrate temperature up to 400 °C, and most of the source complex passed the deposition zone without giving metal deposition. The carrier gas was then changed to H2 in an attempt to lower the deposition temperature; however, this modification caused serious decomposition of the source reagent before it entered the deposition zone. This was indicated by the observation of a black residual solid in the source reservoir and a thin coating of Pd metal along the glass connecting tube, even though the temperatures were maintained as low as 80–100 °C. We believed that the poor thermal stability is caused by the rapid hydrogenolysis of the Pd complexes, which would liberate free ligands and cause metal deposition to occur at lower temperatures.18 Oxygen was then selected as the carrier gas to suppress this premature decomposition before volatilization and subsequent transportation into the deposition zone. This is because the Pd source compounds appeared to be more stable under an O2 atmosphere at temperatures below 100 °C, and because the Pd-catalyzed oxidation occurred at higher temperatures and would ensure the suppression of carbon impurities.19For the experiments carried out using O2 carrier gas, all the as-deposited thin films showed a silver-like metal cast and were lustrous. They gave good adhesion to Si wafers but the thin films would come off more easily while deposited on Pyrex glass or on SiO2 substrates. A summary of the experimental parameters, together with analytical data for the Pd metal thin films, is given in Table 5, and the corresponding SEM images are presented in Fig. 5. A comparison of the SEM photographs suggests an apparent variation according to the deposition temperatures.
 | ||
| Fig. 5 SEM micrographs of Pd films: films (1) (2) and (3) are deposited at 250, 300 and 350 °C, respectively, using precursor 1c; whereas films (4) (5) and (6) are deposited using precursor 2c under similar conditions. | ||
| Thin film (source cpd) | Gas (O2) flow rate/sccm | T S/°C | T D/°C | P S/Torr | Thickness/Å | DR/Å min−1 | Resistivity ρ/µΩ cm | Content (at.%) |
|---|---|---|---|---|---|---|---|---|
| a T S, source temperature; TD, deposition temperature; PS, system pressure; DR, deposition rate. | ||||||||
| 1 (1c) | 30 | 80 | 250 | 0.3 | 1510 | 8.4 | 41 | Pd, ∼96%; C, ∼4%. |
| 2 (1c) | 30 | 80 | 300 | 0.3 | 1680 | 9.3 | 40 | Pd, ∼97%; C, ∼3%. |
| 3 (1c) | 30 | 80 | 350 | 0.3 | 2350 | 13.1 | 30 | Pd, ∼96%; C, ∼4%. |
| 4 (2c) | 30 | 100 | 250 | 0.3 | 550 | 3.1 | 36 | Pd, ∼92%; C, ∼8%. |
| 5 (2c) | 30 | 100 | 300 | 0.3 | 1420 | 7.9 | 27 | Pd, ∼98%; C, ∼2%. |
| 6 (2c) | 30 | 100 | 350 | 0.3 | 1580 | 8.8 | 19 | Pd, ∼99%; C, ∼1%. |
The film deposited using 1c as the source reagent at the lower temperature of 250 °C shows a thickness of 1510 Å with a relatively smooth top surface consisting of aggregates of very small crystallites. Upon increasing the temperature to 300 °C, the grains on the top surface turn much larger and the thickness increases to 1680 Å. It also shows a substantial increase of crystallite sizes, while the thickness become 2350 Å upon further increasing temperature to 350 °C. The electrical resistivities were measured using the four-point probe method. The observed resistivities were in the range 41–30 µΩ cm, showing data that were only 3–4 times greater than that of bulk metal standard (11 µΩ cm). This discrepancy may be attributed to the incorporation of carbon impurities and, in part, to the poor contact between the grains that appeared during SEM analysis.
The as-deposited Pd thin films were examined by XPS to determine their compositional ratio, particularly the content of carbon impurity. A typical XPS spectrum of the Pd thin film deposited at TD = 300 °C is illustrated in Fig. 6. The Pd 3d5/2 peak located at a binding energy (Eb) of 335.2 eV was used to measure the content of Pd metal composition, for which the observed Eb agrees with the literature value of 335.1–335.4 eV.20 The presence of carbon impurities was clearly evidenced by the detection of a weak C 1s signals at Eb 284.2, which was in the range expected for graphitic carbon. Thus, a slow scanning over these regions of interest, followed by least-square curve fitting, was utilized to estimate the relative contents of palladium and carbon. The atomic contents of the thin films calculated using the XPS data are summarized in Table 5. Moreover, no obvious peak was observed between the higher Eb region 690–800 eV, in which a peak due to the fluorine atom would appear at Eb ∼ 684.5 eV, while the oxygen Auger peak would instead occur at Eb ∼ 745 eV, thus we can unambiguously rule out the occurrence of both fluorine and oxygen contaminants within the sputtered thin film. This observation was further confirmed by a parallel AES analysis, which provides unambiguous evidence to eliminate the co-deposition of oxide PdO, even though the CVD experiments were conduced under O2 atmosphere.
 | ||
| Fig. 6 XPS spectrum of the Pd thin film deposited at 300 °C using precursor 1c. | ||
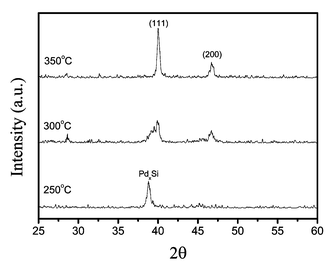
The X-ray diffraction patterns of the Pd films are depicted in Fig. 7. At 350 °C, formation of Pd metal is confirmed using the main diffraction signals of the Pd standard, which are derived from diffraction signals involving the (111) and (200) planes. Their intensity exhibits an approximate ratio of 18 ∶ 1, which is distinctive from the ideal ratio of 5 ∶ 3 reported for the bulk Pd standard, showing the existence of a preferred (111) orientation. No diffraction peak was detected at position 2θ = 33.9°, the main signal that would arise from the (101) planes of PdO.21 Upon lowering the deposition temperature to 300 °C, the preferred (111) texture was found to remain the same, but both the (111) and (200) diffraction signals turned much broader, indicating the formation of much smaller grain sizes, which was consistent with the SEM observation. For the thin film deposited at the lowest limiting temperature TD = 250 °C, the XRD spectrum shows a third signal centered at 2θ = 39.2°, in addition to the previously identified diffractions arising from the (111) and (200) planes of the Pd metal. Identification of this new diffraction signal is unclear at this moment, but it may be attributable to the Pd-rich metal silicides, such as PdxSi x = 2–5, at the interface between the Pd layer and the underneath Si wafer. It was reported that the formation of a Pd2Si thin film would occur at a temperature as low as 160–220 °C by a solid-state reaction between the Si (111) substrate and the Pd overlayer.22 Thus, it is reasonable to expect that the excessive deposition time, i.e. 3 h for each experiment, would facilitate a similar solid-state reaction between the Pd layer and the Si substrate. Moreover, as the Pd thin film deposited at 250 °C is relatively thin, the effective probing depth of the XRD method would allow us to detect this metal silicide component, even though it was buried under a layer of Pd metal. For the depositions conducted at 300–350 °C, the much thicker Pd layer as well as the better crystallinity would enhance the Pd diffraction signals and, thus make the PdxSi diffraction signal less observable.
 | ||
| Fig. 7 XRD pattern of the Pd films deposited using precursor 1c. | ||
Pd metal depositions using 2c as the source reagent were also conducted under similar conditions. The exact CVD experimental parameters, the SEM photographs that showed the surface morphology as well as the XRD pattern of the as-deposited thin films are depicted in Table 5, Fig. 5 and Fig. 8, respectively. The thin film deposited at 250 °C gave a thickness of only 550 Å and a carbon impurity content as high as 8 at.%. Upon increasing the deposition temperature to 300 °C and then to 350 °C, the film thickness and grain sizes of the thin film increase as expected, which is revealed by SEM, and the content of carbon impurities dropped to the lowest level of 1 at.%, which was indicated by routine XPS determination. Moreover, XRD analysis showed the occurrence of a broad peak centered at 2θ = 38.7° for the thin film deposited at the lower limiting temperature of 250 °C, which was again attributed to the formation of metal silicides as discussed in the previous section. It is notable that no diffraction signal due to metallic Pd was observed, showing that the Pd constituent within the thin film is virtually amorphous. At 300 °C, XRD indicated the suppression of the peak due to the silicides PdxSi and the emergence of Pd (111) and (200) diffraction signals. Once the deposition temperature was increased to 350 °C, only the Pd (111) and (200) signals were observable, confirming the successful deposition of the Pd thin film with the preferred (111) orientation..
 | ||
| Fig. 8 XRD pattern of the Pd films deposited using precursor 2c. | ||
Conclusion
Pd complexes with two β-ketoiminate ligands or with two imino-alcoholate ligands were prepared. These metal complexes showed enhanced thermal and chemical stabilities, which were nearly identical to the properties of the fluorinated β-diketonate complex Pd(hfac)2, but which were superior to the non-fluorinated analogue Pd(acac)2.23 TGA experiments confirmed that all of them can be fully vaporized in the temperature range 150–250 °C. For structural characterization, the β-ketoiminate complex 1c showed a bent basal plane consisting of two chelate hexagons, which was probably induced by a stacking interaction of the individual molecules in the solid state. In contrast, the imino-alcoholate complex 2c showed a regular chelate conformation, in which the alkyl substituent on each of the imino nitrogen atoms is pointing in the opposite direction. Deposition of a Pd thin film was possible using source complexes 1c and 2c as well as a reactive O2 carrier gas. The composition and texture (amorphous or polycrystalline) of the thin films can be engineered by varying the deposition temperature or by choice of the CVD source reagent. A Pd thin film with the preferred (111) orientation was obtained on a Si substrate at 350 °C using 1c as the source reagent. This implies that both complexes 1c and 2c are suitable for the preparation of Pd-containing alloys or for depositing metal-oxide thin films with lower carbon content, using the recently developed concepts involving catalyst-enhanced chemical vapor deposition methods.24Acknowledgements
We thank the National Science Council and the Ministry of Education for financial support (NSC 91-2113-M-007-006) and (MOE program 89-FA04-AA).References
- (a) E. Feurer and H. Suhr, Thin Solid Films, 1988, 157, 81 CrossRef CAS; (b) R. R. Thomas and J. M. Park, J. Electrochem. Soc., 1989, 136, 1661 CAS; (c) V. Bhaskaran, M. J. Hampden-Smith and T. T. Kodas, Chem. Vap. Deposition, 1997, 3, 85 CAS; (d) W. Lin, T. H. Warren, R. G. Nuzzo and G. S. Girolami, J. Am. Chem. Soc., 1993, 115, 11644 CrossRef CAS; (e) W. Lin, R. G. Nuzzo and G. S. Girolami, J. Am. Chem. Soc., 1996, 118, 5988 CrossRef CAS; (f) N. L. Jeon, W. Lin, M. K. Erhardt, G. S. Girolami and R. G. Nuzzo, Langmuir, 1997, 13, 3833 CrossRef CAS.
- (a) C. Dossi, R. Psaro, A. Bartsch, E. Brivio, A. Galasco and P. Losi, Catal. Today, 1993, 17, 527 CrossRef CAS; (b) L. Sordelli, G. Martra, R. Psaro, C. Dossi and S. Coluccia, J. Chem. Soc., Dalton Trans., 1996, 765 RSC; (c) J.-C. Hierso, R. Feurer, J. Poujardieu, Y. Kihn and P. Kalck, J. Mol. Catal. A: Chem., 1998, 135, 321 CrossRef CAS; (d) J.-C. Hierso, P. Serp, R. Feurer and P. Kalck, Appl. Organomet. Chem., 1998, 12, 161 CrossRef CAS; (e) J.-C. Hierso, R. Feurer and P. Kalck, Coord. Chem. Rev., 1998, 178–180, 1811 CrossRef CAS; (f) P. Atanasova, J. Wise, M. Fallbach, T. Kodas and M. Hampden-Smith, Stud. Surf. Sci. Catal., 1998, 118, 73 Search PubMed.
- (a) W. L. Gladfelter, Chem. Mater., 1993, 5, 1372 CrossRef CAS; (b) R. J. Puddephatt, Polyhedron, 1994, 13, 1233 CrossRef CAS.
- (a) J. E. Gozum, D. M. Pollina, J. A. Jensen and G. S. Girolami, J. Am. Chem. Soc., 1988, 110, 2688 CrossRef CAS; (b) Z. Yuan, D. Jiang, S. J. Naftel, T.-K. Sham and R. J. Puddephatt, Chem. Mater., 1994, 6, 2151 CrossRef CAS.
- (a) Z. Yuan and R. J. Puddephatt, Adv. Mater., 1994, 6, 51 CrossRef CAS; (b) Z. Yuan and R. J. Puddephatt, Chem. Vap. Deposition, 1997, 3, 81; (c) Y. Zang, Z. Yuan and R. J. Puddephatt, Chem. Mater., 1998, 10, 2293 CrossRef.
- Y.-L. Tung, W.-C. Tseng, C.-Y. Lee, Y. Chi, S.-M. Peng and G.-H. Lee, Organometallics, 1999, 18, 864 CrossRef CAS.
- (a) J. A. Samuels, E. B. Lobkovsky, W. E. Streib, K. Folting, J. C. Hoffman, J. W. Zwanziger and K. G. Caulton, J. Am. Chem. Soc., 1993, 115, 5093 CrossRef CAS; (b) J. A. Samuels, K. Folting, J. C. Huffman and K. G. Caulton, Chem. Mater., 1995, 7, 929 CrossRef CAS; (c) Y. Chi, S. Ranjan, P.-W. Chung, C.-S. Liu, S.-M. Peng and G.-H. Lee, J. Chem. Soc., Dalton Trans., 2000, 343 RSC.
- H. Takahasi, K. Ohe, S. Uemura and N. Sugita, J. Organomet. Chem., 1988, 350, 227 CrossRef CAS.
- M. E. F. Braibante, H. S. Braibante, L. Missio and A. Andricopulo, Synthesis, 1994, 898 CrossRef CAS.
- (a) J. W. L. Martin and C. J. Willis, Can. J. Chem., 1977, 55, 2459 CAS; (b) I.-S. Chang and C. J. Willis, Can. J. Chem., 1977, 55, 2465 CAS.
- H.-K. Shin, M. J. Hampden-Smith, T. T. Kodas and A. L. Rheingold, J. Chem. Soc., Chem. Commun., 1992, 217 RSC.
- H. Lida, Y. Yuasa and C. Kibayashi, J. Chem. Soc., Dalton Trans., 1981, 2212 RSC.
- R. Claverini, P. Ganis and C. Pedone, J. Organomet. Chem., 1973, 50, 327 CrossRef CAS.
- G. J. Baker, J. B. Raynor, J. M. M. Smits, P. T. Beurskens, H. Vergoossen and C. P. Keijzers, J. Chem. Soc., Dalton Trans., 1986, 2655 RSC.
- (a) J. H. Timmons, J. W. L. Martin, A. E. Martell, P. Rudolf, A. Clearfield, S. J. Loeb and C. J. Willis, Inorg. Chem., 1981, 20, 181 CrossRef CAS; (b) J. H. Timmons, J. W. L. Martin, A. E. Martell, P. Rudolf, A. Clearfield and R. C. Buckley, Inorg. Chem., 1981, 20, 3056 CrossRef CAS.
- (a) Y. Chi, S. Ranjan, T.-Y. Chou, C.-S. Liu, S.-M. Peng and G.-H. Lee, J. Chem. Soc., Dalton Trans., 2001, 2462 RSC; (b) Y. Chi, S. Ranjan, P.-W. Chung, H.-Y. Hsieh, S.-M. Peng and G.-H. Lee, Inorg. Chim. Acta, 2002, 334, 172 CrossRef CAS.
- (a) F. Maury, J. Phys. IV, 1995, C5, 449 Search PubMed; (b) F. Maury, Chem. Vap. Deposition, 1996, 2, 113 CAS.
- (a) J.-C. Hierso, R. Feurer and P. Kalck, Chem. Mater., 2000, 12, 390 CrossRef CAS; (b) A. E. Kaloyeros, A. Feng, J. Garhart, K. C. Brooks, S. K. Ghosh, A. N. Saxena and F. Luehrs, J. Electron. Mater., 1990, 19, 271 Search PubMed; (c) N. S. Borgharkar and G. L. Griffin, J. Electrochem. Soc., 1998, 145, 347 CAS; (d) F.-J. Lee, Y. Chi, P.-F. Hsu, T.-Y. Chou, C.-S. Liu, S.-M. Peng and G.-H. Lee, Chem. Vap. Deposition, 2001, 7, 99 CrossRef CAS; (e) Y. Chi, H.-L. Yu, W.-L. Ching, C.-S. Liu, Y.-L. Chen, T.-Y. Chou, S.-M. Peng and G.-H. Lee, J. Mater. Chem., 2002, 12, 1363 RSC.
- (a) J.-C. Hierso, C. Satto, R. Feurer and P. Kalck, Chem. Mater., 1996, 8, 2481 CrossRef CAS; (b) Y.-L. Chen, C.-S. Liu, Y. Chi, A. J. Carty, S.-M. Peng and G.-H. Lee, Chem. Vap. Deposition, 2002, 8, 17 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, PerkinElmer, Eden Prairie, MN, 1992 Search PubMed.
- JCPDS-International Center for Diffraction Data, PCPDFWIN v.1.30, 1997.
- (a) B. Coulman and H. Chen, J. Appl. Phys., 1986, 59, 3467 CrossRef CAS; (b) J. F. Chen and L. J. Chen, Mater. Chem. Phys., 1995, 39, 229 CrossRef CAS.
- (a) A. R. Siedle, R. A. Newmark and L. H. Pignolet, Inorg. Chem., 1980, 19, 2052 CrossRef CAS; (b) V. Cominos and A. Gavriilidis, Eur. Phys. J.: Appl. Phys., 2001, 15, 23 Search PubMed.
- (a) S. W.-K. Choi and R. J. Puddephatt, Chem. Mater., 1997, 9, 1191 CrossRef CAS; (b) Y. Zhang, S. W.-K. Choi and R. J. Puddephatt, J. Am. Chem. Soc., 1997, 119, 9295 CrossRef CAS; (c) K. D. Pollard and R. J. Puddephatt, Chem. Mater., 1999, 11, 1069 CrossRef CAS; (d) Y. Zhang and R. J. Puddephatt, Chem. Mater., 1999, 11, 148 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |